Abstract
Using state-of-the-art first-principles electronic-band-structure calculations alongside density functional theory, we investigated the structural, elastic, electronic, and magnetic properties of LiMnZ2 (Z = Se, Te) compounds with a trigonal structure. Initially, we determined the equilibrium lattice structure and atomic positions, which aligned well with experimental values. Ferromagnetism was shown to be more favorable than the non-magnetic state. The elastic constants, cohesive energies, and formation energies indicated that the studied compounds were mechanically stable in the experimentally determined trigonal lattice. The analysis of spin-polarized band structures and density of states revealed that both LiMnZ2 compounds exhibited perfect half-metallic characters. The total spin magnetic moment per formula unit adhered to the Slater–Pauling rule, being exactly 4 μΒ, mainly concentrated at the Mn atoms due to the strong spin polarization of the Mn d orbitals. We anticipate that our results will prompt further experimental and computational studies for the application of these layered materials in practical devices.
1. Introduction
The ever-growing demand for data storage devices has driven researchers to explore materials with additional degrees of freedom. This need is particularly critical in advanced technological fields that require smaller and more efficient devices. Over the past few decades, the quest to harness the spin of electrons has given rise to a new technological field known as spintronics, which relies on the electron’s spin rather than its charge [1,2,3]. The manipulation of the spin degree of freedom is the key goal in the field of spintronics [4]. The rapid development was spurred by the discovery of Giant Magnetoresistance and the concept of spin valves, which revolutionized the capacity of hard disks, paving the way for consequential spintronic applications such as magnetic random access memories (MRAMs) [5].
Among the materials studied for spintronic applications, the so-called half-metals (HMs) are of particular interest [6]. Half-metals are ferromagnetic or ferrimagnetic materials for which the band structure behaves like metallics for the one spin channel, whereas the other spin band structure exhibits an energy gap at the Fermi level. The presence of the gap in one spin direction results in perfect spin polarization at the Fermi level, maximizing the efficiency of relevant spintronic devices [5]. Due to the integer number of states in the semiconducting spin band structure, the total spin magnetic moment per formula unit adopts integer values, a behavior known as the Slater–Pauling rule [7].
Recently, there has been noteworthy advancement in the discovery of new spintronic materials. Some notable reported or predicted half-metals are magnetic semiconductors, in which transition metal elements alloy with III–V or II–VI semiconductors [8,9]; oxides in metastable phases such as CrO2, which is verified experimentally as an HM at low temperatures [10]; and Heusler alloys [11]. Succeeding the experimental work carried out by Kim and collaborators [12,13] on the structural properties of layered ternary manganese chalcogenides, recent ab initio studies [14,15,16,17,18] have predicted half-metallicity in such compounds having the chemical formula AMnQ2, where A and Q denote alkali and chalcogenide atoms, respectively.
Among the AMnQ2 materials, the ternary-based lithium materials LiMnSe2 and LiMnTe2 were experimentally grown using solid-state reaction and cation exchange techniques by Kim and collaborators [12,13]. These new layered materials were found to crystallize in the non-centrosymmetric space group P3m1 (no. 156), adopting a trigonal structure similar to NaMnZ2 compounds [12,13]. Kim and collaborators reported the single-crystal structure and temperature-dependent magnetic susceptibility of these two compounds, but they were unable to determine the exact positions of the Li atoms [12,13]. Recently, ab initio calculations successfully demonstrated the stability of LiMnTe2 and predicted the half-metallic nature of this compound [17], but the authors assumed for LiMnTe2 a tetragonal structure, like the one adopted by KMnTe2 [12,13], and not the experimental trigonal one.
Our understanding is limited with regard to the physical properties of LiMnSe2 and LiMnTe2 compounds, and they have been overlooked in research. Consequently, the objective of this investigation was to explore the physical characteristics of these trigonal LiMnZ2 structures. This was accomplished through a computational analysis of the electronic, elastic, magnetic, and structural properties of these ternary manganese chalcogenides using advanced first-principles electronic-band-structure calculations. The outcome of this study is expected to offer the foundations for future experimental and computational studies and enable the development of materials for spintronic technology applications.
2. Calculation Details
The first step of our study was dedicated to the structural and elastic properties of the LiMnSe2 and LiMnTe2 materials. We employed the density-functional-based plane-wave pseudopotential method, as implemented in the CASTEP code [19]. On-the-fly-generated (OTFG) ultrasoft pseudopotentials were used to model the Coulomb interactions between the valence electrons and the rest of the atom (nucleus and core electrons) [20]. The cut-off energy for the plane wave basis was set to 700 eV. Integrations over the Brillouin zone (BZ) were performed using a 9 × 9 × 4 Monkhorst–Pack k-points grid [21]. To obtain the equilibrium lattice including the atomic equilibrium positions, the Broyden–Fletcher–Goldfarb–Shanno minimization scheme was used [22]. Finally, the elastic properties were determined using the finite-strain method, using a maximum amplitude of 0.003 Å for each of the four deformation steps [23].
The second step of our study involved the determination of the electronic and magnetic properties using electronic-band-structure calculations. To achieve the highest possible accuracy, we employed an all-electron method and performed self-consistent density-functional-based electronic-band-structure calculations at the equilibrium lattice structure. The method we chose was the full-potential linearized augmented plane-wave (FPLAPW) method, as implemented in the Wien2k code [24]. To account for the exchange-correlation potential, we employed the generalized gradient approximation (GGA), as parameterized by the Perdew–Burke–Ernzerhof (PBE) method [25]. The muffin-tin (MT) sphere radii 1.6, 1.8, 1.8, and 1.8 were chosen for Li, Mn, Se, and Te, respectively. The convergence of the basis set was determined by a cutoff parameter, defined as Rmt × Kmax = 8, where Rmt represents the smallest among the MT sphere radii and Kmax is the largest reciprocal lattice vector employed in the plane wave expansion. The cutoff energy, which delineated the distinction between valence and core states, was set to −6 Ry. Energy convergence was set at 0.0001 Ry for self-consistency iterations. In the context of Brillouin zone (BZ) integration, 1500 k-points spanning the entire BZ were employed to generate the charge density at each step of the self-consistency process. In the case of non-magnetic calculations, the total electronic charge was kept constant and, simultaneously, the spin-up and spin-down electronic charges were constrained to be equal. On the contrary, in the case of spin-polarized calculations, only the total electronic charge in the unit cell was kept constant and the spin-up and spin-down charge densities were allowed to converge. The atomic (total) spin magnetic moments were calculated as the difference between the integrated spin-up and spin-down charge densities in the muffin-tin sphere (unit cell). Although the atomic spin magnetic moments depended slightly on the choice of the MT radius, the total spin magnetic moment did not. The difference between the total and the sum of the atomic spin magnetic moments was the so-called “intertitial spin magnetic moment” which referred to the electrons in the interstitial region.
3. Results
3.1. Structural Properties
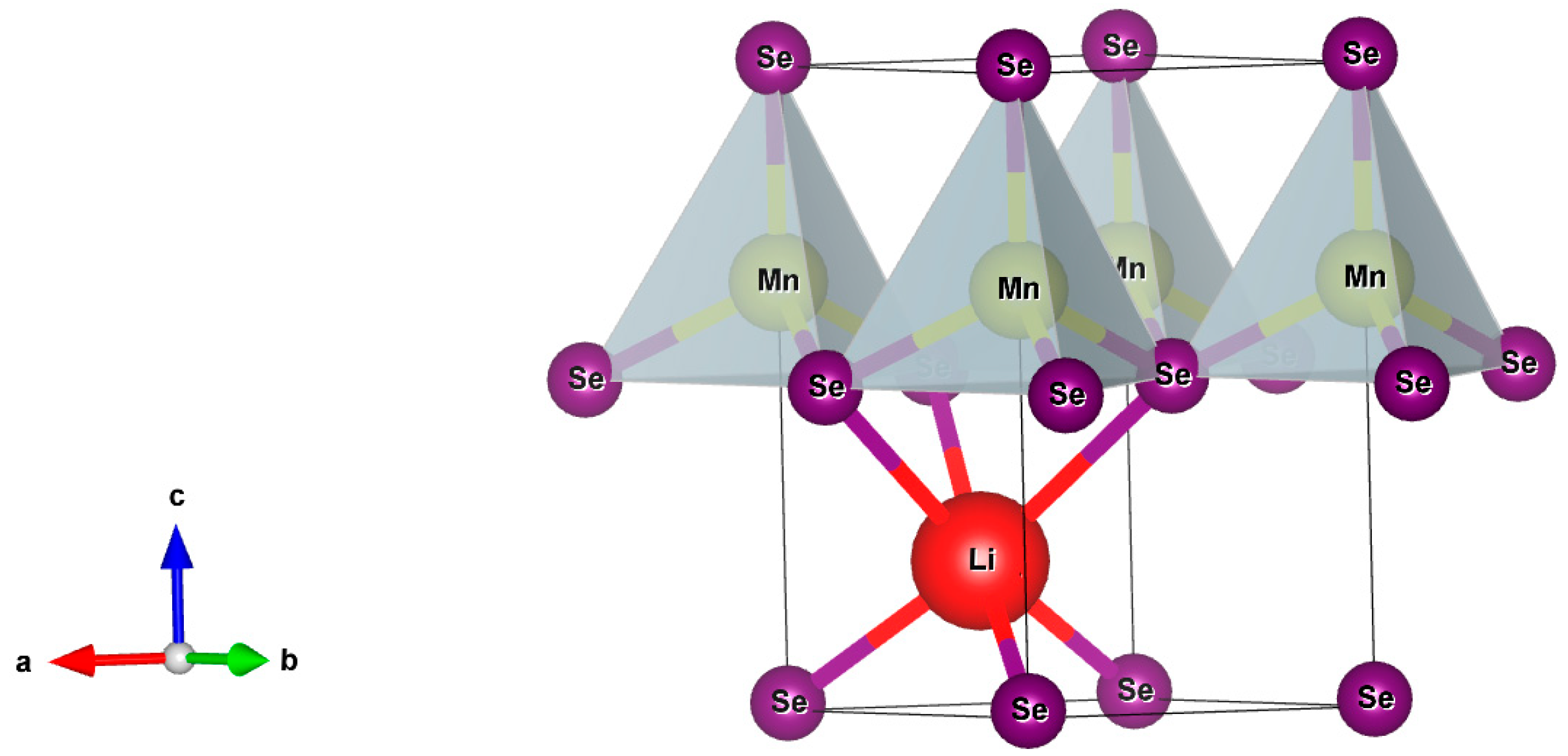
Analyses based on X-ray diffraction measurements revealed that synthetized LiMnSe2 and LiMnTe2 crystallized in a layered trigonal structure [12,13]. The crystal structure was described by the P3m1 space group (N° 156) and had a crystal system with a = b ≠ c, α = β = 90°, and γ = 120°. The conventional cell, shown in Figure 1, was identical to the one for the NaMnZ2 compounds [18] and could be viewed as consisting of periodic two-dimensional layers that stretched along the ab planes. The MnSe4 and MnTe4 tetrahedra formed the fundamental building blocks, which were separated by a Van der Waals (VDW) gap in between planes. Each lithium atom was bonded to six Se(Te) atoms to form distorted LiSe6(LiTe6) octahedra, which shared corners with six equivalent MnSe4(MnTe4) tetrahedra. Each LiMnZ2 structure was characterized by three shorter and three longer Li-Z bonds, as well as one shorter and three longer Mn-Z bonds, resulting in two inequivalent Z atoms per unit cell.
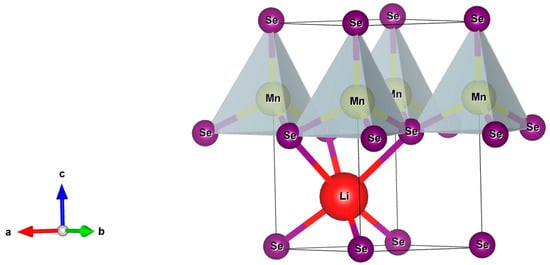
Figure 1.
A conventional cell of the LiMnZ2 (Z = Se, Te) trigonal structure. The purple circles represent the Se(Te) atoms, the red circles represent the Li atoms, and the rest represent the Mn atoms. The MnSe(Te)4 tetrahedra were the fundamental building blocks of the LiMnZ2 crystals. The colored arrows denote the cartesian coordinate system.
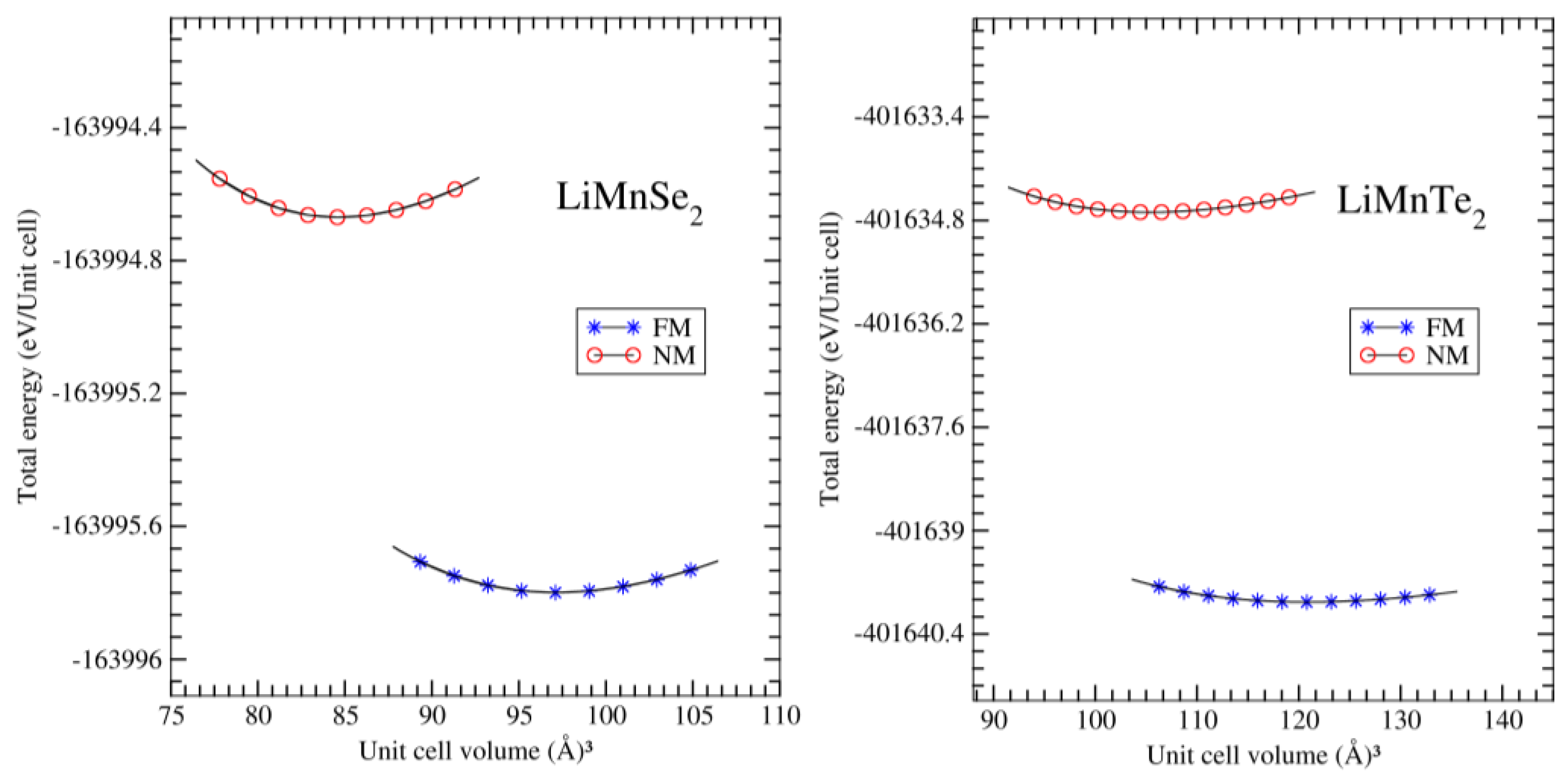
Employing the aforementioned computational method, the structural properties of the trigonal LiMnZ2 materials were examined in the non-magnetic (NM) and ferromagnetic (FM) configurations. The total energy for various volumes was computed, and these energies were used to fit a third-order Birch Murnaghan equation of state (EOS), as described below [26]:
The results are presented in Figure 2, indicating that the ferromagnetic order was the energetically preferred ground state for both investigated materials. The position of the minimum of the EOS defined the equilibrium lattice parameter and unit cell volume at zero pressure. Furthermore, Table 1 and Table 2 list the calculated crystal data and the experimental data, including the equilibrium lattice constants and atomic positions using Wyckoff coordinates. Notably, our computed lattice constants a and c for LiMnZ2 crystals are in satisfactory agreement with the experimentally determined lattice parameters reported by J. Kim et al. [12,13]. It can be observed that the lattice parameters a and c of the LiMnZ2 structures increased as the atomic number of chalcogenides atoms increased. This is in agreement with the trend observed in other compounds with similar crystal structures [18]. The relative uncertainty between the calculated and measured lattice constants a and c for LiMnSe2 and LiMnTe2 are reported in Table 1. It was found that the relative uncertainties were relatively low, being lower than 3.28% (3.13%) and 4.82% (4.99%) for a and c for LiMnSe2 and LiMnTe2, respectively. This indicated the reliability of the DFT calculations in predicting the lattice parameters of the LiMnZ2 structures. The observation that the error on the lattice constant c (corresponding to the direction of the VDW forces between ab planes) was similar to that measured in a direction is noteworthy. In our previous study on NaMnZ2 compounds [19] using the same (GGA-PBE) approach, it was found that the relative uncertainty between the calculated and the measured results became more significant concerning the c parameter compared to the a parameter. Note here that studied compounds AMnZ2 consist of layers extending in the ab planes perpendicular to the c-axis and bound by weak Van der Walls interactions. The difference between LiMnZ2 and NaMnZ2 compounds can be attributed to the fact that the VDW forces are more negligible in LiMnZ2 compounds compared to the other bonding forces, while they become important when Li atoms are replaced by Na atoms.
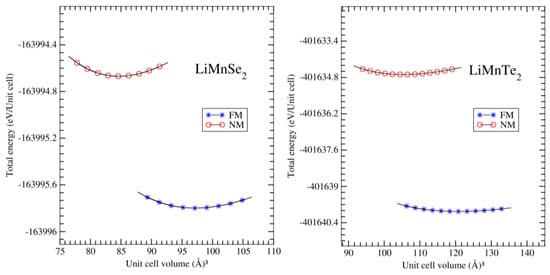
Figure 2.
For LiMnSe2 (left panel) and LiMnTe2 (right panel), we present the total energy per unit cell as a function of the unit cell volume. We considered for each compound both the ferromagnetic alignment of the Mn spin magnetic moments (FM) and the case where we obliged our atoms to carry no net spin magnetic moments (denoted as non-magnetic, NM).

Table 1.
For both compounds under study, we present the characteristics of the unit cell (lattice parameters a and c and unit-cell volume V) at the equilibrium. Using the results in Figure 2 for the FM case, we then present the calculated bulk modulus B and the pressure derivative of the bulk modulus B′. Finally, in the last four lines, we present the calculated cohesive Ecoh and formation Eform energies expressed per unit cell (one unit cell contained exactly one formula unit) and per atom. We compare our calculated values with the experimental ones in References [12,13]. d% corresponds to the relative deviation of the calculated values from the corresponding experimental ones.
Table 2.
Calculated atomic Wyckoff coordinates of the LiMnZ2 trigonal structures compared with available experimental data in References [12,13]; for Li atoms, the experiments were inconclusive regarding their exact position in the unit cell.
Based on the information provided, it appears that the experimental attempts to refine the position of the Li atom in both LiMnZ2 compounds were inconclusive due to the weak scattering power of the Li atom [12,13]. The calculated internal atomic coordinates of the Mn, Se, and Te atoms were reported to be close to the experimental values, which suggests that the theoretical method used to calculate these values was reliable. Table 3 lists selected interatomic distances, and the calculated shorter and longer distances reported agreed well with the measured ones.
Table 3.
Calculated selected interatomic distances compared to available experimental data in References [12,13] for LiMnZ2 trigonal structures.
The bulk modulus (B) plays a fundamental role in characterizing the physical properties of a material system. It provides a measure of stiffness and represents the energy required to induce volume deformation. Moreover, the bulk modulus reflects the bonding characteristics within the material and serves as an important indicator of its strength and hardness [27]. Several theoretical methods are available for evaluating the bulk modulus of materials. In this study, we employed two commonly used methods: the equation of state (EOS) method and the elasticity theory. EOS involves fitting volume–pressure data to an equation of state to estimate B. On the other hand, the elasticity theory provides a framework to calculate the bulk modulus based on the elastic constants [16]. Utilizing the EOS method, we employed a fitting procedure to derive the values of the bulk modulus (B) and its derivative (B’) by analyzing the energy–volume relationship of the LiMnZ2 compounds, as depicted in Figure 2. This fitting process involved the utilization of Equation (1). Although there were no experimental data available for comparison, the bulk modulus values obtained using GGA for both LiMnSe2 and LiMnTe2 listed in Table 1 demonstrated similarities to the calculated B values reported for other isostructural compounds [18].
Cohesive energy plays a crucial role in assessing the thermodynamic stability of crystalline materials, serving as a measure of the material’s ability to maintain its crystal structure under specific conditions. A higher cohesive energy absolute value suggests greater stability, indicating the presence of strongly bonded atoms within the material. To evaluate the thermodynamic stability of LiMnZ2 materials, we employed Equation (2), outlined below:
In Equation (2), Ecoh represents the combined energy of LiMnZ2 in its trigonal crystal structure. Ea represents the energies of the free atoms. As depicted in Table 1, the negative values of Ecoh signified the thermodynamic stability of both investigated LiMnZ2 crystals. We should note here that the cohesive energy is the opposite of the so-called atomization energy, which is the energy required to break a crystal in separate free atoms.
The formation energy Eform was given by the following expression [28]:
It described the crystal’s total energy minus the sum of the chemical potentials of the Li, Mn, and Z atoms (μLi, μMn, and μZ, respectively). The chemical potential of an element was considered equal to the total energy at its reference state (body-centered cubic for Li and Mn crystals, simple monoclinic for Se crystals, and simple trigonal for Te crystals), a widely used assumption [28]. In Table 1, we summarize the obtained values Eform. We should note here that negative Eform suggested the stability of the trigonal crystalline structure.
Often, the cohesion and formation energies are provided per atom instead of per formula unit. In this case, the two aforementioned expressions become and , respectively.
3.2. Elastic Properties
The elastic properties of crystals are elucidated through the utilization of single-crystal elastic constants, denoted as Cij. In the case of LiMnZ2, which crystallized in the trigonal system, the description of elastic properties necessitated the determination of six distinct elastic constants, namely, C11, C33, C44, C12, C13, and C14. Within the context of the studied crystals, the computed values of the elastic constants Cij are presented in Table 4.
Table 4.
Calculation-derived single-crystal elastic constants (Cij) in GPa for the LiMnSe2 and LiMnTe2 compounds.
The values of C33 were significantly greater compared to the C11 constants for LiMnSe2 and LiMnTe2, suggesting that these materials are less compressible along the c-axis compared to the a-axis. This result implies that the interatomic bonds along the crystal direction [001] were substantially greater in magnitude than the bonds along the [100] and [010] directions in both LiMnZ2 compounds. The low values of the C44 constants indicated a relatively lower resistance to shear deformation.
Mechanical stability is a crucial aspect of material studies and is determined by certain conditions imposed on their elastic constants. To ensure mechanical stability at zero pressure, a set of necessary and sufficient conditions must be fulfilled. In the context of materials exhibiting trigonal symmetry, the following conditions are crucial [29]:
The computed values of Cij for the materials under investigation, as presented in Table 4, satisfied the aforementioned criteria, providing evidence of their mechanical stability.
At the microscopic scale, materials can be conceptualized as consisting of individual monocrystals that possess unique crystallographic orientations and exhibit anisotropic mechanical properties. However, in reality, when viewed from a macroscopic perspective, the majority of materials are polycrystalline, consisting of multiple crystal grains with different orientations. Macroscopic analysis considers the statistical distribution of grain sizes, orientations, and grain boundary characteristics to describe the overall behavior of polycrystalline materials. Consequently, to accurately characterize these materials, it becomes necessary to determine their macroscopic properties using alternative parameters known as polycrystalline elastic moduli, including the bulk modulus, shear modulus, Young’s modulus, and Poisson’s ratio [15]. In theoretical terms, the polycrystalline isotropic bulk modulus B and shear modulus G can be estimated by employing Voigt–Reuss–Hill approximations [30]. These methods provide a means to relate the properties of a polycrystalline material to the single elastic constants Cij. Subsequently, the estimation of the Young’s modulus (E) and Poisson’s ratio (σ) becomes possible using the established equations [31]:
The B/G ratio, proposed by Pugh [32], is a parameter used to assess the brittleness and ductility of materials. According to Pugh’s empirical criterion, materials with B/G > 1.75 are considered ductile, while those with B/G < 1.75 are classified as brittle. A higher B/G ratio indicates better ductility in a material. In Table 5 we present our calculated values for the above mentioned parameters for both compounds under study. In the context of our study, both compounds demonstrated an exceptionally high B/G ratio, indicating remarkable ductility.
Table 5.
Calculation-derived polycrystalline elastic moduli for the two compounds under study. The bulk modulus (B), shear modulus (G), and Young’s modulus (E) are given in GPa units. The Poisson’s ratio (σ), Pugh’s ratio B/G, and universal anisotropy factor AU are dimensionless.
3.3. Electronic Properties
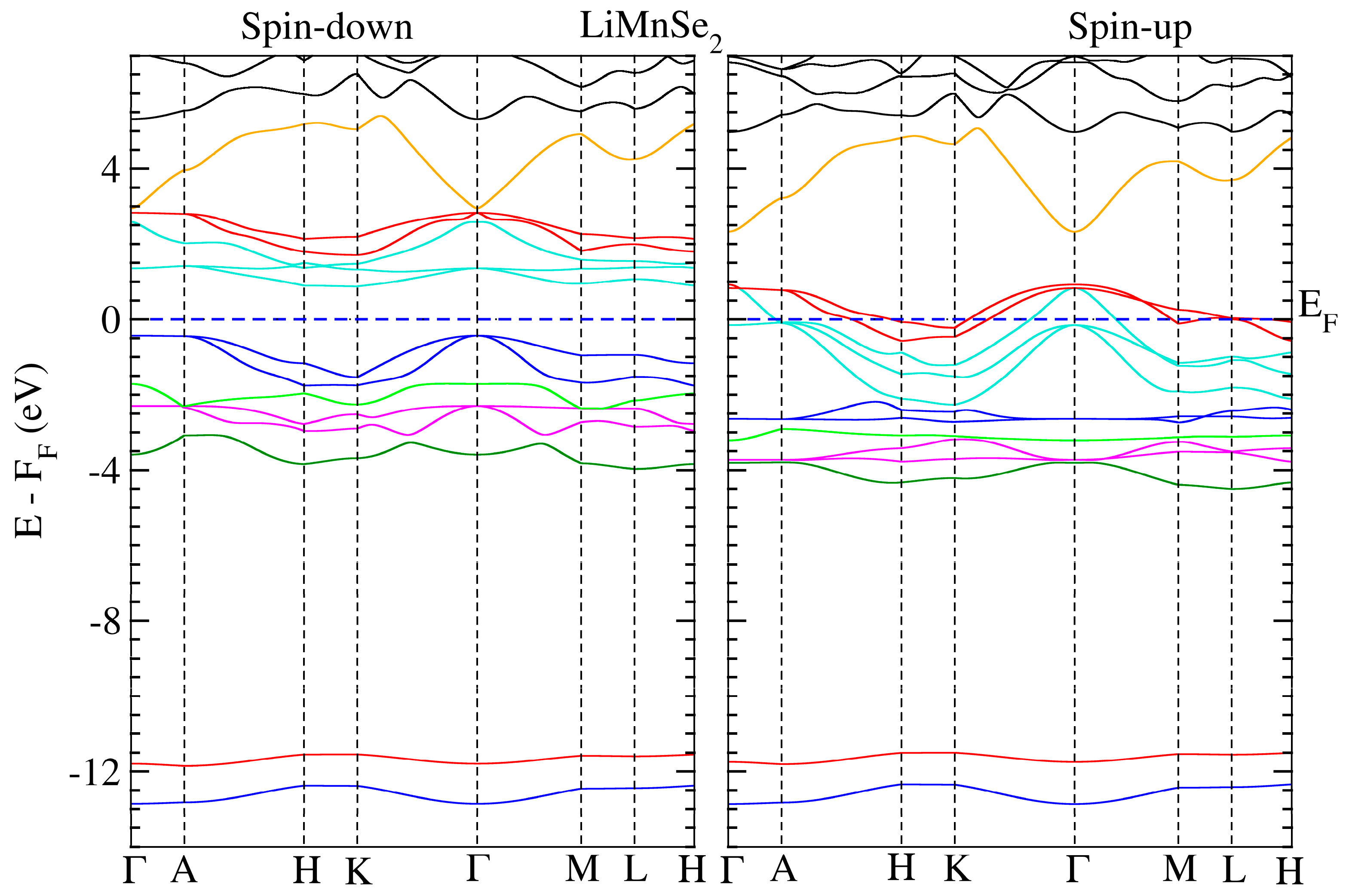
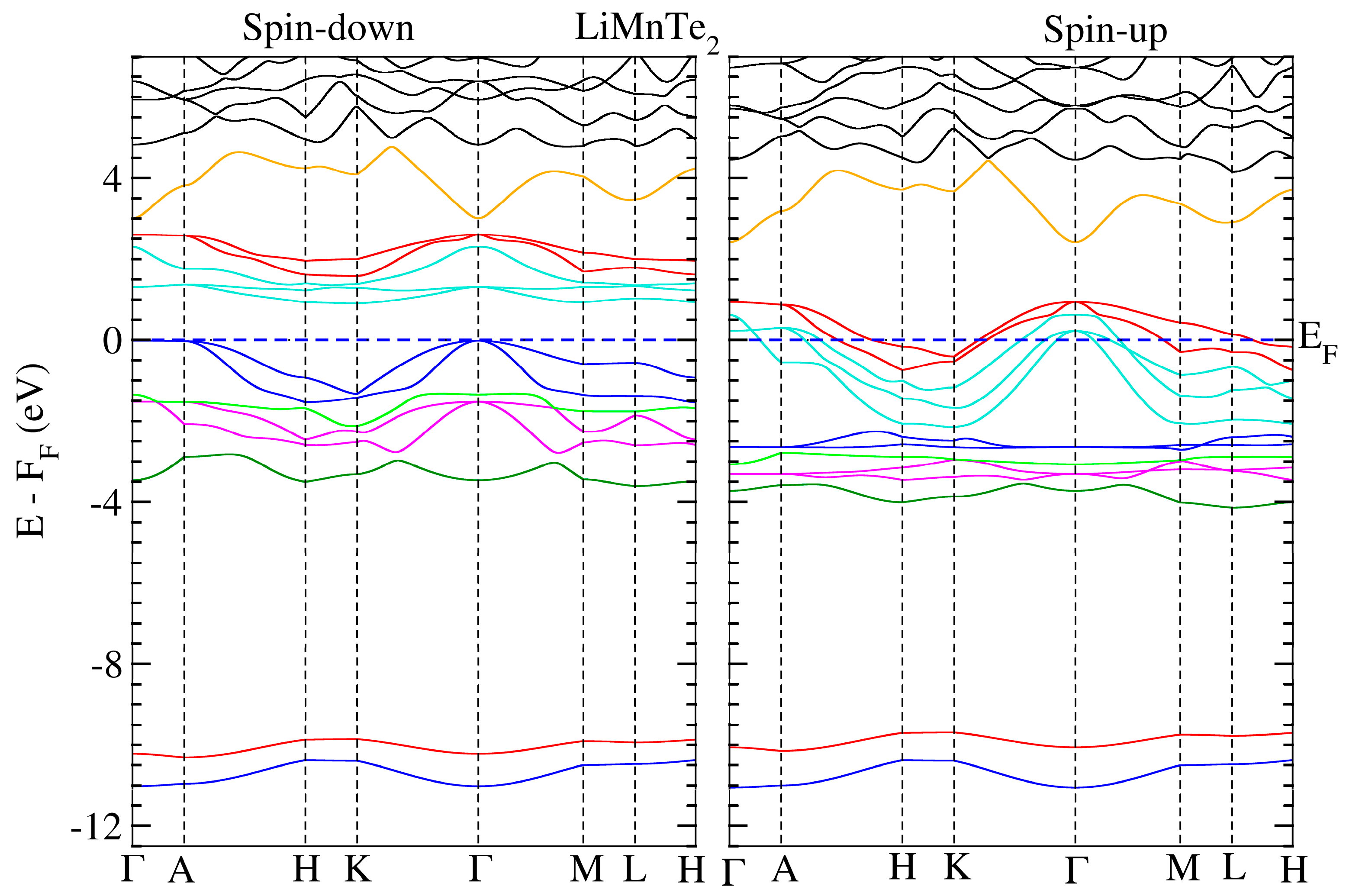
We conducted electronic structure calculations on the crystals under consideration. The optimum parameters were used for these computations. The calculated spin-polarized energy band dispersion is depicted in Figure 3 and Figure 4 for LiMnZ2 compounds, along the lines connecting the high symmetry points in the Brillouin Zone. LiMnZ2 alloy band topologies showed similarities in their characteristics. In each compound, the energy bands corresponding to the majority spin states (spin-up) intersected at the Fermi level. However, the situation for the minority spin states (spin-down) was not the same. The band structure of the minority spin channel revealed a band gap of approximately 1.2 eV and 0.89 eV for LiMnSe2 and LiMnTe2, respectively. The contrasting behavior observed between the spin-up and spin-down band structures indicated the half-metallic nature of the LiMnZ2 alloys.
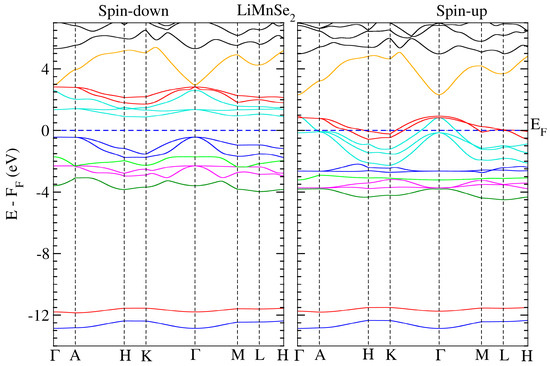
Figure 3.
Calculated band structure for LiMnSe2 along several high-symmetry axes for both spin-down (left panel) and spin-up (right panel) directions. The dashed line denotes the Fermi level.
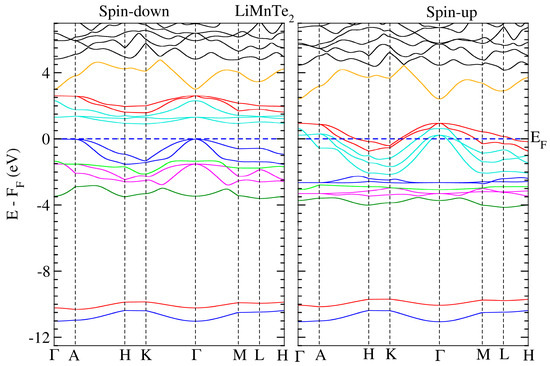
Figure 4.
Similar to Figure 2 for LiMnTe2.
The properties and effectiveness of half-metallic materials can be characterized by two significant factors: the half-metallic gap EHM and the band gap Eg. These parameters provide important details regarding the electronic and magnetic characteristics of these materials. The so-called half-metallic gap EHM represents the energy needed for an electron at the Fermi level to flip its spin. It is determined by taking the energy difference between the lowest energy of the spin-down conduction band and the Fermi level. On the other hand, the spin-down band gap, Eg, quantifies the energy separation between the minimum of the conduction band (CBM) and the maximum of the valence band (VBM) in the spin-down channel. The estimated values of EHM and Eg for LiMnSe2 and LiMnTe2 are presented in Table 6. The values of EHM and Eg are crucial in evaluating the suitability of half-metallic materials for various applications, such as spintronic and magneto-electronics. A large EHM indicates a more pronounced half-metallicity, offering better spin polarization and enhanced efficiency in spin-dependent devices. Typically, the half-metallic behavior of compounds based on d transition elements is unstable and disrupted when a lattice undergoes slight strain, primarily due to the narrow half-metallic gap EHM [33]. Similarly, a wider Eg signifies a larger energy gap, leading to improved insulation properties and reduced leakage currents. Consequently, between the two studied materials, LiMnSe2 compounds can be considered a promising candidate for practical spintronic applications. This is attributed to its remarkable half-metallic behavior characterized by wide EHM and Eg gaps.
Table 6.
Calculated total (μtot) and atomic (μLi, μMn, μZ) spin magnetic moments in μΒ. μinter is the spin magnetic moment in the interstitial region. We also present in the last three lines (a) the spin-down energy gap (Eg) in eV, (b) the half-metallic gap (EHM) in eV, and (c) the density of states of the spin-up states at the Fermi level N(up,EF) in states/eV/spin/atom units. Z1 and Z2 correspond to the two inequivalent Se(Te) atoms in the unit cell. We also present the experimental results for the total spin magnetic moment.
Considering that the band gap is a crucial factor in half metals, understanding its source becomes significant. Currently, numerous half-metallic materials have been identified, and they can be classified into three categories based on the type of band gap. The first category is characterized by a covalent band gap—a well-known example is NiMnSb [11]. In this category, the band gap arises from the covalent bonding between atoms. The second category exhibits a charge transfer band gap, commonly found in strongly magnetic compounds. In these materials, the minority spin bands of the transition metal are unoccupied, while the itinerant s and p electrons of the transition metal become localized on the anions. Examples of half metals in this category include CrO2 and double perovskites [10]. Naturally, compounds in this category tend to exhibit strong magnetism. The third category encompasses a class of half metals characterized by d-d band gaps due to the crystal field. In this case, the energy bands are relatively narrow. In these materials, the exchange splitting effect results in the Fermi level being positioned within a gap only for one spin direction. As a result, materials falling into this category are inherently weak magnets. Prominent examples of such half metals include Fe3O4, FexCo1−xS2, and Mn2VAl [34]. The compounds under study here presented a spin-down energy gap of similar origin as the NaMnZ2 compounds studied in Reference [18]. The gap, as discussed below, was created between the occupied valence p states of the Se(Te) atoms and the unoccupied valence d states of the Mn atom. Thus, it was of a covalent nature, like in the first case where NiMnSb belongs.
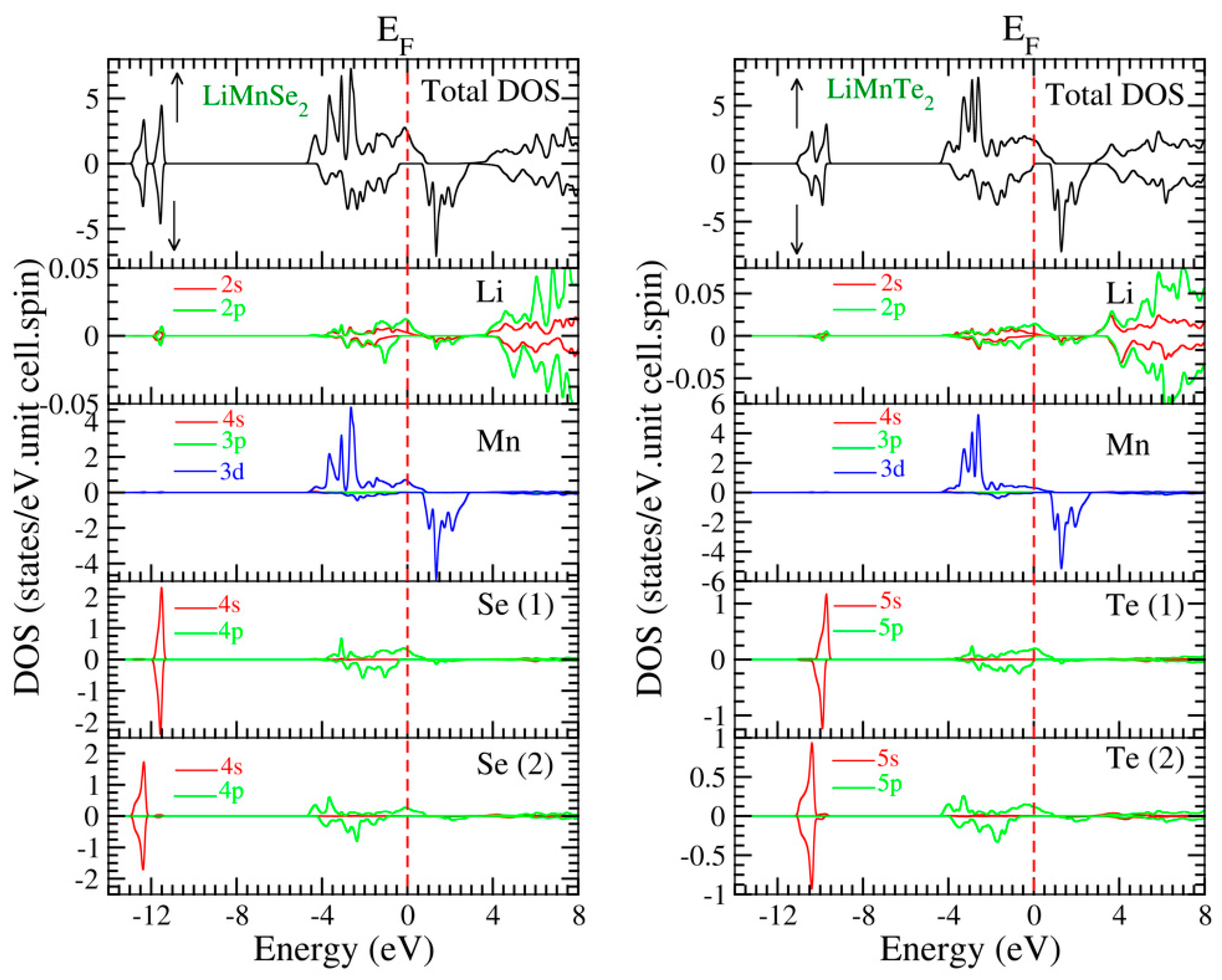
To further study the electronic properties, we examined the spin-polarized total and atom-resolved density of states (TDOS and ADOS, respectively). Figure 5 illustrates the DOS spectra of the mentioned materials. The asymmetry in the density of states (DOS) between the two spin channels is evident in Figure 5 for both compounds LiMnSe2 and LiMnTe2. This asymmetry explains the magnetic properties observed in both materials.
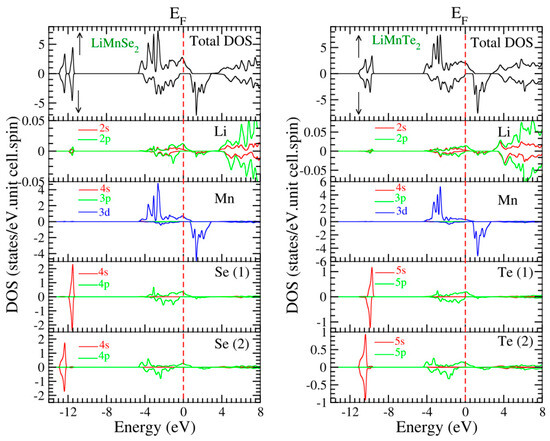
Figure 5.
Calculated spin-projected total and atom-resolved density of states for the two compounds under study (LiMnSe2 in the (left panel) and LiMnTe2 in the (right panel)). The Fermi level, depicted by a vertical dashed line, is adjusted to zero for reference. Note that the scale of the vertical DOS axis is different for the various atoms.
The TDOS spectra for each structural configuration can be subdivided into two regions below the Fermi level and two regions above the Fermi level. The first region, characterized by symmetrical spin-up and spin-down patterns, is situated at a lower energy, significantly distant from the Fermi level, and is primarily composed of Z-s orbitals. The second region, located just below the Fermi level, displays antisymmetric spin-up and spin-down profiles, primarily stemming from Mn-d and Z-p states, with a small contribution from Li-s, Li-p, and Mn-p states (note that in Figure 5 the scale for the vertical DOS axis is different). The third region just above the Fermi level is characterized by the spin-down Mn d states, with a small admixture of Se(Te) p-states. The fourth region well above the Fermi level is primarily composed of a blend of antibonding s and p states originating from the Li atom.
We will focus on the two regions just above and just below the Fermi level to discuss the origin of the spin-down energy gap. The occurrence of covalent bonding between the Z and Mn atoms was due to the hybridization between the Mn-d and the Se(Te)-p orbitals. The resulting bonds had most of their weight on the Se(Te) atoms below the Fermi level and on the Mn atoms above the energy gap. This covalent p-d bonding was due to the triple degenerate t2g d-orbitals of Mn, which transformed following the same irreducible representation with the Se(Te) p-orbitals, and thus could hybridize between them [16,18]. Li atoms provided one valence electron to the compounds and kept the LiMnZ2 (Z = Se, Te) sandwiches apart, resulting in vanishing ADOS in the valence band region [17].
3.4. Magnetic Properties
In the case of transition metal compounds, one can ignore the orbital part of the total magnetic moment, which stems almost exclusively from the spin degree of freedom and, thus, (in μB units) equals the number of spin-up valence electrons minus the number of spin-down valence electrons [14]. Table 6 shows the calculated atomic and total spin magnetic moments for the LiMnSe2 and LiMnTe2 compounds. Both studied materials had a total spin magnetic moment per unit cell of exactly 4 μB, in accordance with their half-metallic character discussed above. These calculated values for LiMnSe2 and LiMnTe2 deviated from the measured ones by about 14.5% and 18.4%, respectively, exhibiting behavior similar to the NaMnZ2 compounds [18]. There is a wide range of reasons for the observed discrepancy between the calculations and the experiments, such as impurities, defects, or incomplete crystallinity in the synthesized samples.
The data in Table 6 suggest that Mn carried most of the spin magnetic moment in the LiMnZ2 materials, as was the case also for other Mn-based chalcogenides [14,15,16,17,18]. Li atoms had a negligible magnetic moment, while Se and Te atoms carried small negative or positive spin magnetic moments. This behavior was due to the large exchange splitting between the unoccupied minority-spin and occupied majority-spin states of the Mn atom. Interestingly, a large portion of the spin magnetic moment was located at the interstitial region. To elucidate the origin of this spin moment, we also performed ab initio calculations using the full-potential nonorthogonal local-orbital minimum-basis band structure approach (FPLO) in conjunction with the same GGA-PBE exchange–correlation potential [35]. FPLO is well known to produce almost identical results with FLAPW, without the need for an interstitial region. FPLO for both compounds under study produced total and atom-resolved DOS identical to the ones presented above in Figure 5. With respect to the spin magnetic moments, FPLO also produced half-metallicity, with a total spin magnetic moment of 4 μB and similar spin magnetic moments for the Li and Se(Te) atoms. The spin magnetic moment at the Mn site computed using FPLO equaled the sum of the Mn and interstitial spin magnetic moments using the FPLAPW method. Thus, the spin magnetic moment at the interstitial region calculated using FLAPW was actually distributed at the region around the muffin-tin sphere of the Mn atom.
In the case of half-metallic Heusler compounds, it was initially shown that the electronic (total number of valence electrons Zt in the formula unit) and magnetic (total magnetic moment Mt) properties are connected through the so-called Slater–Pauling rules [11,14]. This concept was later generalized for the rest of the half-metallic compounds. In the case of the two LiMnZ2 compounds under study, the total spin magnetic moment per formula unit was exactly 4 μB, as mentioned above. The total number of valence electrons in the unit cell was 20; the Li atom had one valence electron in the 2s orbital, the Mn atom had seven valence electrons (two in the Mn-4s states and five in the spin-up Mn-3d states), and each Se(Te) atom had six valence electrons, two in the Se-4s(Te-5s) states and four in the Se-4p(Te-5p) states. Thus, the linear relationship between Mt and Zt should be Mt = (Zt − 16) μB. This form of the Slater–Pauling rule implies that the spin-down states below the Fermi level accommodate exactly eight valence electrons. This is confirmed if we examine in detail the character of the occupied spin-down states in Figure 5; there are 1 × s (Se,Te), 1 × s (Se,Te), 3 × p (Se,Te), and 3 × p (Se,Te) states for each considered compound (there are two inequivalent Se or Te atoms per unit cell, as discussed above). This expression of the Slater–Pauling rule is slightly different from that for NaMnZ2 in Reference [18]: Mt = (Zt − 22) μB, where Na also contributed to the spin-down occupied states, with 3 × p states, and the number of spin-down occupied states was 11 instead of 8.
4. Conclusions
Mn-based ternary chalcogenides have been synthesized experimentally, but they have received limited theoretical attention. Utilizing advanced first-principles electronic band structure methods rooted in density functional theory, we investigated the structural, elastic, electronic, and magnetic properties of the LiMnSe2 and LiMnTe2 compounds in their experimentally observed trigonal lattice structure. Our findings indicate that these two compounds are structurally stable, aligning well with existing experimental data. Both materials are predicted to be half-metallic ferromagnets, characterized by a spin-down band with semiconducting properties and a spin-up band with metallic properties. In each compound, the total spin magnetic moment per formula unit (or per unit cell), Mt, adopted an integer value of 4 μB, consistent with half-metal behavior. The Slater–Pauling relationship between the total number of valence electrons in the unit cell, Zt, and the magnetic moment, Mt = (Zt − 16) μB, is supported by symmetry considerations and the permissible interactions between the orbitals of neighboring atoms.
We expect our results to pave the way for further experimental studies on the properties of the Mn-based chalcogenides and their implementation in realistic spintronic and magneto-electronic devices.
Author Contributions
Conceptualization, A.B. (Abdennour Benmakhlouf) and A.B. (Abdelmadjid Bouhemadou); methodology, A.B. (Abdennour Benmakhlouf); validation, A.B. (Abdennour Benmakhlouf), F.F., N.G., K.Ö. and T.H.; formal analysis, A.B. (Abdennour Benmakhlouf), F.F., N.G., K.Ö., T.H. and A.B. (Abdelmadjid Bouhemadou); data curation, A.B. (Abdennour Benmakhlouf), F.F., N.G., K.Ö., T.H. and A.B. (Abdelmadjid Bouhemadou); writing—original draft preparation, A.B. (Abdennour Benmakhlouf); writing—review and editing, I.G.; supervision, A.B. and I.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors on request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Al-Qhtani, M.; Mustafa, G.M.; Mazhar, N.; Bouzgarrou, S.; Mahmood, Q.; Mera, A.; Zaki, Z.I.; Mostafa, N.Y.; Alotaibi, S.H.; Amin, M.A. Half Metallic Ferromagnetism and Transport Properties of Zinc Chalcogenides ZnX2Se4 (X = Ti, V, Cr) for Spintronic Applications. Materials 2021, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Awschalom, D.D.; Kikkawa, J.M. Electron spin and optical coherence in semiconductors. Phys. Today 1999, 52, 33–38. [Google Scholar] [CrossRef]
- Dey, P.; Roy, J.N. (Eds.) Spintronics Applications, Spintronics: Fundamentals and Applications; Springer: Singapore, 2021. [Google Scholar]
- Žutić, I.; Fabian, J.; Sarma, S.D. Spintronics: Fundamentals and applications. Rev. Mod. Phys. 2004, 76, 323. [Google Scholar] [CrossRef]
- Kalyani, V.L.; Agrawal, V. Spintronics—A vision for future in electronics and computers. J. Manag. Eng. Inf. Technol. 2015, 2, 30–36. [Google Scholar]
- De Groot, R.; Mueller, F.; van Engen, P.; Buschow, K. New class of materials: Half-metallic ferromagnets. Phys. Rev. Lett. 1983, 50, 2024. [Google Scholar] [CrossRef]
- Galanakis, I. Slater–Pauling Behavior in Half-Metallic Heusler Compounds. Nanomaterials 2023, 13, 2010. [Google Scholar] [CrossRef] [PubMed]
- Munekata, H.; Ohno, H.; Von Molnar, S.; Segmüller, A.; Chang, L.; Esaki, L. Diluted magnetic III-V semiconductors. Phys. Rev. Lett. 1989, 63, 1849. [Google Scholar] [CrossRef]
- Tanaka, M.; Higo, Y. Large tunneling magnetoresistance in GaMnAs/AlAs/GaMnAs ferromagnetic semiconductor tunnel junctions. Phys. Rev. Lett. 2001, 87, 026602. [Google Scholar] [CrossRef]
- Ji, Y.; Strijkers, G.; Yang, F.; Chien, C.; Byers, J.; Anguelouch, A.; Xiao, G.; Gupta, A. Determination of the spin polarization of half-metallic CrO2 by point contact Andreev reflection. Phys. Rev. Lett. 2001, 816, 5585. [Google Scholar] [CrossRef]
- Tavares, S.; Yang, K.; Meyers, M.A. Heusler alloys: Past, properties, new alloys, and prospects. Prog. Mater. Sci. 2023, 132, 101017. [Google Scholar] [CrossRef]
- Kim, J.; Hughbanks, T. Synthesis and structures of new layered ternary manganese selenides: AMnSe2 (A = Li, Na, K, Rb, Cs) and Na2Mn2Se3. J. Sol. State Chem. 1999, 146, 217–225. [Google Scholar] [CrossRef]
- Kim, J.; Wang, C.; Hughbanks, T. Synthesis and Structures of New Layered Ternary Manganese Tellurides: AMnTe2 (A = Li, Na). Inorg. Chem. 1998, 37, 1428–1429. [Google Scholar] [CrossRef]
- Benmakhlouf, A.; Bentabet, A.; Bouhemadou, A.; Benghia, A. Prediction of half-metallic properties for the AMnSe2 (A = Rb, Cs) compounds from first-principles calculations. J. Magn. Magn. Mater. 2016, 399, 179–184. [Google Scholar] [CrossRef]
- Benmakhlouf, A.; Bentabet, A.; Bouhemadou, A.; Maabed, S.; Benghia, A.; Khenata, R.; Bin-Omran, S. Structural, half-metallic magnetism and elastic properties of the KMnQ2 (Q = O, S, Se, Te) chalcogenides from first-principles calculations. J. Magn. Magn. Mater. 2016, 408, 199–205. [Google Scholar] [CrossRef]
- Benmakhlouf, A.; Bourourou, Y.; Bouhemadou, A.; Bentabet, A.; Khemloul, F.; Maabed, S.; Bouchenafa, M.; Galanakis, I. Structural, electronic and magnetic properties of the manganese telluride layers AMnTe2 (A = K, Rb, Cs) from first-principles calculations. J. Magn. Magn. Mater. 2018, 465, 430–436. [Google Scholar] [CrossRef]
- Yaqoob, N.; Sabir, B.; Murtaza, G.; Khalil, R.M.A.; Muhammad, N.; Laref, A. Structural, electronic, magnetic, optical and thermoelectric response of half-metallic AMnTe2 (A = Li, Na, K): An ab-initio calculations. Phys. B Condens. Matter 2019, 574, 311656. [Google Scholar] [CrossRef]
- Ghermoul, N.; Benmakhlouf, A.; Faid, F.; Bourourou, Y.; Bouhemadou, A.; Fakhreddine, K.; Maabed, S.; Bouchenafa, M.; Bentabet, A.; Galanakis, I. Ab initio prediction of half-metallicity in the NaMnZ2 (Z = S, Se, Te) ternary layered compounds. Comput. Condens. Matter 2022, 33, e00754. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist.-Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Fischer, T.H.; Almlof, J. General methods for geometry and wave function optimization. J. Phys. Chem. 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Murnaghan, F. Finite Deformation of an Elastic Solid; Wiley: New York, NY, USA; Chapman and Hall: London, UK, 1951. [Google Scholar]
- Blaha, P.; Schwarz, K.; Tran, F.; Laskowski, R.; Madsen, G.K.; Marks, L.D. WIEN2k: An APW+ lo program for calculating the properties of solids. J. Chem. Phys. 2020, 152, 074101. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Birch, F. Finite elastic strain of cubic crystals. Phys. Rev. 1947, 71, 809. [Google Scholar] [CrossRef]
- Kim, E.; Chen, C. Calculation of bulk modulus for highly anisotropic materials. Phys. Lett. A 2004, 326, 442–448. [Google Scholar] [CrossRef]
- Emery, A.A.; Wolverton, C. High-throughput dft calculations of formation energy, stability and oxygen vacancy formation energy of abo 3 perovskites. Sci. Data 2017, 4, 170153. [Google Scholar] [CrossRef]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Hill, R. Proceedings of the Physical Society. Section A 1952, 65, 349. [Google Scholar]
- Ravindran, P.; Fast, L.; Korzhavyi, P.A.; Johansson, B.; Wills, J.; Eriksson, O. Density functional theory for calculation of elastic properties of orthorhombic crystals: Application to TiSi2. J. Appl. Phys. 1998, 84, 4891–4904. [Google Scholar] [CrossRef]
- Pugh, S. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1954, 45, 823–843. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, Z.; Wang, J.; Wang, L.; Yu, Z.; Fang, C.; Yang, J.; Liu, G. Origin of the half-metallic band-gap in newly designed quaternary Heusler compounds ZrVTiZ (Z = Al, Ga). RSC Adv. 2016, 6, 57041–57047. [Google Scholar] [CrossRef]
- Fang, C.M.; de Wijs, G.A.; de Groot, R.A. Spin-polarization in half-metals. J. Appl. Phys. 2002, 91, 8340–8344. [Google Scholar] [CrossRef]
- Koepernik, K.; Eschrig, H. Full-potential nonorthogonal local-orbital minimum-basis band-structure scheme. Phys. Rev. B 1999, 59, 1743. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).