
An Electrochemical Approach to the Recovery of Metals Typical of Battery Waste

 , and
, and
Abstract
:1. Introduction
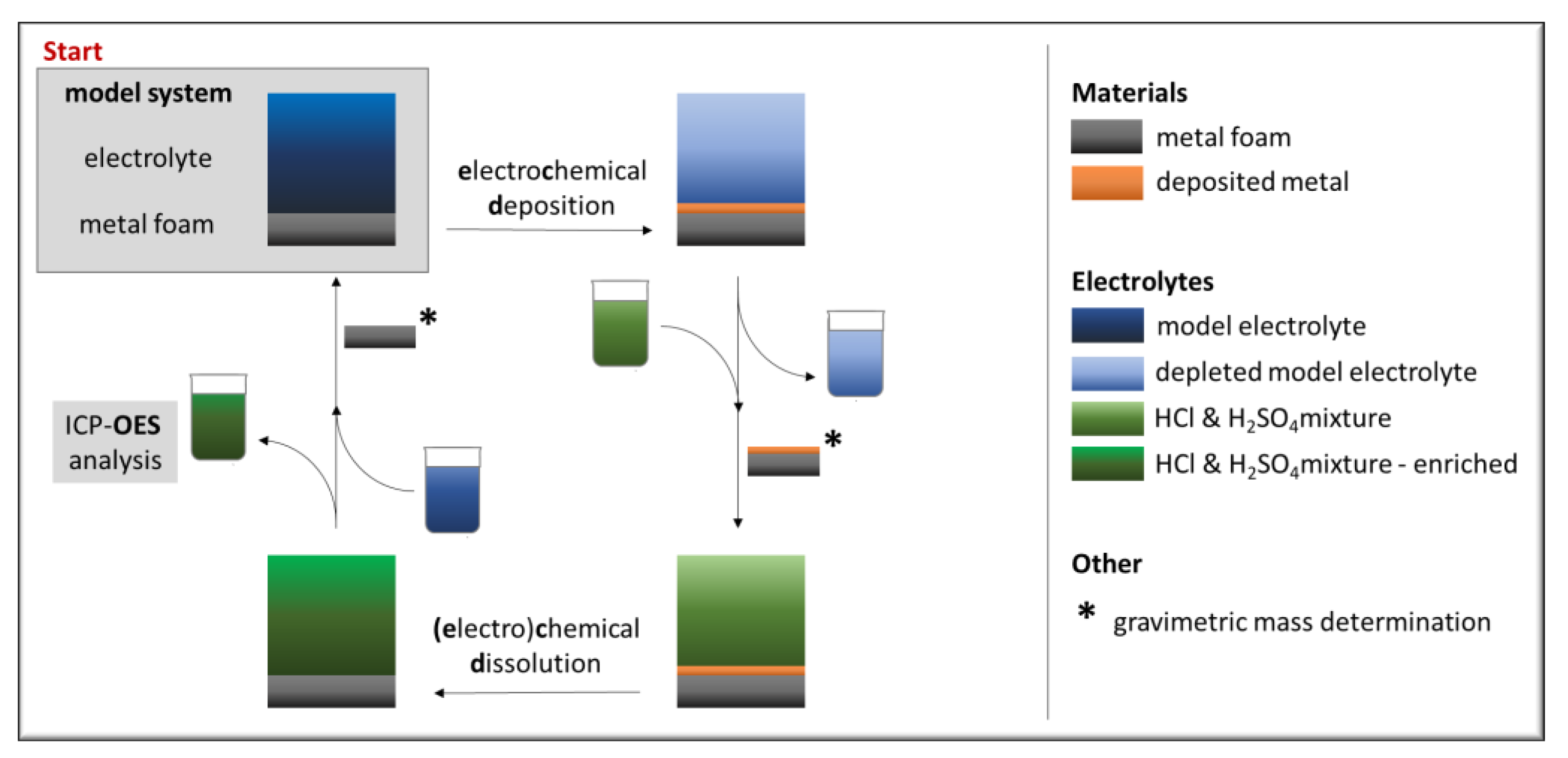
2. Materials and Methods
2.1. Electrolytes and Materials
2.2. Methods
3. Results
3.1. Stationary Electrochemical Deposition
3.2. Metal Recovery and Electrolyte Depletion
4. Conclusions
- Copper starts at 0 V (vs. SHE).
- Cobalt starts at −1.1 V (vs. SHE).
- Nickel starts at −1.4 V (vs. SHE).
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthes, S.; Grewe, V.; Sausen, R.; Roelofs, G.-J. Global impact of road traffic emissions on tropospheric ozone. Atmos. Chem. Phys. 2007, 7, 1707–1718. [Google Scholar] [CrossRef]
- Matthes, S. Globale Auswirkung des Straßenverkehrs auf die Chemische Zusammensetzung der Atmosphäre. Ph.D. Thesis, Ludwig-Maximilians-Universität, München, Germany, 2023. [Google Scholar]
- McDuffie, E.E.; Smith, S.J.; O’Rourke, P.; Tibrewal, K.; Venkataraman, C.; Marais, E.A.; Zheng, B.; Crippa, M.; Brauer, M.; Martin, R.V. A global anthropogenic emission inventory of atmospheric pollutants from sector- and fuel-specific sources (1970–2017): An application of the Community Emissions Data System (CEDS). Earth Syst. Sci. Data 2020, 12, 3413–3442. [Google Scholar] [CrossRef]
- Situ, L. Electric Vehicle Development: The Past, Present & Future. In Proceedings of the 3rd International Conference on Power Electronics Systems and Applications (PESA), Hong Kong, China, 20 May 2009; Available online: https://www.researchgate.net/publication/224585932_Electric_Vehicle_development_The_past_present_future (accessed on 3 September 2023).
- Harper, G.; Sommerville, R.; Kendrick, E.; Driscoll, L.; Slater, P.; Stolkin, R.; Walton, A.; Christensen, P.; Heidrich, O.; Lambert, S.; et al. Recycling lithium-ion batteries from electric vehicles. Nature 2019, 575, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.T.; Iyer-Raniga, U. Lithium-Ion Battery Recycling in the Circular Economy: A Review. Recycling 2022, 7, 33. [Google Scholar] [CrossRef]
- Doose, S.; Mayer, J.K.; Michalowski, P.; Kwade, A. Challenges in Ecofriendly Battery Recycling and Closed Material Cycles: A Perspective on Future Lithium Battery Generations. Metals 2021, 11, 291. [Google Scholar] [CrossRef]
- Mousa, E.; Hu, X.; Ånnhagen, L.; Ye, G.; Cornelio, A.; Fahimi, A.; Bontempi, E.; Frontera, P.; Badenhorst, C.; Santos, A.C.; et al. Characterization and Thermal Treatment of the Black Mass from Spent Lithium-Ion Batteries. Sustainability 2023, 15, 15. [Google Scholar] [CrossRef]
- Peschel, C.; van Wickeren, S.; Preibisch, Y.; Naber, V.; Werner, D.; Frankenstein, L.; Horsthemke, F.; Peuker, U.; Winter, M.; Nowak, S. Comprehensive Characterization of Shredded Lithium-Ion Battery Recycling Material. Chem. Eur. J. 2022, 28, e202200485. [Google Scholar] [CrossRef] [PubMed]
- Schwich, L.; Schubert, T.; Friedrich, B. Early-Stage Recovery of Lithium from Tailored Thermal Conditioned Black Mass Part I: Mobilizing Lithium via Supercritical CO2-Carbonation. Metals 2021, 11, 177. [Google Scholar] [CrossRef]
- Brückner, L.; Frank, J.; Elwert, T. Industrial Recycling of Lithium-Ion Batteries—A Critical Review of Metallurgical Process Routes. Metals 2020, 10, 1107. [Google Scholar] [CrossRef]
- Mohr, M.; Weil, M.; Peters, J.; Wang, Z. Recycling of Lithium-Ion Batteries. In Encyclopedia of Electrochemistry; Bard, A.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020. [Google Scholar] [CrossRef]
- Makuza, B.; Tian, Q.; Guo, X.; Chattopadhyay, K.; Yu, D. Pyrometallurgical options for re-cycling spent lithium-ion batteries: A comprehensive review. J. Power Sources 2021, 491, 229622. [Google Scholar] [CrossRef]
- Ciez, R.E.; Whitacre, J.F. Examining different recycling processes for lithium-ion batteries. Nat. Sustain. 2019, 2, 148–156. [Google Scholar] [CrossRef]
- Asadi Dalini, E.; Karimi, G.; Zandevakili, S.; Goodarzi, M. A Review on Environ-mental, Economic and Hydrometallurgical Processes of Recycling Spent Lithium-ion Batteries. Miner. Process. Extr. Met. Rev. 2020, 42, 451–472. [Google Scholar] [CrossRef]
- Arshad, F.; Li, L.; Amin, K.; Fan, E.; Manurkar, N.; Ahmad, A.; Yang, J.; Wu, F.; Chen, R. A Comprehensive Review of the Advancement in Recycling the Anode and Electrolyte from Spent Lithium Ion Batteries. ACS Sustain. Chem. Eng. 2020, 8, 13527–13554. [Google Scholar] [CrossRef]
- Fan, X.; Song, C.; Lu, X.; Shi, Y.; Yang, S.; Zheng, F.; Huang, Y.; Liu, K.; Wang, H.; Li, Q. Separation and recovery of valuable metals from spent lithium-ion batteries via concen-trated sulfuric acid leaching and regeneration of LiNi1/3Co1/3Mn1/3O2. J. Alloys Compd. 2021, 863, 158775. [Google Scholar] [CrossRef]
- Vieceli, N.; Casasola, R.; Lombardo, G.; Ebin, B.; Petranikova, M. Hydrometallurgical recycling of EV lithium-ion batteries: Effects of incineration on the leaching efficiency of metals using sulfuric acid. Waste Manag. 2021, 125, 192–203. [Google Scholar] [CrossRef]
- Walther, G.; Klöden, B.; Büttner, T.; Weißgärber, T.; Kieback, B.; Böhm, A.; Naumann, D.; Saberi, S.; Timberg, L. A New Class of High Temperature and Corrosion Resistant Nickel-Based Open-Cell Foams. Adv. Eng. Mater. 2008, 10, 803–811. [Google Scholar] [CrossRef]
- Harald Pihl, Inconel 625. Available online: https://www.haraldpihl.com/de/products/Nickellegierungen/inconel-625 (accessed on 5 September 2023).
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions; Pergamon Press: New York, NY, USA, 1966; ISBN -13. [Google Scholar]
- Bockris, J.O.M. Electrolytic polarisation—I. The overpotential of hydrogen on some less common metals at high current densities. Influence of current density and time. Trans. Faraday Soc. 1947, 43, 417–429. [Google Scholar] [CrossRef]
- Heard, D.M.; Lennox, A.J.J. Electrode Materials in Modern Organic Electrochemistry. Angew. Chem. Int. Ed. 2020, 59, 18866–18884. [Google Scholar] [CrossRef]
- Haynes, W.M. Crc Handbook of Chemistry and Physics, 97th ed.; CRC Press LLC Taylor & Francis Group: Boca Raton, FL, USA, 2017; ISBN -13: 978-1-4987-5429-3. [Google Scholar]
- Han, P.; Bartels, D.M. Temperature Dependence of Oxygen Diffusion in H2O and D2O. J. Phys. Chem. 1996, 100, 5597–5602. [Google Scholar] [CrossRef]
- The Engineering ToolBox. Oxygen–Solubility in Fresh and Sea Water vs. Temperature. Available online: https://www.engineeringtoolbox.com/oxygen-solubility-water-d_841.html (accessed on 6 September 2023).
- Schneider, M.; Langklotz, U.; Labus, M.; Arnold, B.; Michaelis, A. Investigation of corrosion products formed on C35–AlMgSi0·5 friction welds during natural weathering in marine climate. Corros. Eng. Sci. Technol. 2012, 47, 312–319. [Google Scholar] [CrossRef]
- Rode, S.; Henninot, C.; Vallières, C.; Matlosz, M. Complexation Chemistry in Copper Plating from Citrate Baths. J. Electrochem. Soc. 2004, 151, C405–C411. [Google Scholar] [CrossRef]
- Zelenin, O.Y. Interaction of the Ni2+ ion with citric acid in an aqueous solution. Russ. J. Coord. Chem. 2007, 33, 346–350. [Google Scholar] [CrossRef]
- Irving, H.; Williams, R.J.P. Order of Stability of Metal Complexes. Nature 1948, 162, 746–747. [Google Scholar] [CrossRef]
- Daniele, P.G.; Ostacoli, G.; Zerbinati, O.; Sammartano, S.; De Robertis, A. Mixed metal complexes in solution. Thermodynamic and spectrophotometric study of copper(II)-citrate heterobinuclear complexes with nickel(II), zinc(II) or cadmium(II) in aqueous solution. Transit. Met. Chem. 1988, 13, 87–91. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Element | ρ (Element) g L−1 | (Metallic) Salt Used | ρ (Salt) g L−1 | c mol L−1 |
|---|---|---|---|---|
| Co | 10 | CoSO4 7H2O | 47.7 | 0.17 |
| Ni | 9 | NiSO4 6H2O | 40.3 | 0.15 |
| Mn | 8 | MnSO4 H2O | 24.61 | 0.15 |
| Cu | 2 | CuSO4 5H2O | 7.86 | 0.03 |
| P | 2 | H3PO4 (85 wt.%) | 4.41 cm3 L−1 | 0.07 |
| Al | 0.3 | Al2(SO4)3 16H2O | 3.5 | 0.01 |
| Li | 0.1 | LiCl | 0.61 | 0.01 |
| H2SO4 (96 wt.%) | 5.6 cm3 L−1 | 0.1 | ||
| citric acid | 50 | 0.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kutzer-Schulze, C.; Schmidt, H.; Weiser, M.; Büttner, T.; Schneider, M.; Michaelis, A. An Electrochemical Approach to the Recovery of Metals Typical of Battery Waste. Metals 2024, 14, 109. https://doi.org/10.3390/met14010109
Kutzer-Schulze C, Schmidt H, Weiser M, Büttner T, Schneider M, Michaelis A. An Electrochemical Approach to the Recovery of Metals Typical of Battery Waste. Metals. 2024; 14(1):109. https://doi.org/10.3390/met14010109
Chicago/Turabian StyleKutzer-Schulze, Claudia, Hannes Schmidt, Mathias Weiser, Tilo Büttner, Michael Schneider, and Alexander Michaelis. 2024. "An Electrochemical Approach to the Recovery of Metals Typical of Battery Waste" Metals 14, no. 1: 109. https://doi.org/10.3390/met14010109