
Laboratory-Scale Implementation of Standardized Reconstituted Geothermal Water for Electrochemical Investigations of Carbon Steel Corrosion


Abstract
:1. Introduction
2. Materials and Methods
2.1. Modus Operandi for the Reconstitution of a Representative Medium of the DAPB Waters
2.1.1. Analysis of Dissolved Species and Gases of Actual Geothermal DAPB Waters
2.1.2. Physical and Chemical Characteristics of Actual Geothermal DAPB Waters
2.1.3. Modeling
2.1.4. Reconstitution
2.2. Carbon Steel Working Electrode
2.2.1. Carbon Steel Grade
2.2.2. CS-XC38 Working Electrodes
2.3. Experimental Setup
2.3.1. The Electrochemical Reactor
- -
- A refrigerant, into which water flowing at 1 °C was used to condense vapors and minimize any losses of SRGW;
- -
- Six essential electrodes which were used to monitor the physical and chemical parameters of the fluid and to investigate the reactivity of CS-XC38:
- ○
- A commercial combined pH glass electrode (InLab Reach, Mettler Toledo, Colombus, OH, USA) that was systematically calibrated between each experiment using commercial standard buffer solutions (pH 4.006 (NIST/DIN), pH 7.00 (ANA) from Mettler Toledo® Colombus, OH, USA);
- ○
- A platinum wire electrode, the open circuit potential (OCP) of which was monitored and compared to the internal reference of the pH electrode;
- ○
- An electrochemical triplet used only for electrochemical corrosion measurement and composed of the following: a CS-XC38 working electrode (WE); a saturated calomel reference electrode (SCE), which consists of a commercial SCE (K0077 from AMETEK, Inc., Berwyn, PA, USA) protected with a KCl 3 mol L−1 junction (K0065 from AMETEK, Inc., Berwyn, PA, USA); and a 6 cm high cylindrical Pt/Ir grid counter electrode (CE) with a diameter of 6 cm;
- ○
- One CS-XC38, referred to as the free electrode and used to monitor the reactor’s OCP; this item was not exposed to external electrochemical disturbances;
- ○
- A Pyrex® tube glass bubbler comprising a dip tube and a diffuser. This was used for gas equilibration, i.e., of the humidified N2 (79.2%), CO2 (20%), and H2S (0.8%) gas mixture, as otherwise stated.
2.3.2. Electrochemical Apparatuses, pH Meter, and Data Logger
2.4. Electrochemical Techniques
2.5. Gravimetric Analyses
2.6. Electrochemical Study
2.7. SEM Characterization
2.8. Petrosourced (PS) and Biosourced (BS) Organic Corrosion Inhibitors (OCIs); PS-OCI or BS-OCI
3. Results and Discussion
3.1. Implementation of the SRGW
3.2. CCD in the Absence of Inhibitor
3.3. SEM Characterization in the Absence of Inhibitor
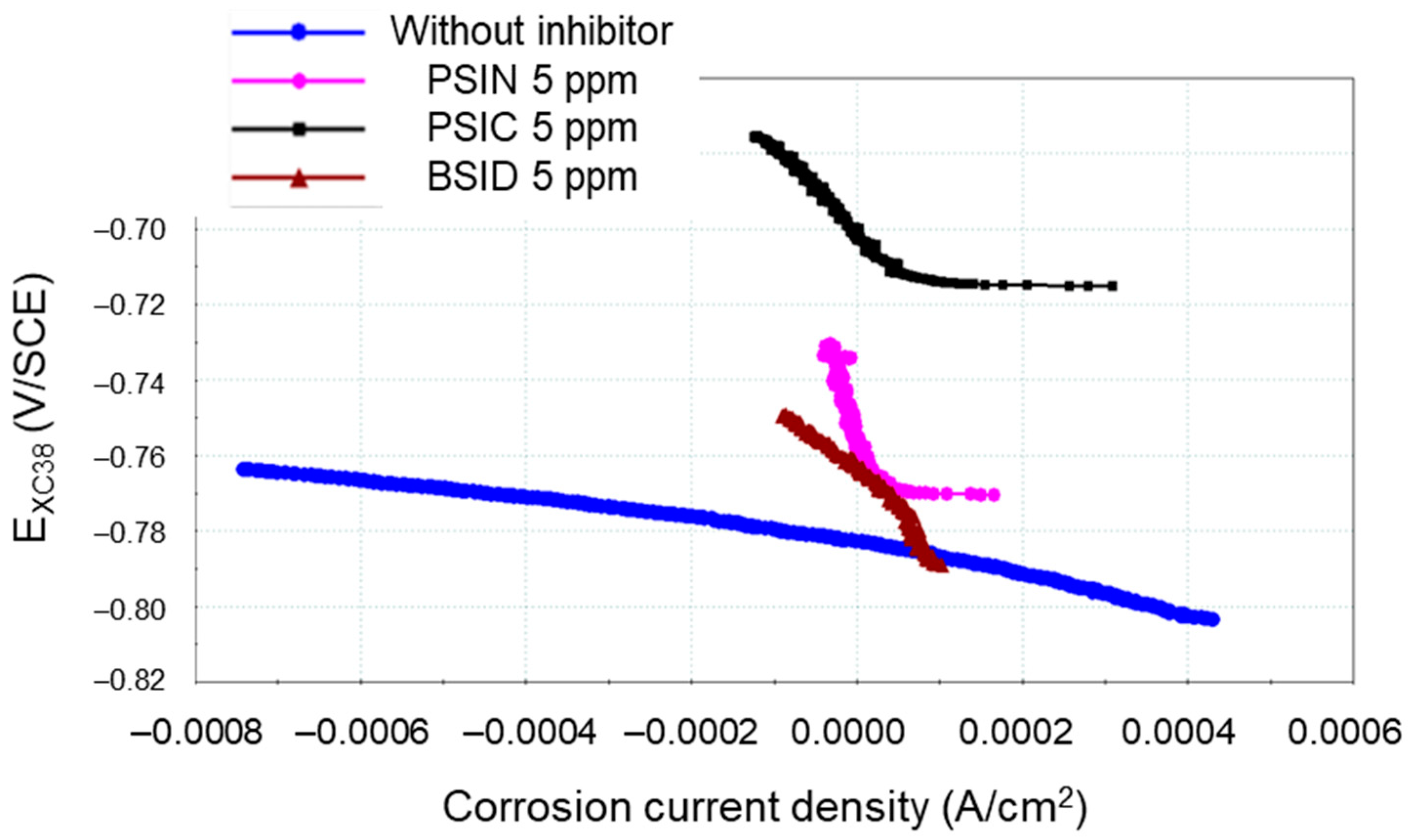
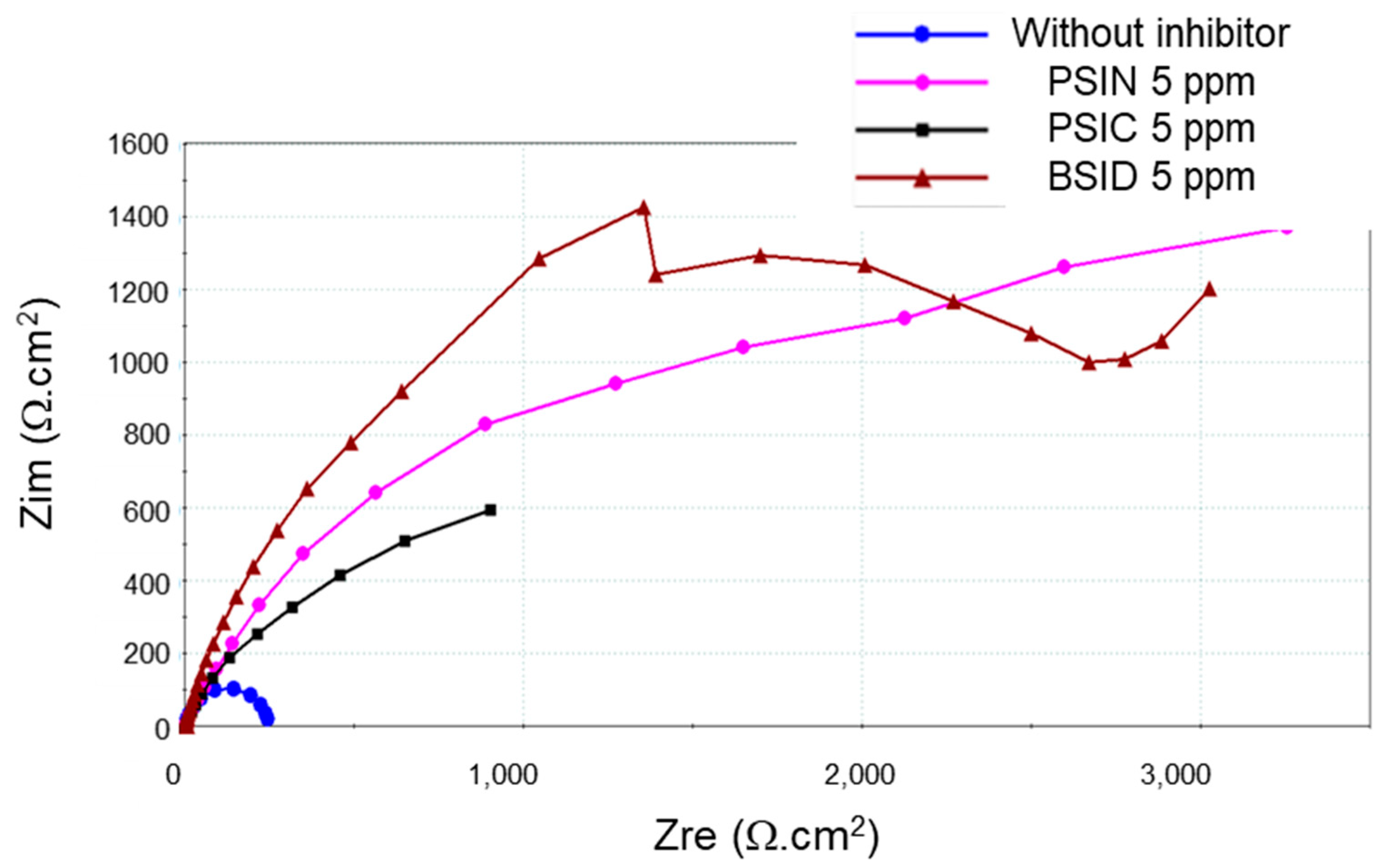
3.4. CCD in the Presence of Inhibitors
3.5. SEM Characterization in the Presence of BSID
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Production Well | T | pH | Eh | Ω | P.Bulle | Salinity | Alcaline Reserve | Equiv HCO3− | S−II | Fe2+ | Na | K | Ca | Mg | NH4+ | HCO3− | Cl− | SO42− | B | SiO2 | F | Sr | Ba | Li |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (mV/SHE) | (mS/cm) | (bar) | (eq. NaCl) | (mmol L−1) | (mg L−1) | ||||||||||||||||||
| ADP(GADP-1) | 72.4 | 6.44 | −317 | 20.38 | 8.15 | 12.0 | 5.18 | 316.0 | 4.83 | 0.32 | 3916.12 | 63.59 | 472.53 | 107.77 | 15.54 | 322.61 | 6768.67 | 842.33 | 11.31 | 37.43 | 4.55 | 27.46 | 0.16 | 1.54 |
| Alfortville (GAL-2) | 69.9 | 6.34 | −337 | 29.80 | 8.06 | 20.1 | 5.24 | 319.4 | 17.60 | 0.12 | 6335.53 | 92.98 | 942.20 | 211.25 | 21.77 | 299.85 | 11,453.95 | 858.49 | 12.34 | 41.77 | 4.20 | 52.12 | 0.29 | 2.44 |
| Champigny GCHM-3 | 73.1 | 6.31 | −300 | 41.83 | 9.2 | 28.0 | 4.75 | 290.0 | 9.91 | 1.43 | 8012.63 | 152.50 | 1229.28 | 239.70 | 26.70 | 280.00 | 15,633.75 | 905.00 | 17.34 | 38.48 | 2.70 | 67.05 | 0.35 | 3.14 |
| Champigny GCHM-1 | 74.6 | 6.26 | −329 | 36.06 | 8.39 | 26.6 | 5.36 | 327.1 | 12.52 | 0.27 | 8329.13 | 123.92 | 1297.32 | 264.08 | 25.22 | 290.14 | 15,357.67 | 886.25 | 13.48 | 41.70 | 3.96 | 67.13 | 0.35 | 2.77 |
| Chevilly Larue GCHL-2 | 69.4 | 6.36 | −339 | 23.03 | 7.48 | 16.7 | 5.09 | 310.3 | 14.00 | 0.06 | 5022.28 | 79.57 | 757.69 | 171.39 | 20.09 | 301.67 | 9414.16 | 796.55 | 11.05 | 40.34 | 4.52 | 42.63 | 0.24 | 1.94 |
| Créteil GCRT-1 | 74.0 | 6.41 | −300 | 35.79 | 7.79 | 21.8 | 4.86 | 296.3 | 18.18 | 1.15 | 6892.25 | 97.86 | 1074.44 | 222.08 | 23.31 | 291.26 | 12,433.91 | 841.08 | 12.06 | 42.16 | 4.83 | 54.64 | 0.28 | |
| Fresnes GFR- 2 | 72.3 | 6.40 | −336 | 20.84 | 7.77 | 13.0 | 5.23 | 319.0 | 13.27 | 0.16 | 4135.22 | 66.06 | 528.89 | 126.77 | 16.27 | 334.05 | 7157.26 | 901.26 | 9.58 | 38.53 | 4.12 | 28.74 | 0.18 | 1.61 |
| L’Haÿ-les-Roses GHLR-2 | 70.5 | 6.38 | −338 | 24.1 | 7.51 | 16.3 | 4.65 | 283.9 | 13.65 | 0.04 | 5150.74 | 79.55 | 731.32 | 169.65 | 19.48 | 307.47 | 9138.23 | 789.29 | 10.81 | 39.92 | 4.12 | 40.60 | 1.99 | |
| Orly 1 Gazier GORY-2 | 71.4 | 6.39 | −323 | 23.77 | 6.4 | 15.9 | 5.20 | 317.5 | 8.06 | 0.28 | 5043.33 | 76.69 | 719.50 | 171.56 | 18.71 | 308.20 | 8833.70 | 806.47 | 10.96 | 41.32 | 5.68 | 40.42 | ||
| Orly 2 Nouvelet GORY-3 | 73.9 | 6.31 | −342 | 23.77 | 8.52 | 14.9 | 5.90 | 360.0 | 9.58 | 0.11 | 4750.94 | 76.20 | 629.58 | 151.86 | 17.67 | 294.61 | 8290.49 | 814.77 | 10.14 | 38.96 | 4.18 | 32.64 | 1.77 | |
| Sucy en Brie GSUC-3 | 74.1 | 6.20 | −325 | 39.32 | 9.81 | 25.9 | 4.62 | 282 | 14.97 | 0.96 | 8059.13 | 123.18 | 1278.63 | 245.01 | 26.77 | 284.81 | 1,5068.28 | 913.34 | 16.16 | 41.74 | 4.02 | 63.71 | 3.21 | |
| Thiais GTHI-1 | 73.8 | 6.35 | −337 | 24.71 | 7.61 | 17.0 | 4.94 | 301.63 | 11.17 | 0.09 | 5361.22 | 82.92 | 785.29 | 175.67 | 19.17 | 293.64 | 9657.98 | 795.39 | 10.42 | 40.62 | 4.60 | 43.12 | 1.88 | |
| Average value | 72.5 | 6.35 | −326.9 | 28.62 | 8.06 | 19.0 | 5.1 | 12.31 | 0.42 | 5917.38 | 92.92 | 870.56 | 188.07 | 20.89 | 300.69 | 10,767.34 | 845.85 | 12.14 | 40.25 | 4.29 | 46.69 | 2.23 | ||
| Standard deviation | 1.8 | 0.1 | 14.6 | 7.6 | 0.88 | 5.4 | 0.4 | 3.86 | 0.48 | 1564.46 | 27.02 | 289.12 | 48.56 | 3.86 | 15.56 | 3178.22 | 46.36 | 2.40 | 1.58 | 0.69 | 14.17 | 0.62 | ||
| Geothermal Production Well | CO2.L (%) | CO2.T | AR.L (%) | AR.T | N2.L (%) | N2.T | HE.L (%) | HE.T | H2S.L (%) | H2S.T | CH4.L (%) | CH4.T | C2.L (%) | C2.T | C3.L (%) | C3.T | IC4.L (%) | IC4.T | NC4.L (%) | NC4.T | C5.L (%) | C5.T | C6.L (%) | C6.T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADP (GADP-1) | 16.450 | 0.275 | 24.250 | 0.690 | 0.106 | 48.900 | 3.170 | 2.140 | 0.695 | 0.855 | 0.580 | 0.165 | ||||||||||||
| Alfortville (GAL-2) | 14.427 | 0.674 | 0.259 | 0.012 | 26.443 | 1.325 | 0.646 | 0.035 | 0.547 | 0.382 | 49.450 | 2.682 | 3.408 | 0.173 | 2.199 | 0.119 | 0.296 | 0.017 | 0.777 | 0.047 | 0.389 | 0.024 | 0.229 | 0.014 |
| Champigny GCHM-3 | 17.300 | 0.230 | 29.500 | 0.710 | 0.058 | 42.800 | 3.310 | 2.010 | 0.240 | 0.590 | 0.350 | 0.120 | ||||||||||||
| Champigny GCHM-1 | 16.216 | 0.859 | 0.438 | 0.029 | 29.491 | 1.619 | 0.744 | 0.042 | 0.608 | 0.378 | 45.465 | 2.510 | 3.471 | 0.190 | 2.014 | 0.111 | 0.268 | 0.015 | 0.628 | 0.035 | 0.355 | 0.019 | 0.198 | 0.012 |
| Chevilly Larue GCHL-2 | 14.951 | 0.682 | 0.276 | 0.015 | 24.518 | 1.306 | 0.626 | 0.029 | 0.352 | 0.312 | 51.287 | 2.653 | 3.236 | 0.172 | 2.105 | 0.114 | 0.276 | 0.015 | 0.745 | 0.040 | 0.407 | 0.023 | 0.208 | 0.011 |
| Créteil GCRT-1 | 15.666 | 0.896 | 0.240 | 0.013 | 25.695 | 1.412 | 0.509 | 0.028 | 0.225 | 0.200 | 49.065 | 2.688 | 3.503 | 0.191 | 2.147 | 0.117 | 0.298 | 0.016 | 0.772 | 0.042 | 0.449 | 0.025 | 0.239 | 0.013 |
| Fresnes GFR-2 | 12.953 | 0.663 | 0.344 | 0.018 | 28.971 | 0.902 | 0.634 | 0.039 | 0.271 | 0.317 | 47.583 | 2.310 | 3.294 | 0.235 | 2.186 | 0.140 | 0.302 | 0.022 | 0.884 | 0.051 | 0.513 | 0.031 | 0.211 | 0.021 |
| L’Haÿ-les-Roses GHLR-2 | 13.404 | 0.232 | 0.260 | 0.004 | 23.343 | 0.430 | 0.623 | 0.010 | 0.333 | 0.252 | 47.479 | 0.744 | 3.103 | 0.048 | 1.935 | 0.031 | 0.257 | 0.004 | 0.642 | 0.011 | 0.351 | 0.005 | 0.160 | 0.003 |
| Orly 1 Gazier GORY-2 | 15.936 | 0.705 | 0.341 | 0.012 | 21.893 | 0.905 | 0.531 | 0.018 | 0.308 | 0.192 | 52.536 | 2.258 | 3.108 | 0.127 | 2.058 | 0.082 | 0.284 | 0.012 | 0.820 | 0.035 | 0.541 | 0.022 | 0.276 | 0.011 |
| Orly 2 Nouvelet GORY-3 | 15.317 | 0.935 | 0.293 | 0.020 | 26.300 | 2.311 | 0.767 | 0.036 | 0.390 | 0.273 | 48.483 | 3.033 | 3.185 | 0.196 | 2.053 | 0.113 | 0.282 | 0.018 | 0.787 | 0.045 | 0.487 | 0.033 | 0.195 | 0.015 |
| Sucy en Brie GSUC-3 | 19.360 | 1.173 | 0.226 | 0.014 | 27.480 | 0.108 | 0.720 | 0.006 | 0.578 | 0.341 | 43.780 | 0.018 | 3.266 | 0.080 | 1.980 | 0.047 | 0.240 | 0.006 | 0.630 | 0.015 | 0.368 | 0.009 | 0.128 | 0.000 |
| Thiais GTHI-1 | 14.066 | 0.725 | 0.292 | 0.012 | 28.684 | 1.224 | 0.605 | 0.032 | 0.317 | 0.336 | 46.746 | 2.420 | 2.961 | 0.151 | 1.913 | 0.098 | 0.260 | 0.013 | 0.736 | 0.038 | 0.399 | 0.044 | 0.217 | 0.012 |
| Average value | 15.504 | 0.754 | 0.289 | 0.015 | 26.381 | 1.154 | 0.650 | 0.027 | 0.341 | 0.298 | 47.798 | 2.132 | 3.251 | 0.156 | 2.062 | 0.097 | 0.308 | 0.014 | 0.739 | 0.036 | 0.432 | 0.024 | 0.196 | 0.011 |
| Standard deviation | 1.76 | 0.24 | 0.06 | 0.01 | 2.53 | 0.62 | 0.08 | 0.01 | 0.17 | 0.07 | 2.84 | 0.96 | 0.16 | 0.06 | 0.09 | 0.03 | 0.12 | 0.01 | 0.10 | 0.01 | 0.08 | 0.01 | 0.05 | 0.01 |
| Total gas | CO2 (%) | 16.26 | AR (%) | 0.30 | N2 (%) | 27.53 | HE (%) | 0.68 | H2S (%) | 0.64 | CH4 (%) | 49.93 | C2H6 (%) | 3.41 | C3H8 (%) | 2.16 | iC4H10 (%) | 0.32 | nC4H10 (%) | 0.77 | C5H12 (%) | 0.46 | C6H14 (%) | 0.21 |
| Gases considered for reconstitution | CO2 (%) | 16.26 | H2S (%) | 0.64 | AR (%) + NA(%) + HE (%) + CH4 (%) + C2H6(%) +C3H8 (%) + iC4H10 (%) + nC4H10 (%) + C5H12 (%) + C6H14 (%) | 85.77 | ||||||||||||||||||
| Elements (mM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Mineral or Addition | Ca | Mg | Na | K | NH4+ | HCO3− | Cl | SO42− |
| NaHCO3 | 5.300 | 5.300 | ||||||
| KCl | 2.500 | 2.500 | ||||||
| MgCl2 | 9.000 | 18.00 | ||||||
| (NH4)2SO4 | 1.16 | 0.58 | ||||||
| CaSO4 | 8.226 | 8.226 | ||||||
| CaCl2 | 17.000 | 34.00 | ||||||
| NaCl | 242.371 | 242.371 | ||||||
| HCl 0.1 N 1 mL | 0.1 | |||||||
| Reconstituted value (mM) | 25.226 | 9.000 | 247.671 | 2.500 | 1.160 | 5.300 | 296.971 | 8.805 |
| Targed value (mM) | 21.722 | 7.738 | 197.318 | 2.377 | 1.158 | 4.929 | 303.707 | 8.805 |
| Reconstituted vs targed gap (mM) | 3.5 | 1.3 | 50.4 | 0.1 | 0.0 | 0.4 | -6.7 | 0.0 |
| Phreeqc Interactive 3.7.3–15968 | Gas Mixture Composition Used for Modeling | ||
| Thermodynamic Database Used: | |||
| ThermoddemV1.10_15Dec2020.dat | CO2 (%) | 20.0 | |
| Geothermal Solution 1 | H2S (%) | 0.8 | |
| pH 6.3 charge | N2 (%) | 79.2 | |
| pe 4 | |||
| temp 70 | Main Parameters and Content Obtained after Modeling | ||
| units mmol L−1 | |||
| Ca 25.226 | pH | 6.248 | |
| Mg 9 | pe * | −3.419 | |
| Na 247.3 | |||
| K 2.5 | Elements | [content] (mM) | [content] (mg L−1) |
| N(−3) 1.158 | C(4)tot | 7.19 | 86.32 |
| C(4) 4.929 as HCO3 | HCO3− | 3.70 | 225.52 |
| Cl 296.871 | Ca | 25.69 | 1029.60 |
| S(6) 8.805 | Cl | 302.40 | 10720.99 |
| save geothermal solution 1 | K | 2.55 | 99.54 |
| end | Mg | 9.17 | 222.78 |
| Use geothermal solution 1 | N | 0.94 | 13.14 |
| EQUILIBRIUM_PHASES 1 | Na | 251.90 | 5791.12 |
| CO2(g) –0.698970004 # 20.0% | Stot | 9.18 | 294.21 |
| H2S(g) –2.096910013 # 0.8% | S−II | 0.58 | 18.55 |
| N2(g) –0.101274818 # 79.2% | H2S | 0.35 | 11.84 |
| end | HS− | 0.23 | 7.64 |
Appendix B
| Loop Number | Technique | Date, hour | Time per Technique | Ommesrion Time (h) |
|---|---|---|---|---|
| 1 | EXC38_3_2.5_TESTNAME_161120_1 | 16/01/2024 12:27:00 | 0:00:00 | 0.00 |
| PSXC38_3_2.5_TESTNAME_161120_1 | 16/01/2024 12:38:40 | 0:21:36 | 0.36 | |
| RPXC38_3_2.5_TESTNAME_161120_1 | 16/01/2024 13:00:16 | 0:32:43 | 0.55 | |
| TaXC38_3_2.5_TESTNAME_161120_1 | 16/01/2024 13:11:23 | 0:44:31 | 0.74 | |
| RPXC382_3_2.5_TESTNAME_161120_1 | 16/01/2024 13:23:11 | 2:01:23 | 2.02 | |
| Z1XC38_3_2.5_TESTNAME_161120_1 | 16/01/2024 14:40:03 | 2:13:08 | 2.22 | |
| RPXC383_3_2.5_TESTNAME_161120_1 | 16/01/2024 14:51:48 | 3:34:38 | 3.58 | |
| PSXC382_3_2.5_TESTNAME_161120_1 | 16/01/2024 16:13:18 | 3:46:11 | 3.77 | |
| 2 | EXC38_3_2.5_TESTNAME_161120_2 | 16/01/2024 16:24:58 | 3:57:51 | 3.96 |
| PSXC38_3_2.5_TESTNAME_161120_2 | 16/01/2024 16:46:34 | 4:19:27 | 4.32 | |
| RPXC38_3_2.5_TESTNAME_161120_2 | 16/01/2024 16:57:41 | 4:30:34 | 4.51 | |
| TaXC38_3_2.5_TESTNAME_161120_2 | 16/01/2024 17:09:29 | 4:42:22 | 4.71 | |
| RPXC382_3_2.5_TESTNAME_161120_2 | 16/01/2024 18:26:21 | 5:59:14 | 5.99 | |
| Z1XC38_3_2.5_TESTNAME_161120_2 | 16/01/2024 18:38:06 | 6:10:59 | 6.18 | |
| RPXC383_3_2.5_TESTNAME_161120_2 | 16/01/2024 19:59:36 | 7:32:29 | 7.54 | |
| PSXC382_3_2.5_TESTNAME_161120_2 | 16/01/2024 20:11:09 | 7:44:02 | 7.73 | |
| 3 | EXC38_3_2.5_TESTNAME_161120_3 | 16/01/2024 20:22:49 | 7:55:42 | 7.93 |
| PSXC38_3_2.5_TESTNAME_161120_3 | 16/01/2024 20:44:25 | 8:17:18 | 8.29 | |
| RPXC38_3_2.5_TESTNAME_161120_3 | 16/01/2024 20:55:32 | 8:28:25 | 8.47 | |
| TaXC38_3_2.5_TESTNAME_161120_3 | 16/01/2024 21:07:20 | 8:40:13 | 8.67 | |
| RPXC382_3_2.5_TESTNAME_161120_3 | 16/01/2024 22:24:12 | 9:57:05 | 9.95 | |
| Z1XC38_3_2.5_TESTNAME_161120_3 | 16/01/2024 22:35:57 | 10:08:50 | 10.15 | |
| RPXC383_3_2.5_TESTNAME_161120_3 | 16/01/2024 23:57:27 | 11:30:20 | 11.51 | |
| PSXC382_3_2.5_TESTNAME_161120_3 | 17/01/2024 00:09:00 | 11:41:53 | 11.70 | |
| 4 | EXC38_3_2.5_TESTNAME_161120_4 | 17/01/2024 00:20:40 | 11:53:33 | 11.89 |
| PSXC38_3_2.5_TESTNAME_161120_4 | 17/01/2024 00:42:16 | 12:15:09 | 12.25 | |
| RPXC38_3_2.5_TESTNAME_161120_4 | 17/01/2024 00:53:23 | 12:26:16 | 12.44 | |
| TaXC38_3_2.5_TESTNAME_161120_4 | 17/01/2024 01:05:11 | 12:38:04 | 12.63 | |
| RPXC382_3_2.5_TESTNAME_161120_4 | 17/01/2024 02:22:03 | 13:54:56 | 13.92 | |
| Z1XC38_3_2.5_TESTNAME_161120_4 | 17/01/2024 02:33:48 | 14:06:41 | 14.11 | |
| RPXC383_3_2.5_TESTNAME_161120_4 | 17/01/2024 03:55:18 | 15:28:11 | 15.47 | |
| PSXC382_3_2.5_TESTNAME_161120_4 | 17/01/2024 04:06:51 | 15:39:44 | 15.66 | |
| 5 | EXC38_3_2.5_TESTNAME_161120_5 | 17/01/2024 04:18:31 | 15:51:24 | 15.86 |
| PSXC38_3_2.5_TESTNAME_161120_5 | 17/01/2024 04:40:07 | 16:13:00 | 16.22 | |
| RPXC38_3_2.5_TESTNAME_161120_5 | 17/01/2024 04:51:14 | 16:24:07 | 16.40 | |
| TaXC38_3_2.5_TESTNAME_161120_5 | 17/01/2024 05:03:02 | 16:35:55 | 16.60 | |
| RPXC382_3_2.5_TESTNAME_161120_5 | 17/01/2024 06:19:54 | 17:52:47 | 17.88 | |
| Z1XC38_3_2.5_TESTNAME_161120_5 | 17/01/2024 06:31:39 | 18:04:32 | 18.08 | |
| RPXC383_3_2.5_TESTNAME_161120_5 | 17/01/2024 07:53:09 | 19:26:02 | 19.43 | |
| PSXC382_3_2.5_TESTNAME_161120_5 | 17/01/2024 08:04:42 | 19:37:35 | 19.63 |
Appendix C
| CS-XC38 Sample | CS-XC38/SRGW Interaction Time (h) | Vcorr (mm y−1) |
|---|---|---|
| UD | 118.33 | 0.425 |
| D | 118.33 | 1.248 |
References
- Rojas, J.; Giot, D.; Le Nindre, Y.M.; Criaud, A.; Fouillac, C.; Brach, M.; Menjoz, A.; Martin, J.C.; Lambert, M.; Chiles, J.P.; et al. Caractérisation et Modélisation du Réservoir Géothermique du Dogger, Bassin Pari-Sien, France, Rapport Final; BRGM R 30169 IRG SGN 89; BRGM: Orléans, France, 1989; p. 249. [Google Scholar]
- Castillo, C.; Ignatiadis, I. Sulfate-Reduction State of the Geothermal Dogger Aquifer, Paris Basin (France) after 35 Years of Exploitation: Analysis and Consequences of Bacterial Proliferation in Casings and Reservoir. In Proceedings of the Thirty-Seventh Workshop on Geothermal Reservoir Engineering, Stanford University, Stanford, CA, USA, 30 January–1 February 2012. [Google Scholar]
- Fardeau, M.-L.; Goulhen, F.; Bruschi, M.; Khelifi, N.; Cayol, J.-L.; Ignatiadis, I.; Guyot, F.; Ollivier, B. Archaeoglobus fulgidus and Thermotoga elfii, Thermophilic Isolates from Deep Geothermal Water of the Paris Basin. Geomicrobiol. J. 2009, 26, 119–130. [Google Scholar] [CrossRef]
- Lopez, S.; Hamm, V.; Le Brun, M.; Schaper, L.; Boissier, F.; Cotiche, C.; Giuglaris, E. 40 years of Dogger aquifer management in Ile-de-France, Paris Basin, France. Geothermics 2010, 39, 339–356. [Google Scholar] [CrossRef]
- Peter, F.; Ellis, I. A Geothermal Corrosivity Classification System. GRC Trans. 1981, 5, 463–472. [Google Scholar]
- Ikeda, A.; Mukai, S.; Ueda, M. CO2 corrosion behavior of carbon and Cr steel. Sumitomo Search 1985, 31, 91–102. [Google Scholar]
- Crolet, J.L. Mécanismes de la corrosion uniforme sous dépôt de corrosion. Métaux Corros. Ind. 1998, 63, 279–302. [Google Scholar]
- Xiao, G.; Tan, S.; Yu, Z.; Dong, B.; Yi, Y.; Tian, G.; Yu, H.; Shi, S. CO2 corrosion behaviors of 13Cr steel in the high-temperature steam environment. Petroleum 2020, 6, 106–113. [Google Scholar] [CrossRef]
- Soltis, J. Passivity breakdown, pit initiation and propagation of pits in metallic materials—Review. Corros. Sci. 2015, 90, 5–22. [Google Scholar] [CrossRef]
- Evans, U.R. CXL.—The passivity of metals. Part I. The isolation of the protective film. J. Chem. Soc. 1927, 1020–1040. [Google Scholar] [CrossRef]
- Frankel, G.S. Pitting Corrosion of Metals: A Review of the Critical Factors. J. Electrochem. Soc. 1998, 145, 2186. [Google Scholar] [CrossRef]
- Pou, T.E.; Murphy, O.J.; Young, V.; Bockris, J.O.M.; Tongson, L.L. Passive Films on Iron: The Mechanism of Breakdown in Chloride Containing Solutions. J. Electrochem. Soc. 1984, 131, 1243. [Google Scholar] [CrossRef]
- Traunbenberg, S.; Foley, R. The influence of chloride and sulphate ions on the corrosion of iron in sulphuric acid. J. Electrochem. Soc. 1971, 118, 1066–1070. [Google Scholar] [CrossRef]
- Galvele, J.R. Transport Processes and the Mechanism of Pitting of Metals. J. Electrochem. Soc. 1976, 123, 464. [Google Scholar] [CrossRef]
- Iofa, Z.A.; Batrakov, V.V.; Ngok, B.C. Influence of anion adsorption on the action of inhibitors on the acid corrosion of iron and cobalt. Electrochim. Acta 1964, 9, 1645–1653. [Google Scholar] [CrossRef]
- Ma, H.; Cheng, X.; Chen, S.; Wang, C.; Zhang, J.; Yang, H. An ac impedance study of the anodic dissolution of iron in sulfuric acid solutions containing hydrogen sulfide. J. Electroanal. Chem. 1998, 451, 11–17. [Google Scholar] [CrossRef]
- Ma, H.; Cheng, X.; Li, G.; Chen, S.; Quan, Z.; Zhao, S.; Niu, L. The influence of hydrogen sulfide on corrosion of iron under different conditions. Corros. Sci. 2000, 42, 1669–1683. [Google Scholar] [CrossRef]
- Pound, B.G.; Wright, B.A.; Sharp, R.M. Electrochemical phases for the iron/sulfur/water under geothermal conditions. Aust. J. Chem. 1985, 38, 643–657. [Google Scholar] [CrossRef]
- Pound, B.G.; Abdurrahman, M.H.; Glucina, P.; Wright, G.A.; Sharp, R.M. The corrosion of carbon steel and stainless steel in simulated geothermal media. Aust. J. Chem. 1985, 38, 1133–1140. [Google Scholar] [CrossRef]
- Hardy, J.A. Utilisation of Cathodic Hydrogen by Sulphate-Reducing Bacteria. Br. Corros. J. 1983, 18, 190–193. [Google Scholar] [CrossRef]
- Amalhay, M.; Akar, A.A.; Ignatiadis, I. Study of scale deposition phenomena in geothermal wells in the Paris Basin. In The International Symposium, Geothermics 94 in Europe, from Research to Development; BRGM, Ed.; BRGM: Orléans, France, 1998; pp. 223–232. [Google Scholar]
- Lee, W.; Lewandowski, Z.; Nielsen, P.H.; Hamilton, W.A. Role of sulfate-reducing bacteria in corrosion of mild steel: A review. Biofouling 1995, 8, 165–194. [Google Scholar] [CrossRef]
- Rickard, D. The composition of mackinawite. Am. Min. 2024, 109, 401–407. [Google Scholar] [CrossRef]
- Foroulis, Z.A. Closure to “Discussion of ‘On the Kinetics of the Breakdown of Passivity of Preanodized Aluminum by Chloride Ions’”. J. Electrochem. Soc. 1976, 123, 841a. [Google Scholar] [CrossRef]
- Amalhay, M.; Ignatiadis, I. Comparative Study of the Effectiveness of Various Organic Surfactants in Inhibiting Carbon Steel Corrosion in a Natural Geothermal Environment by Using Rapid Electrochemical Tests. MSF 1998, 289–292, 169–180. [Google Scholar] [CrossRef]
- Blanc, P.; Lassin, A.; Piantone, P.; Azaroual, M.; Jacquemet, N.; Fabbri, A.; Gaucher, E.C. Thermoddem: A geochemical database focused on low temperature water/rock interactions and waste materials. Appl. Geochem. 2012, 27, 2107–2116. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3: A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; Techniques and Methods; U.S. Geological Survey: Reston, VA, USA, 2013; p. 519.
- Marty, B.; Criaud, A.; Fouillac, C. Low enthalpy geothermal fluids from the Paris sedimentary basin—1. Characteristics and origin of gases. Geothermics 1988, 17, 619–633. [Google Scholar] [CrossRef]
- Criaud, A.; Fouillac, C.; Marty, B. Low enthalpy geothermal fluids from the paris basin. 2—Oxidation-reduction state and consequences for the prediction of corrosion and sulfide scaling. Geothermics 1989, 18, 711–727. [Google Scholar] [CrossRef]
- Fouillac, C.; Fouillac, A.M.; Criaud, A. Sulphur and oxygen isotopes of dissolved sulphur species in formation waters from the Dogger geothermal aquifer, Paris Basin, France. Appl. Geochem. 1990, 5, 415–427. [Google Scholar] [CrossRef]
- Thorbjornsson, I.; Kaldal, G.S.; Gunnarsson, B.S.; Ragnarsson, Á. New Approach to Mitigate Casing failures in High-Temperature Geothermal Wells. GRC Trans. 2017, 41, 585–591. [Google Scholar]
- Brioua, S.; Delimi, A.; Ferkous, H.; Boukerche, S.; Allal, H.; Boublia, A.; Djedouani, A.; Berredjem, M.; Kahlouche, A.; Rachedi, K.O.; et al. Enhancing corrosion resistance of XC38 steel using sulfur and nitrogen-containing phenyl thiosemicarbazone: A comprehensive experimental and computational analysis. J. Taiwan Inst. Chem. Eng. 2024, 165, 105718. [Google Scholar] [CrossRef]
- Boulechfar, C.; Ferkous, H.; Djellali, S.; Amin, M.A.; Boufas, S.; Djedouani, A.; Delimi, A.; Amor, Y.B.; Yadav, K.K.; Jeon, B.-H.; et al. DFT/molecular scale, MD simulation and assessment of the eco-friendly anti-corrosion performance of a novel Schiff base on XC38 carbon steel in acidic medium. J. Mol. Liq. 2021, 344, 117874. [Google Scholar] [CrossRef]
- Mouats, N.; Djellali, S.; Ferkous, H.; Sedik, A.; Delimi, A.; Boublia, A.; Rachedi, K.O.; Berredjem, M.; Çukurovali, A.; Alam, M.; et al. Comprehensive Investigation of the Adsorption, Corrosion Inhibitory Properties, and Quantum Calculations for 2-(2,4,5-Trimethoxybenzylidene) Hydrazine Carbothioamide in Mitigating Corrosion of XC38 Carbon Steel under HCl Environment. ACS Omega 2024, 9, 27945–27962. [Google Scholar] [CrossRef]
- Wamba-Tchio, O.R.; Pengou, M.; Teillout, A.-L.; Baumier, C.; Mbomekallé, I.M.; De Oliveira, P.; Nanseu-Njiki, C.P.; Ngameni, E. Electrochemical study and experimental simulation of the synergistic effect of a formulation based on Ficus pumila Linn. Leaves extract and zinc sulfate on the XC38 steel corrosion inhibition in NaCl solution. J. Electroanal. Chem. 2022, 919, 116553. [Google Scholar] [CrossRef]
- Ichchou, I.; Larabi, L.; Rouabhi, H.; Harek, Y.; Fellah, A. Electrochemical evaluation and DFT calculations of aromatic sulfonohydrazides as corrosion inhibitors for XC38 carbon steel in acidic media. J. Mol. Struct. 2019, 1198, 126898. [Google Scholar] [CrossRef]
- Moyeme, Y.C.S.; Betelu, S.; Bertrand, J.; Serrano, K.G.; Ignatiadis, I. Corrosion Current Density of API 5L X65 Carbon Steel in Contact with Natural Callovian-Oxfordian Clay Pore Water, Assessed by Various Electrochemical Methods over 180 Days. Metals 2023, 13, 966. [Google Scholar] [CrossRef]
- Orazem, M.E.; Tribollet, B. Electrochemical Impedance Spectroscopy; The Electrochemical Society Series; John Wiley & Sons, Inc.: Pennington, NJ, USA, 2008; p. 526. [Google Scholar]
- Barsoukov, E.; Macdonald, J.R. Impedance Spectroscopy: Theory, Experiment, and Applications; The Electrochemical Society Series; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018; p. 606. [Google Scholar]
- Stern, M.; Geary, A.L. Electrochemical Polarization: I. A Theoretical Analysis of the Shape of Polarization Curves. J. Electrochem. Soc. 1957, 104, 56. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; Wiley: New York, NY, USA, 1980. [Google Scholar]
- ASTM G1-90; Standard Practice for Preparing, Cleaning, and Evaluating Corrosion Test Specimens. ASTM International: West Conshohocken, PA, USA, 2017.
- Panchenko, Y.M.; Marshakov, A.I.; Igonin, T.N.; Kovtanyuk, V.V.; Nikolaeva, L.A. Long-term forecast of corrosion mass losses of technically important metals in various world regions using a power function. Corros. Sci. 2014, 88, 306–316. [Google Scholar] [CrossRef]
- Skoog, D.; West, D.M.; Holler, F.J.; Fort, W. Gravimetric Analysis. Fundamentals of Analytical Chemistry, 7th ed.; Saunders College Publishing: Fort Worth, TX, USA, 1996; p. 870. [Google Scholar]
- Daoudi, J.; Betelu, S.; Tzedakis, T.; Bertrand, J.; Ignatiadis, I. A Multi-Parametric Device with Innovative Solid Electrodes for Long-Term Monitoring of pH, Redox-Potential and Conductivity in a Nuclear Waste Repository. Sensors 2017, 17, 1372. [Google Scholar] [CrossRef]
- Shoesmith, D.W.; Taylor, P.; Bailey, M.G.; Owen, D.G. The Formation of Ferrous Monosulfide Polymorphs during the Corrosion of Iron by Aqueous Hydrogen Sulfide at 21 °C. J. Electrochem. Soc. 1980, 127, 1007. [Google Scholar] [CrossRef]
- Chivot, J. Thermodynamique des Produits de Corrosion: Fonctions Thermodynamiques, Diagrammes de Solubilité, Diagrammes E-ph des Systèmes Fe-H2O, Fe-CO2-H2O, Fe-S-H2O, Cr-H2O et Ni-H2O en Fonction de la Temperature; Andra: Chatenay-Malabry, France, 2004; 142p, ISBN 2-9510108-6. [Google Scholar]
- Brigham, R.J.; Tozer, E.W. Temperature as a Pitting Criterion. Corrosion 2013, 29, 33–36. [Google Scholar] [CrossRef]
- Ovarfort, R. Critical pitting temperature measurements of stainless steels with an improved electrochemical method. Corros. Sci. 1989, 29, 987–993. [Google Scholar] [CrossRef]



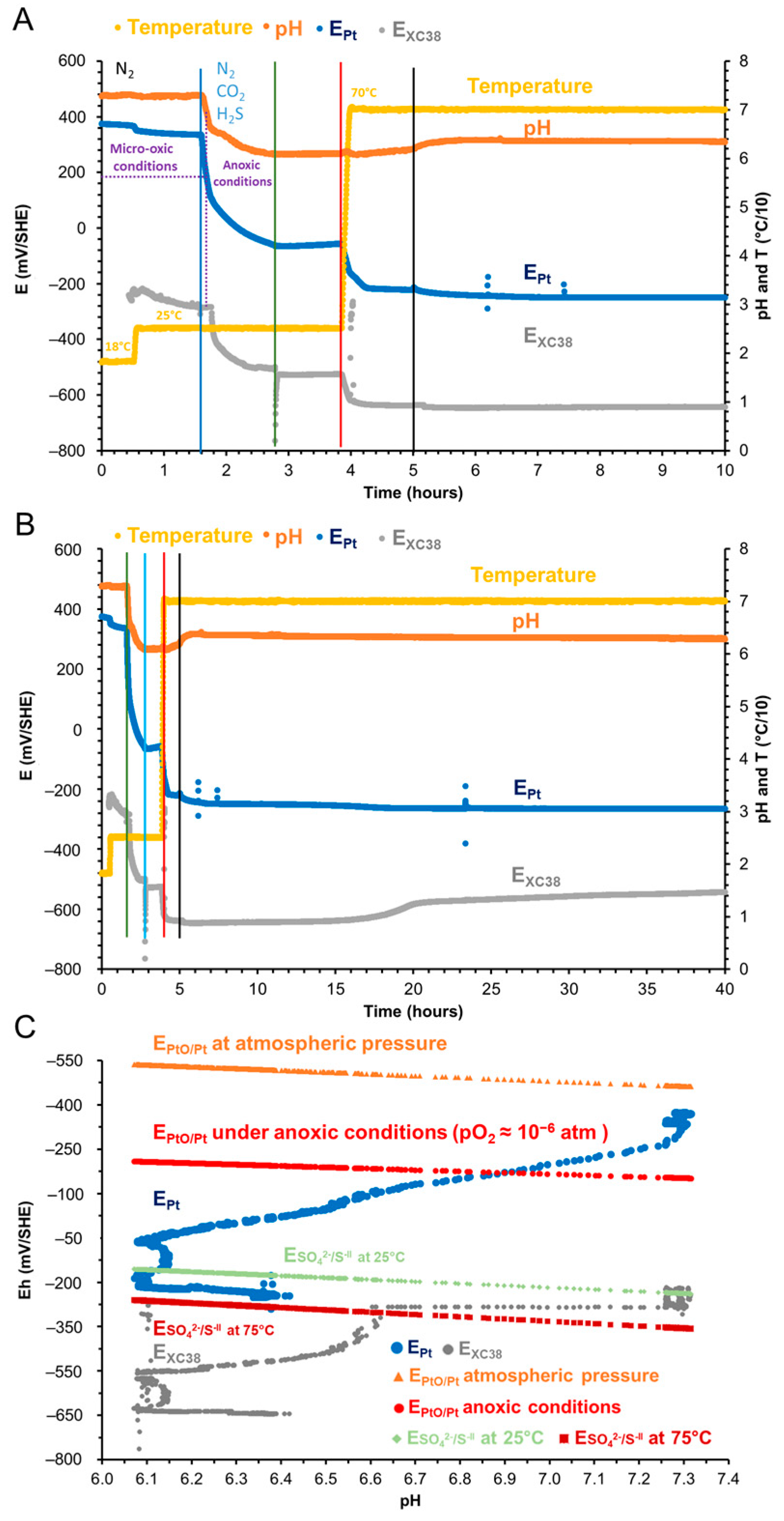
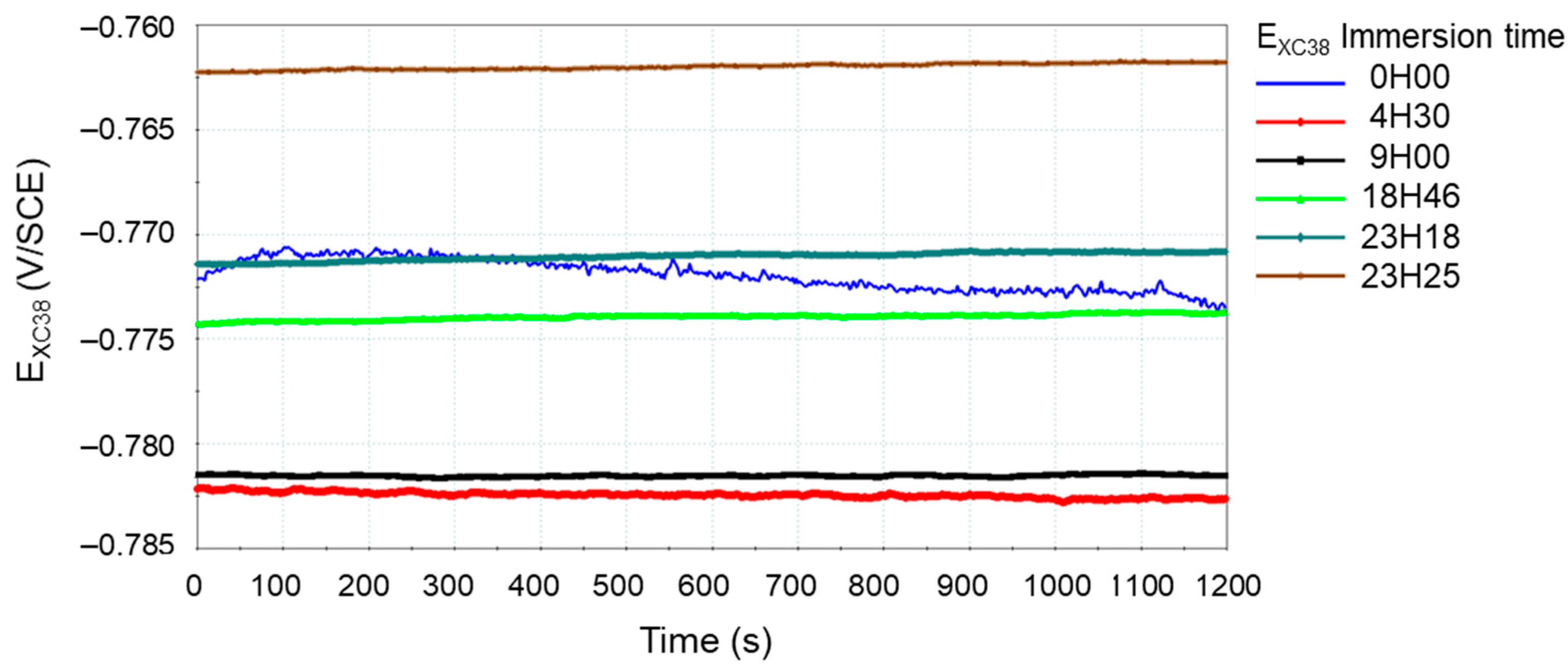
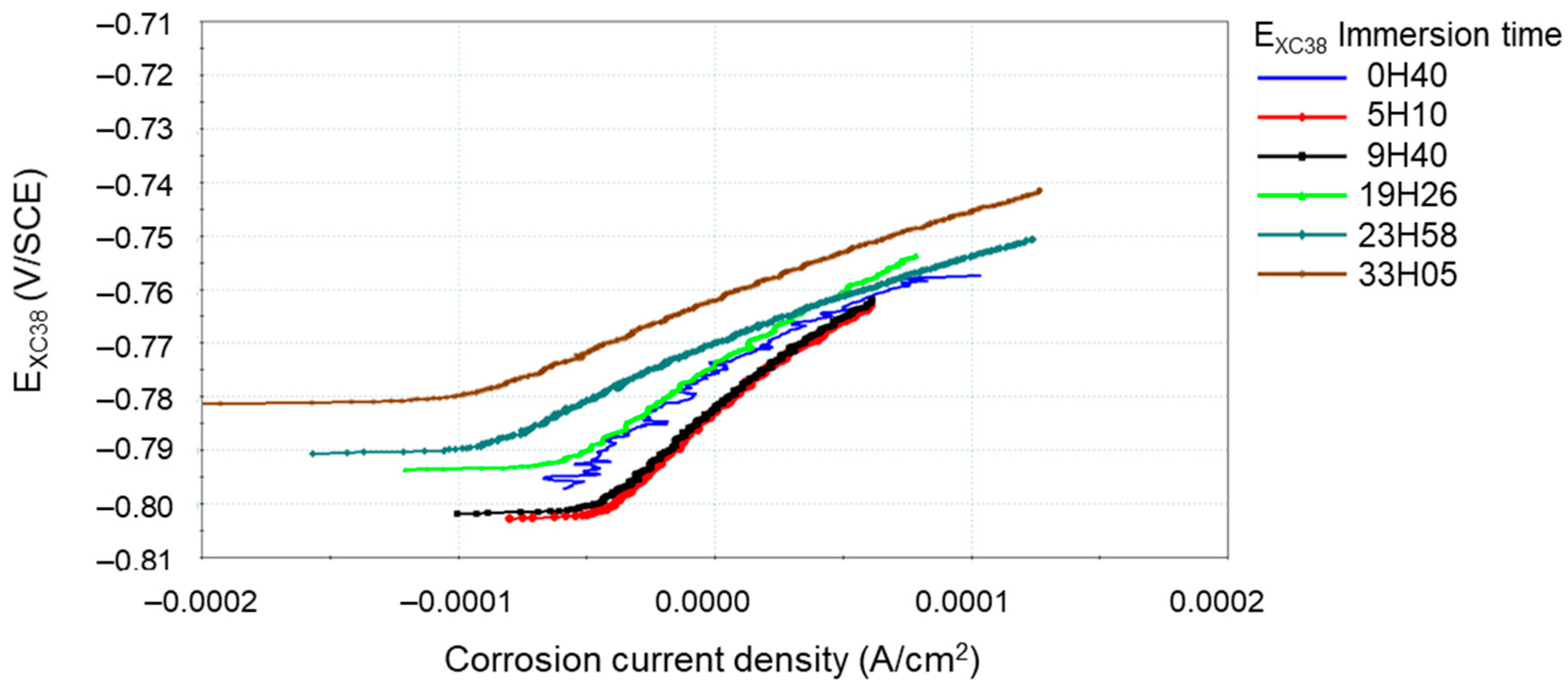



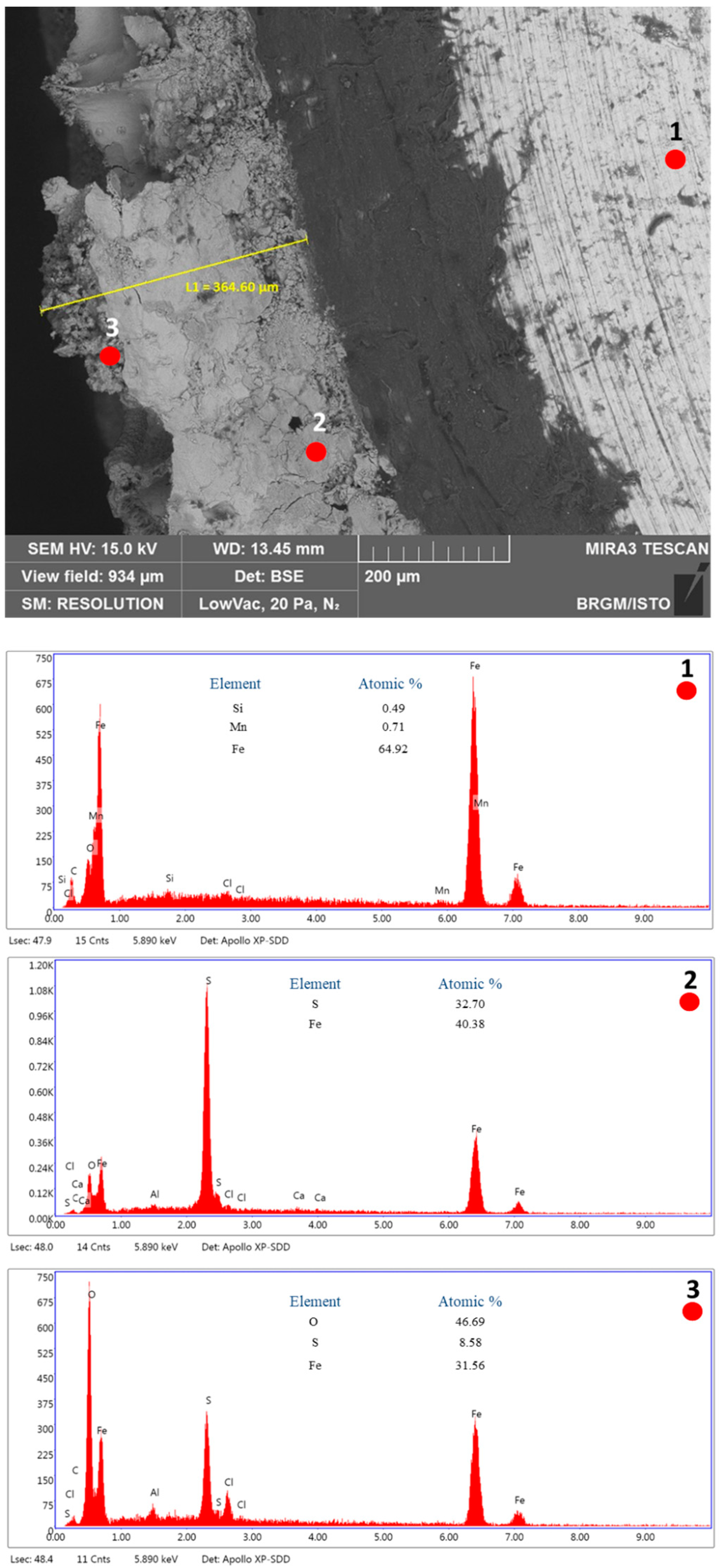



| Mineral Salts’ Names and Formulas | Mass (g) for 1 L of Deionized Water |
|---|---|
| Sodium bicarbonate, NaHCO3 | 0.44523 |
| Potassium chloride, KCl | 0.18637 |
| Magnesium chloride hexahydrate, MgCl2 6H2O | 1.8298 |
| Ammonium sulfate, (NH4)2SO4 | 0.07326 |
| Calcium sulfate, CaSO4 2H2O | 1.41632 |
| Calcium chloride, CaCl2 (anhydrous) | 1.8868 |
| Sodium chloride, NaCl | 14.1645 |
| Elements | Chemical Composition of CS-XC38 (% Mass) | Chemical Composition of CS-API K55 (% Mass) |
|---|---|---|
| C | 0.38 | 0.42 |
| Mn | 0.65 | 1.08 |
| Si | 0.10 | 0.34 |
| P | 0.017 | - |
| S | 0.0116 | 0.004 |
| Ni | 0.050 | 0.092 |
| Cr | 0.03 | 0.10 |
| Cu | 0.062 | - |
| Al | 0.022 | - |
| Fe | ~98.6753 | ~98.3840 |
| EXC38 (mV/SCE) | Immersion Time (h) | Jcorr (LRP) (µA cm−2) | Rp(LRP) (Ω cm2) | Rp(EIS) (Ω cm2) |
|---|---|---|---|---|
| −776 | 2 | 73 | 296 | 350 |
| −784 | 6 | 57 | 383 | 371 |
| −775 | 20 | 75 | 290 | 185 |
| −772 | 25 | 117 | 186 | 193 |
| −762 | 34 | 123 | 177 | 198 |
| Inhibitor | Content (mg L−1) | Rp(LRP) (Ω) | Jcorr(LRP) (µA cm−2) | IE(LRP) (%) | Jcorr (TP) (µA cm−2) | IE(TP) (%) | Rp(EIS) (Ω) | Jcorr(EIS) (µA cm−2) | IE(EIS) (%) |
|---|---|---|---|---|---|---|---|---|---|
| WI | 0 | 234.9 | 102.2 | / | 65.7 | / | 243.5 | 96.7 | / |
| PSIC | 5 | 2002.3 | 10.6 | 90 | 1.3 | 98 | 2809.2 | 7.7 | 92 |
| PSIN | 5 | 2632.9 | 9.5 | 91 | 1.3 | 98 | 2560.4 | 8.9 | 91 |
| BSID | 5 | 2045.6 | 18.7 | 81 | 20.3 | 79 | 1757.1 | 21.1 | 78 |
| BSID | 20 | 1240.4 | 19.6 | 80 | 5.6 | 94 | 1987.0 | 11.0 | 92 |
| BSID | 160 | 5108.5 | 7.59 | 92 | 1.4 | 98 | 6284.6 | 4.2 | 95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Betelu, S.; Helali, C.; Ignatiadis, I. Laboratory-Scale Implementation of Standardized Reconstituted Geothermal Water for Electrochemical Investigations of Carbon Steel Corrosion. Metals 2024, 14, 1216. https://doi.org/10.3390/met14111216
Betelu S, Helali C, Ignatiadis I. Laboratory-Scale Implementation of Standardized Reconstituted Geothermal Water for Electrochemical Investigations of Carbon Steel Corrosion. Metals. 2024; 14(11):1216. https://doi.org/10.3390/met14111216
Chicago/Turabian StyleBetelu, Stephanie, Chahinez Helali, and Ioannis Ignatiadis. 2024. "Laboratory-Scale Implementation of Standardized Reconstituted Geothermal Water for Electrochemical Investigations of Carbon Steel Corrosion" Metals 14, no. 11: 1216. https://doi.org/10.3390/met14111216
APA StyleBetelu, S., Helali, C., & Ignatiadis, I. (2024). Laboratory-Scale Implementation of Standardized Reconstituted Geothermal Water for Electrochemical Investigations of Carbon Steel Corrosion. Metals, 14(11), 1216. https://doi.org/10.3390/met14111216