
Bacteria–Cancer Interface: Awaiting the Perfect Storm



Abstract
:1. Introduction
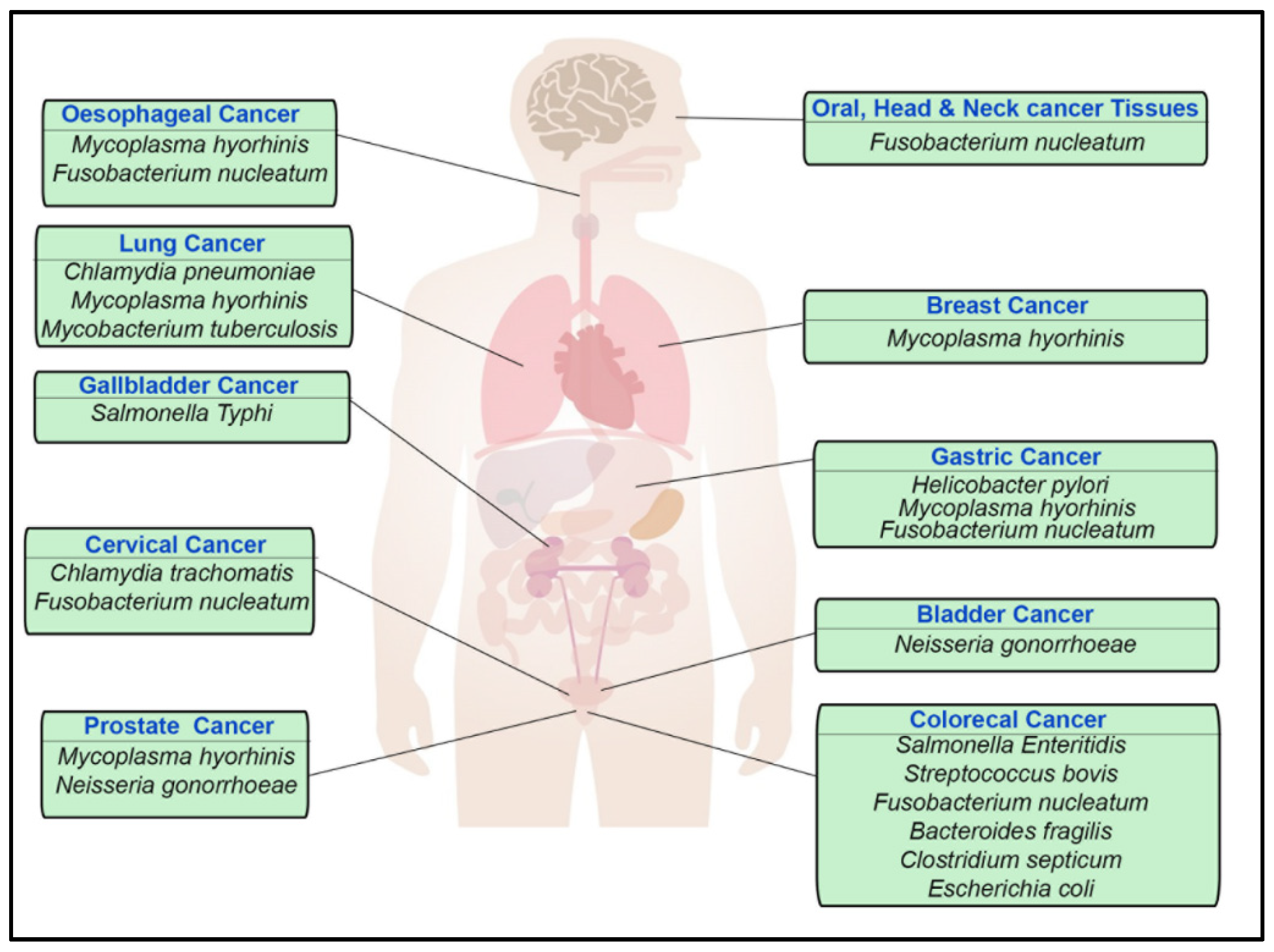
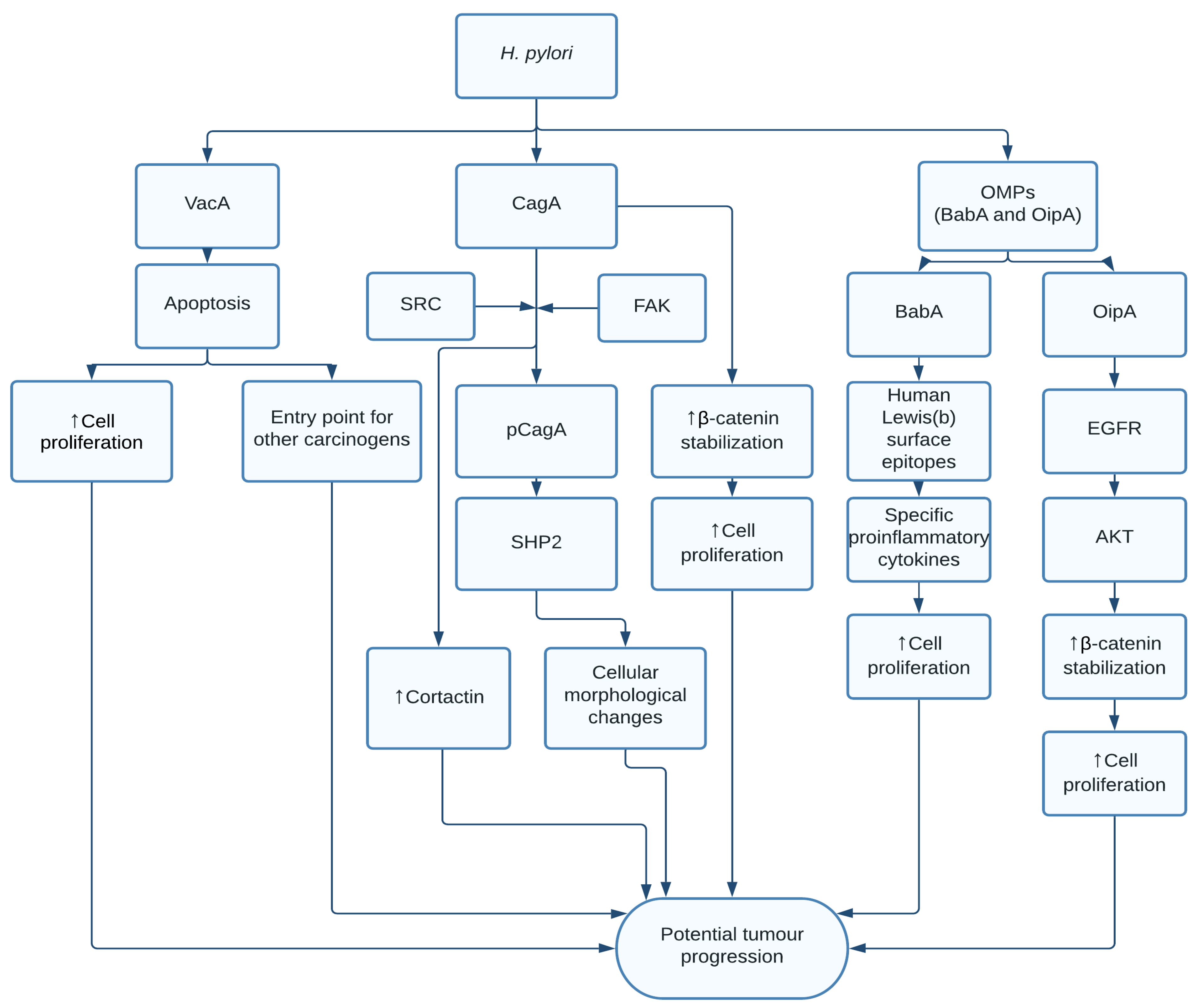
1.1. Helicobacter pylori
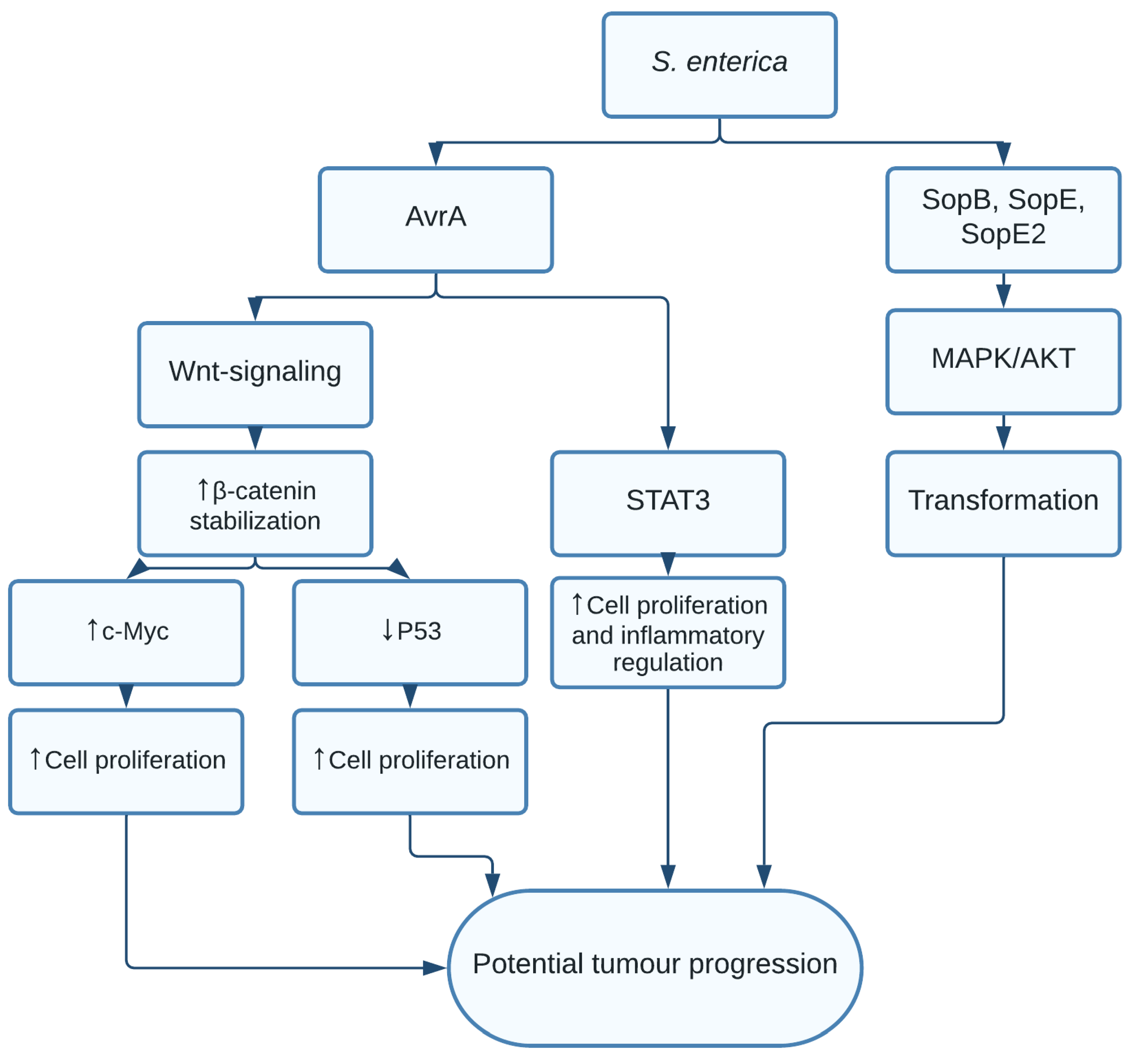
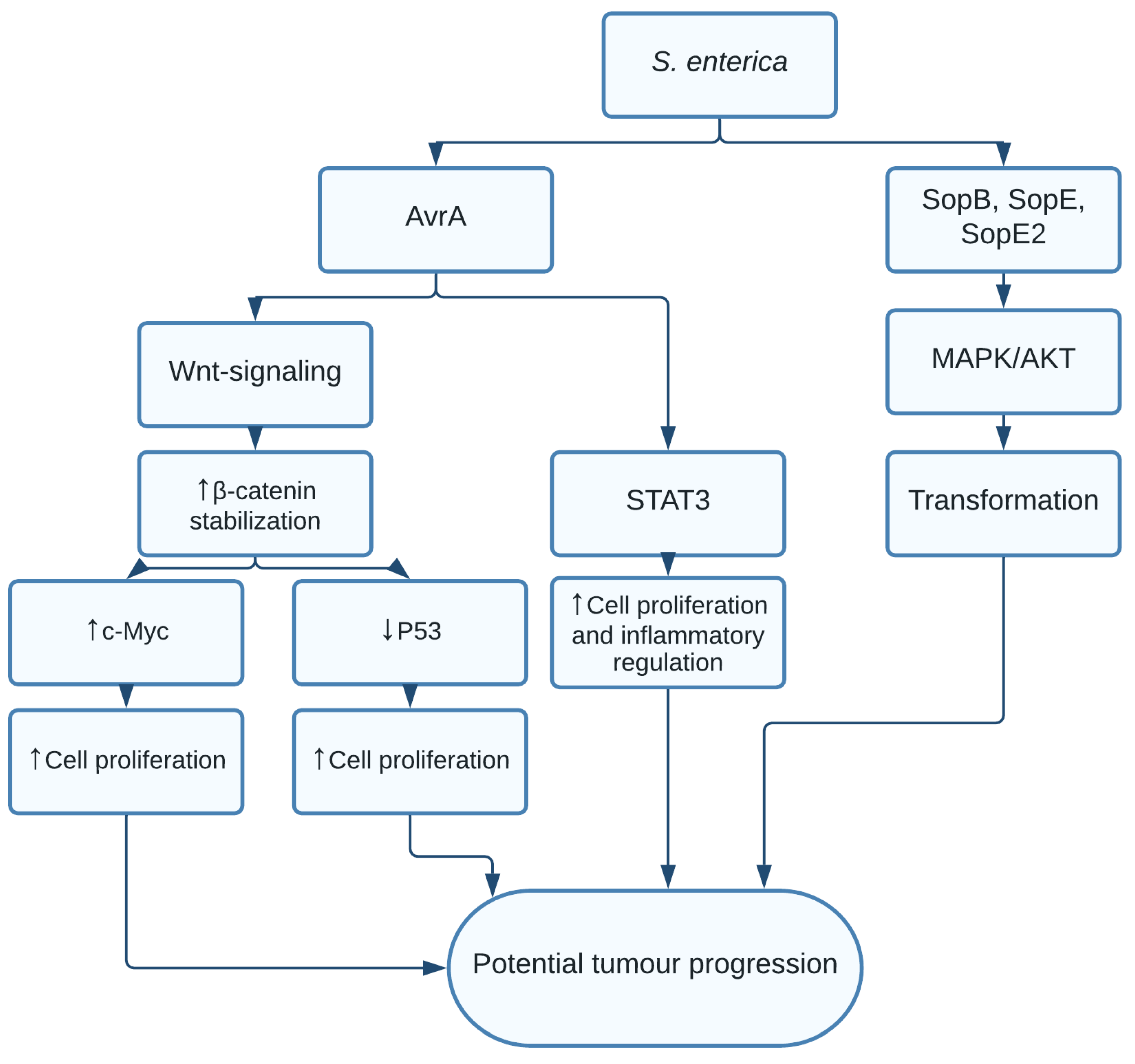
1.2. Salmonella enterica and Salmonella typhi
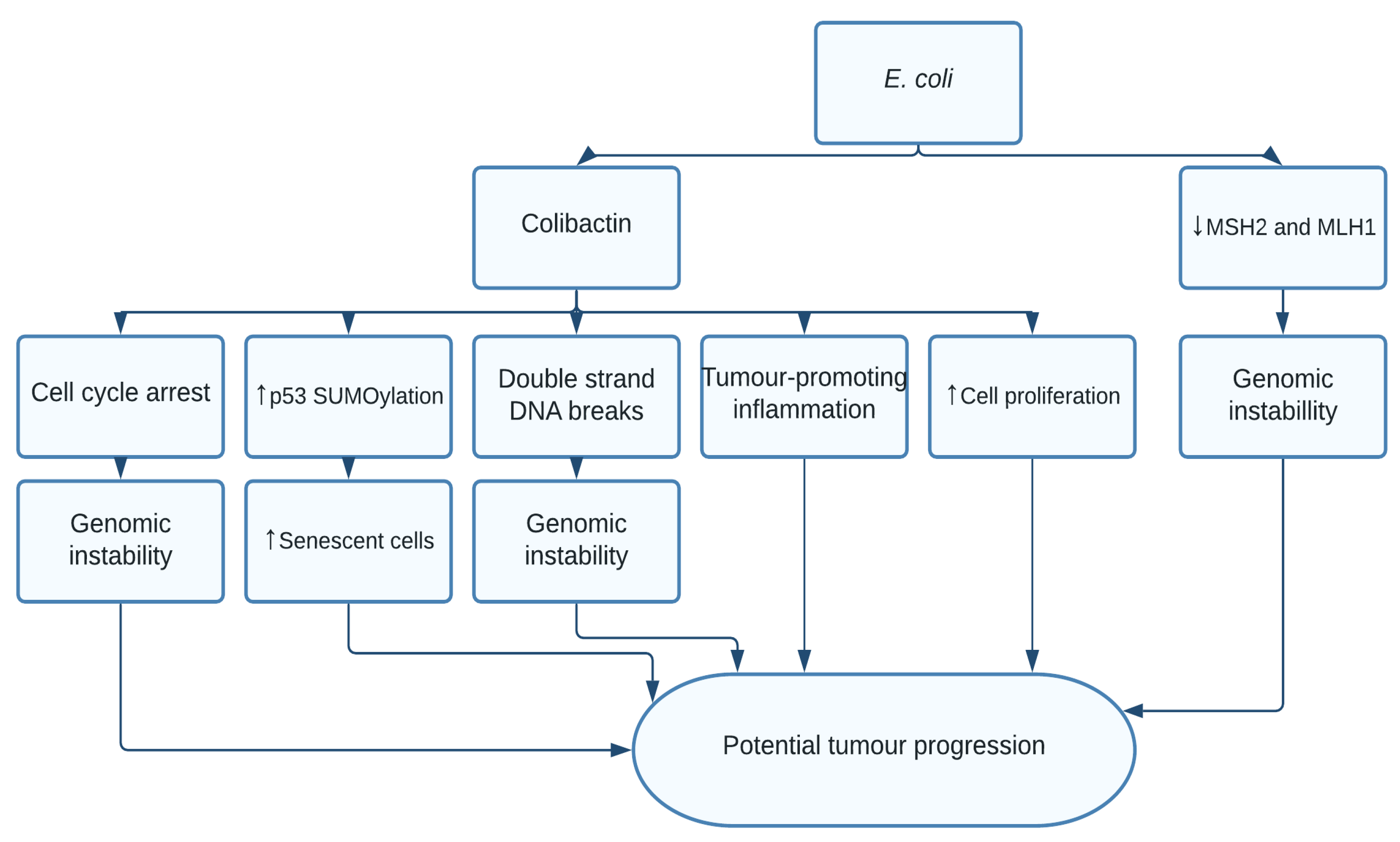
1.3. Escherichia coli
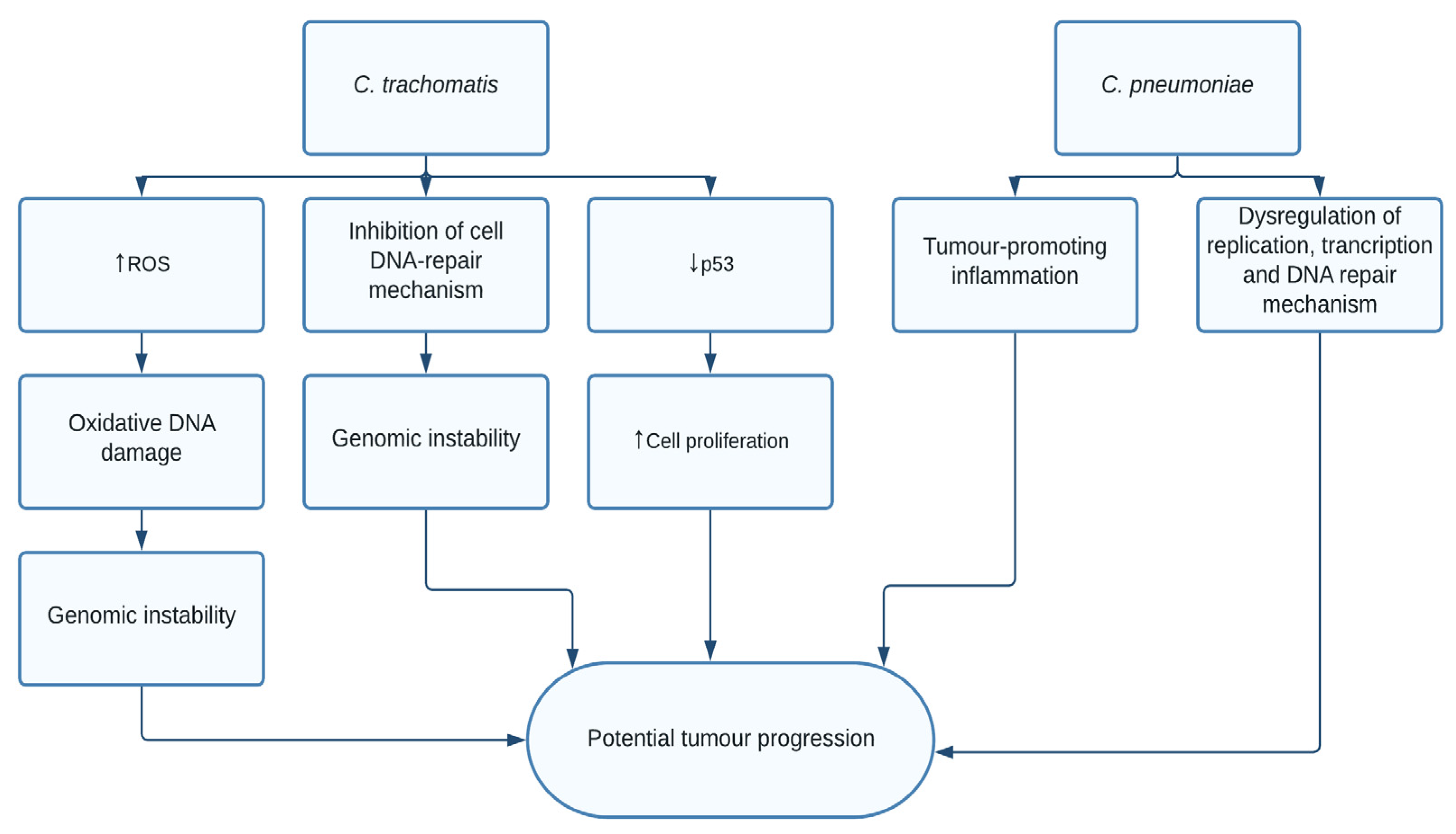
1.4. Chlamydia trachomatis and pneumoniae
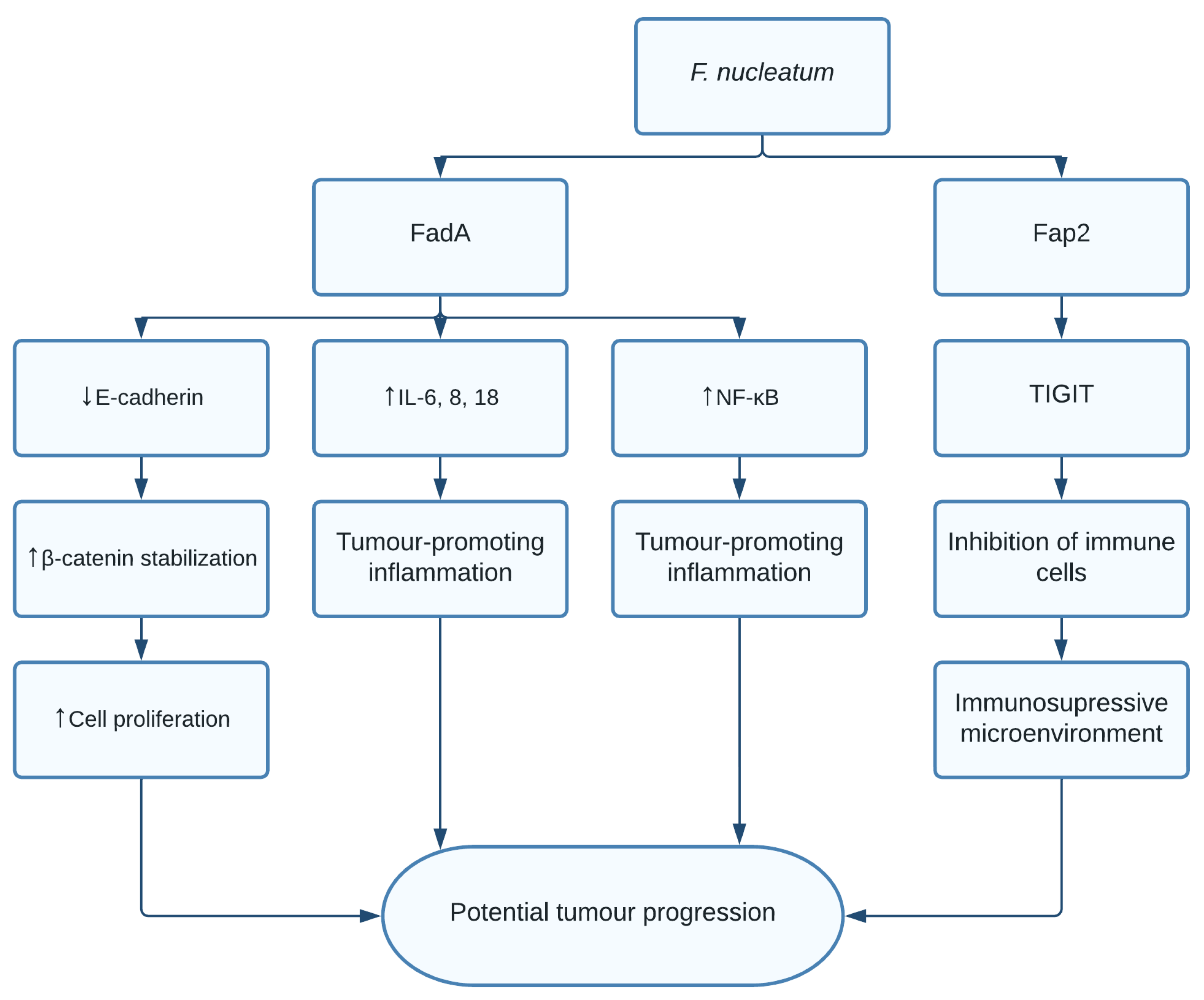
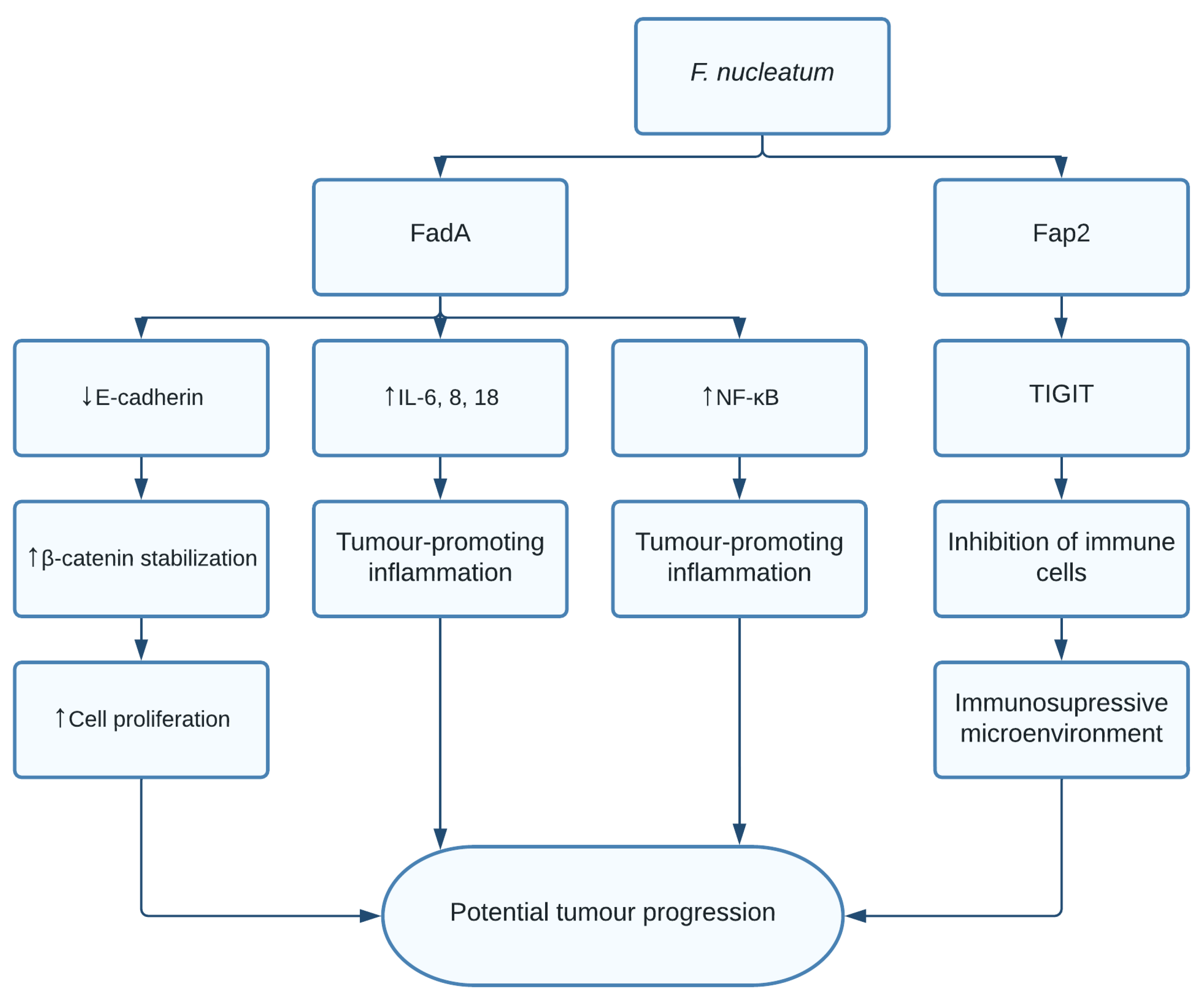
1.5. Fusobacterium nucleatum
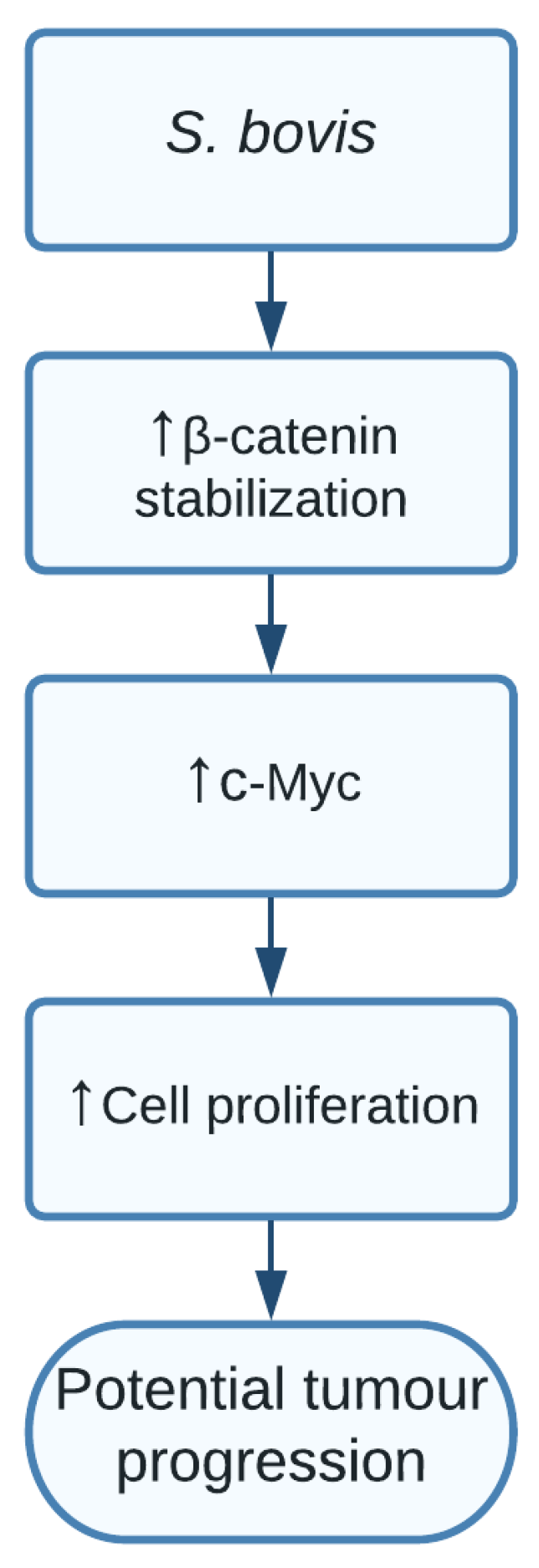
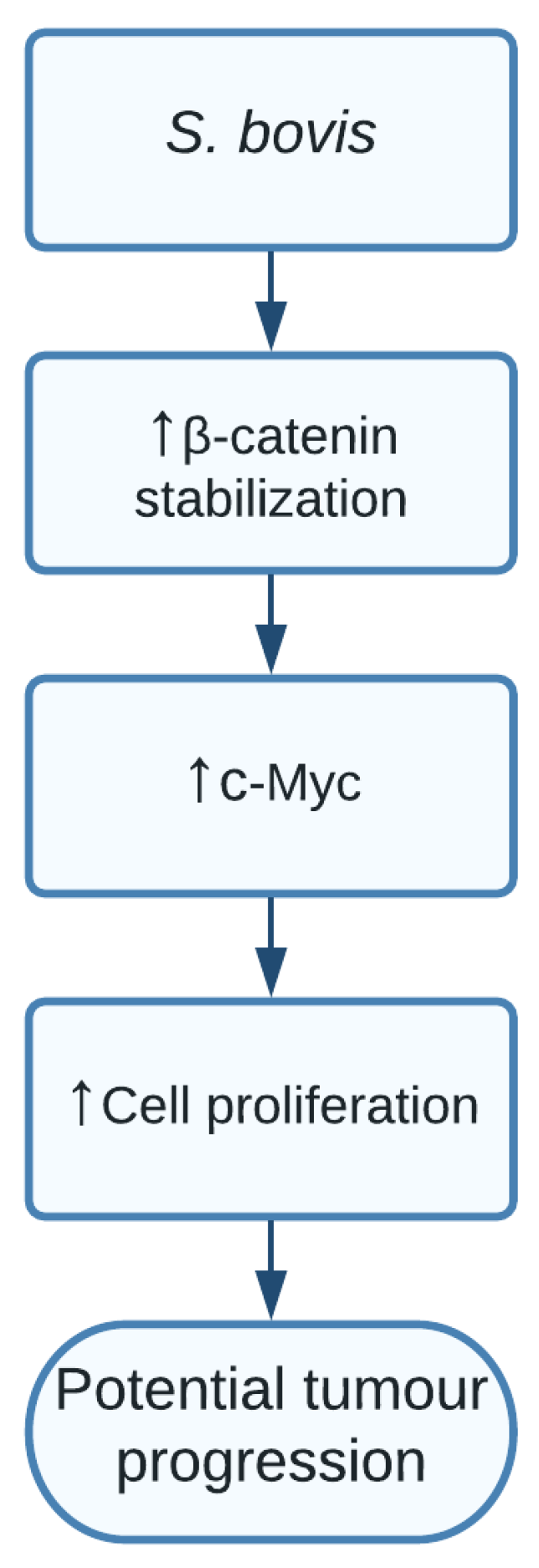
1.6. Streptococcus bovis
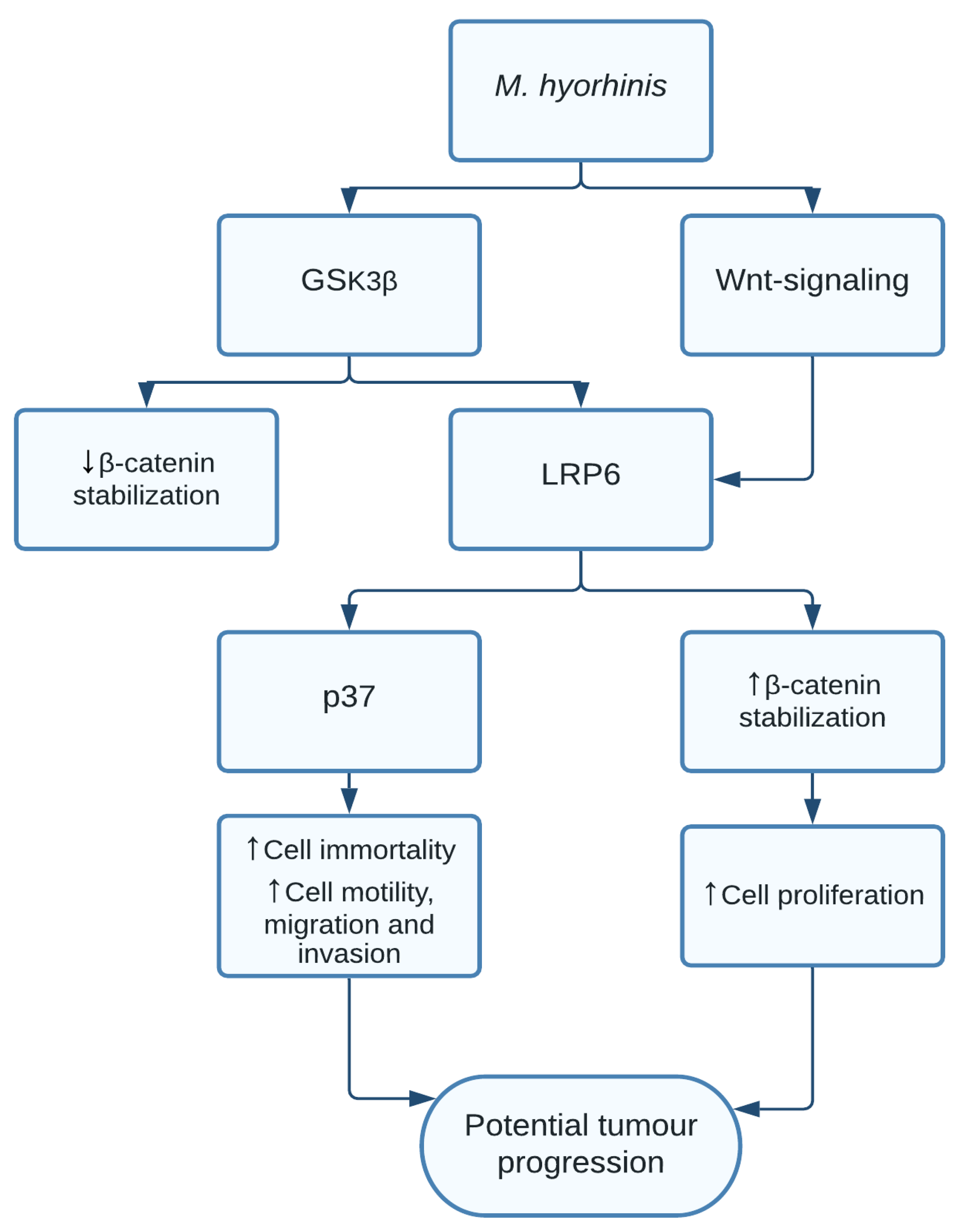
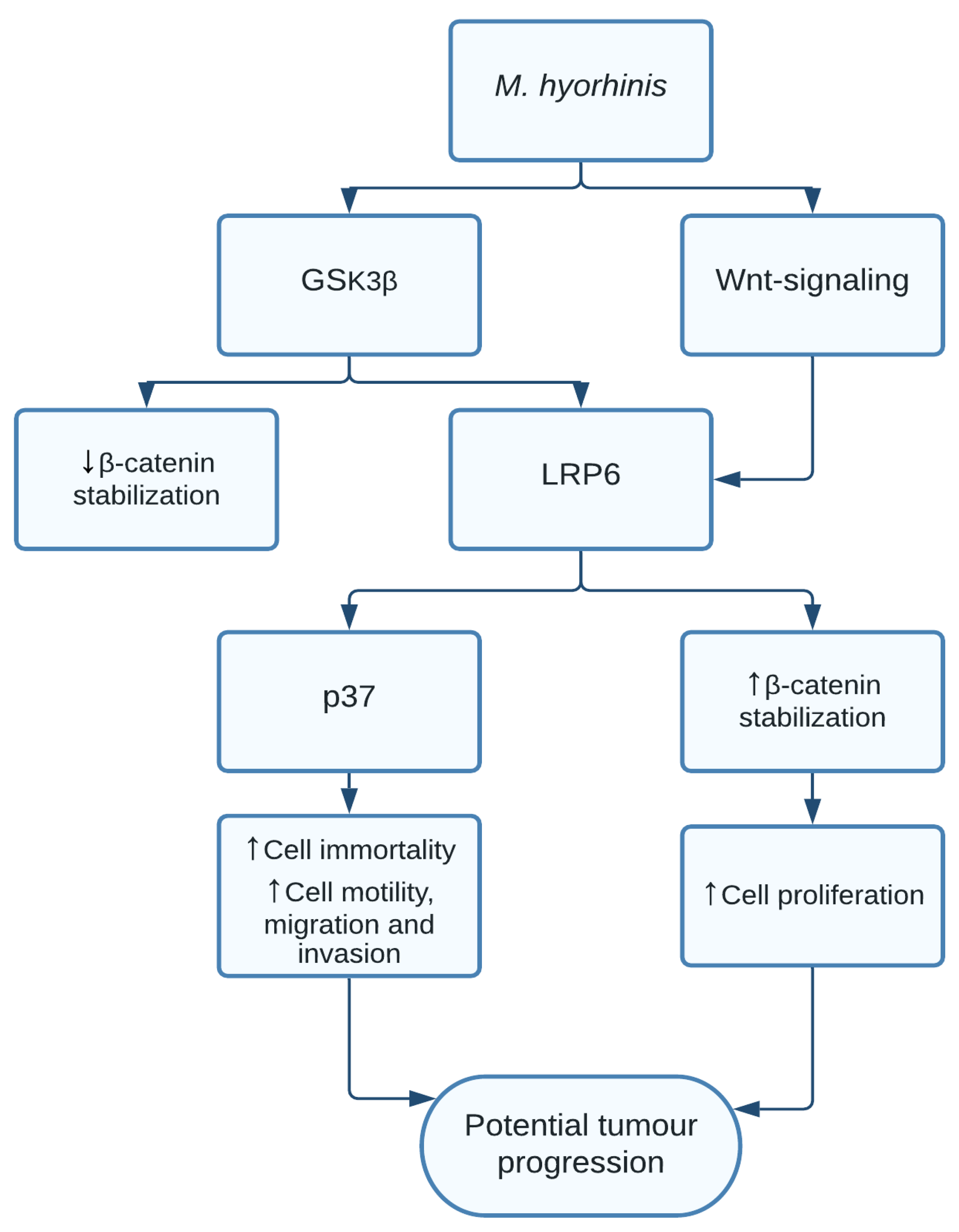
1.7. Mycoplasma
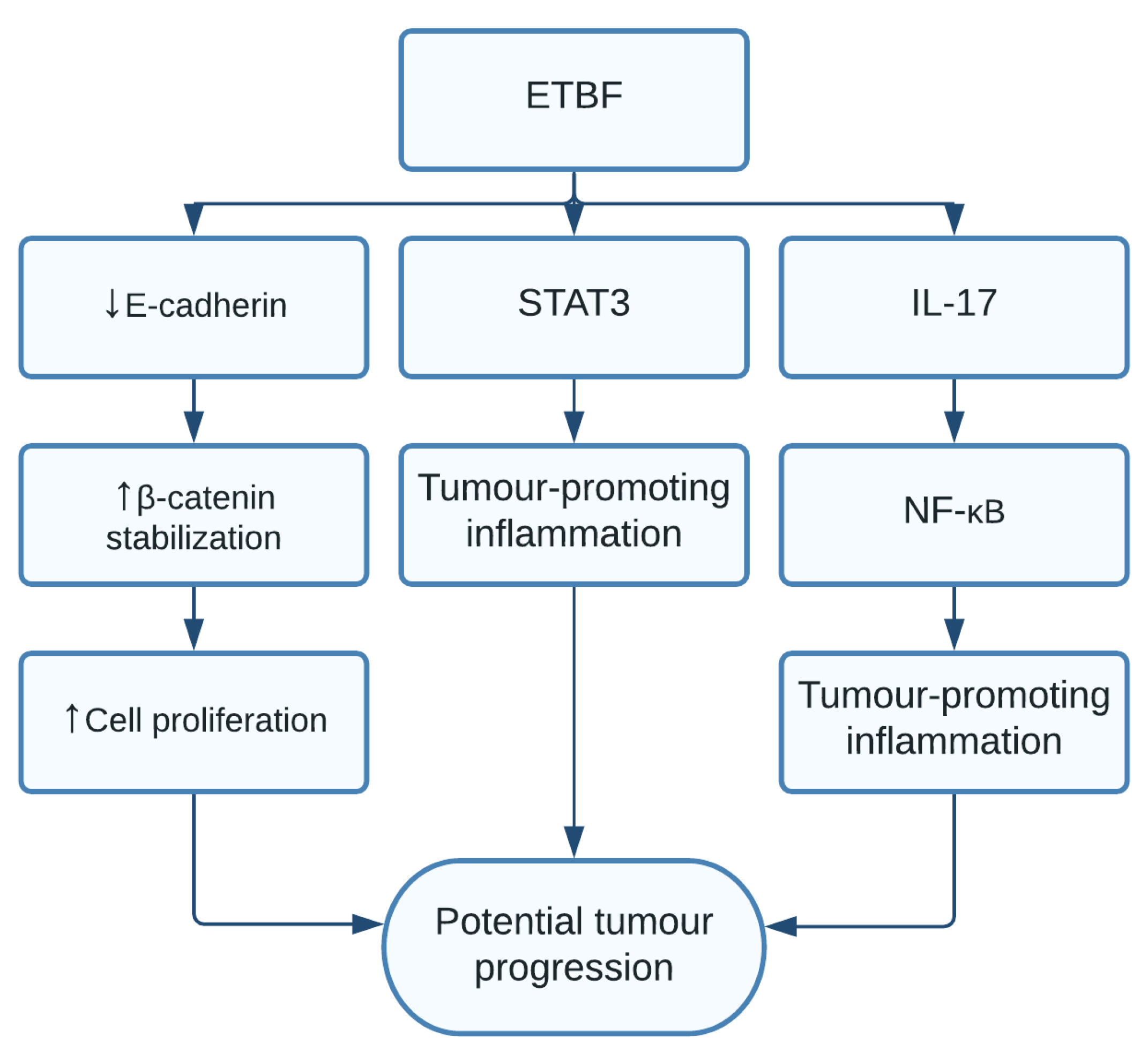
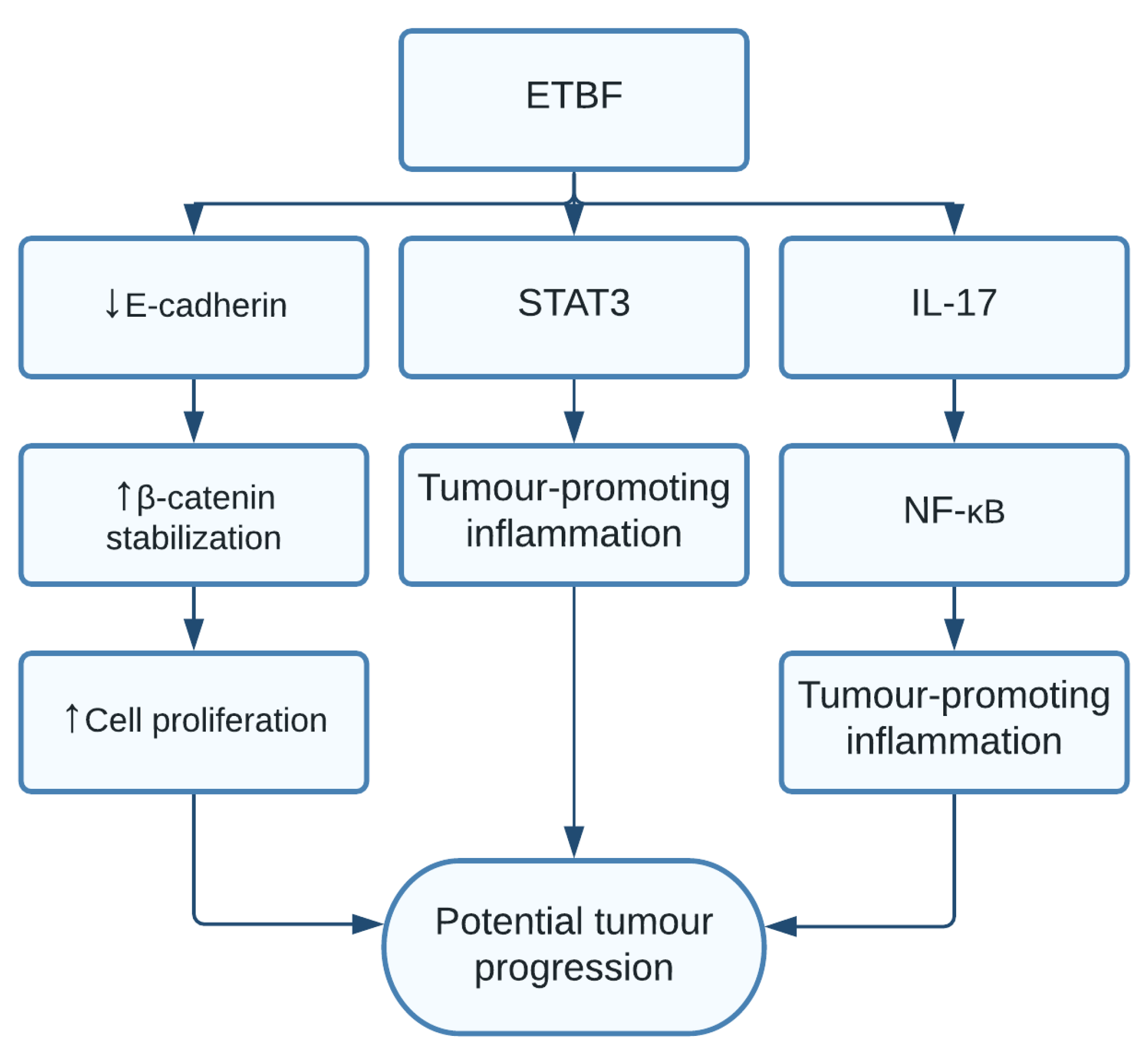
1.8. Bacteroides fragilis
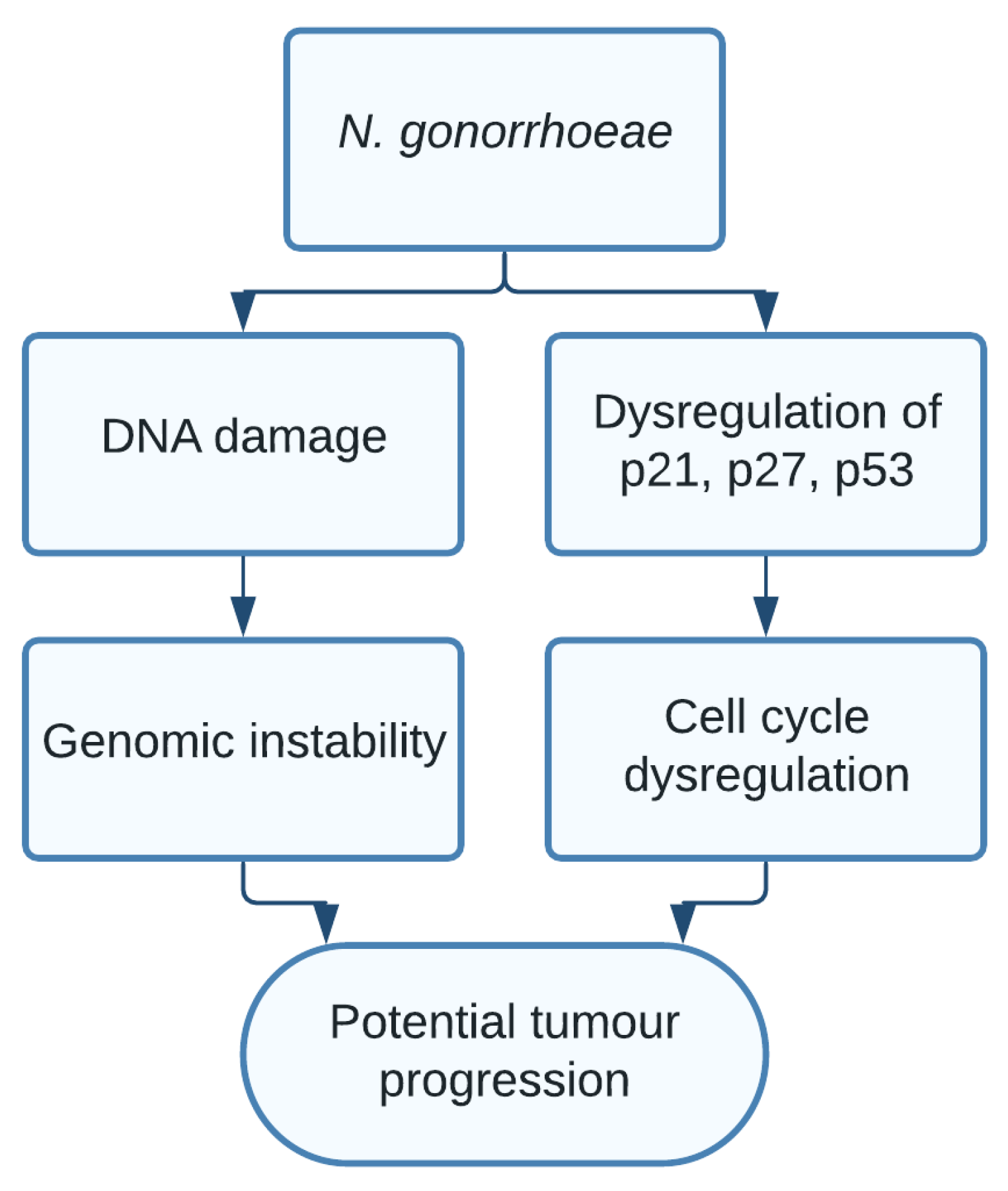
1.9. Neisseria gonorrhoeae
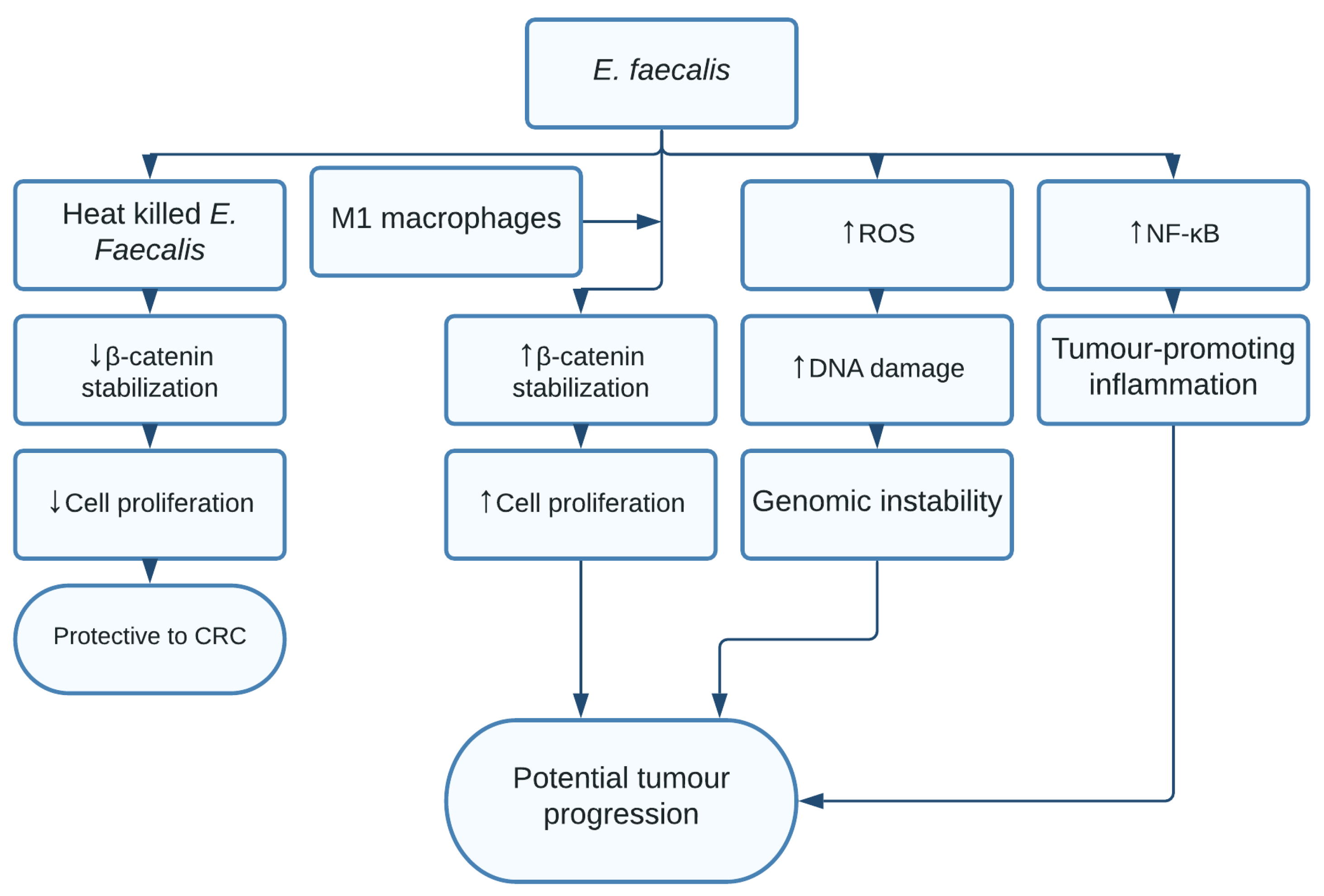
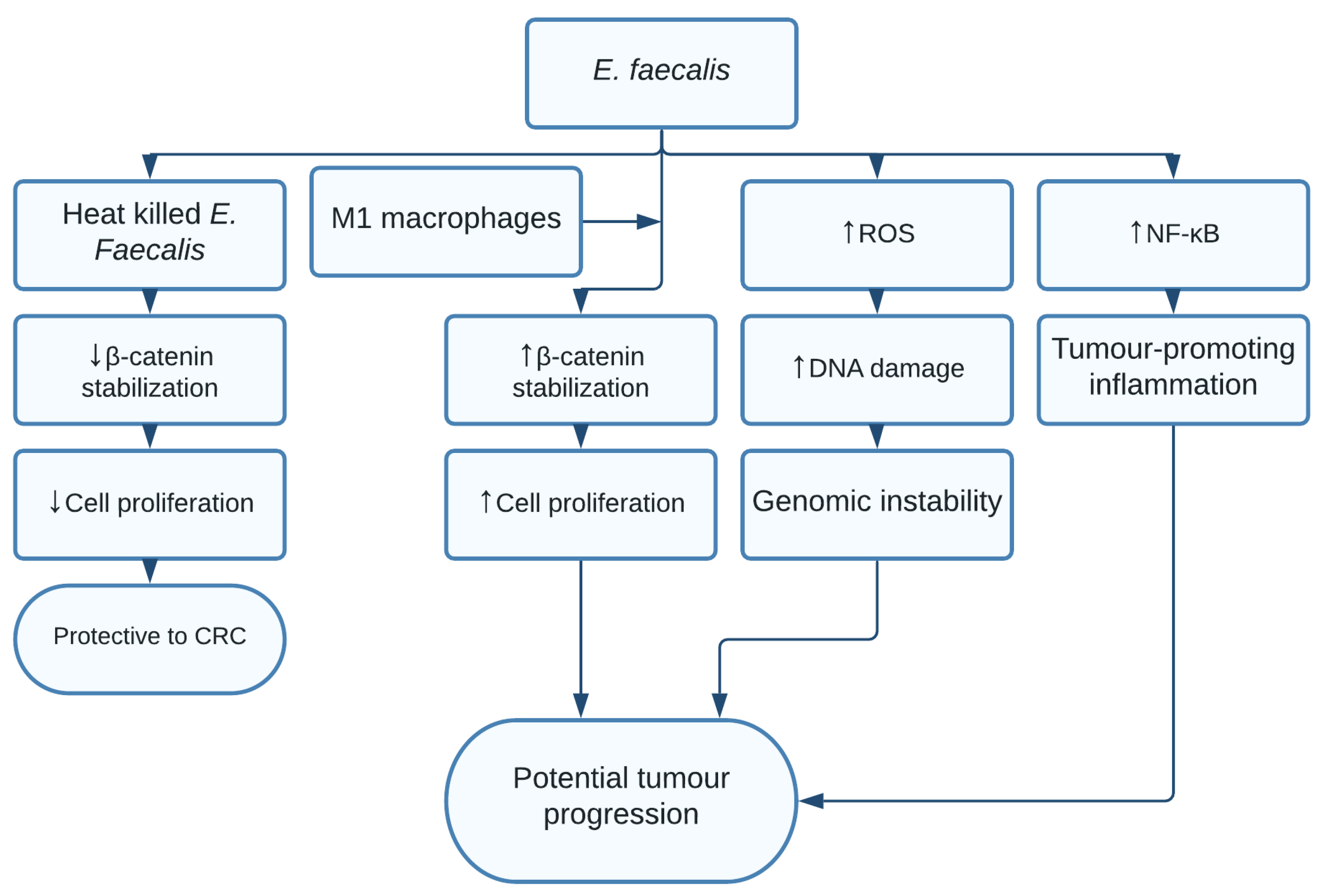
1.10. Enterococcus faecalis
1.11. Clostridium septicum
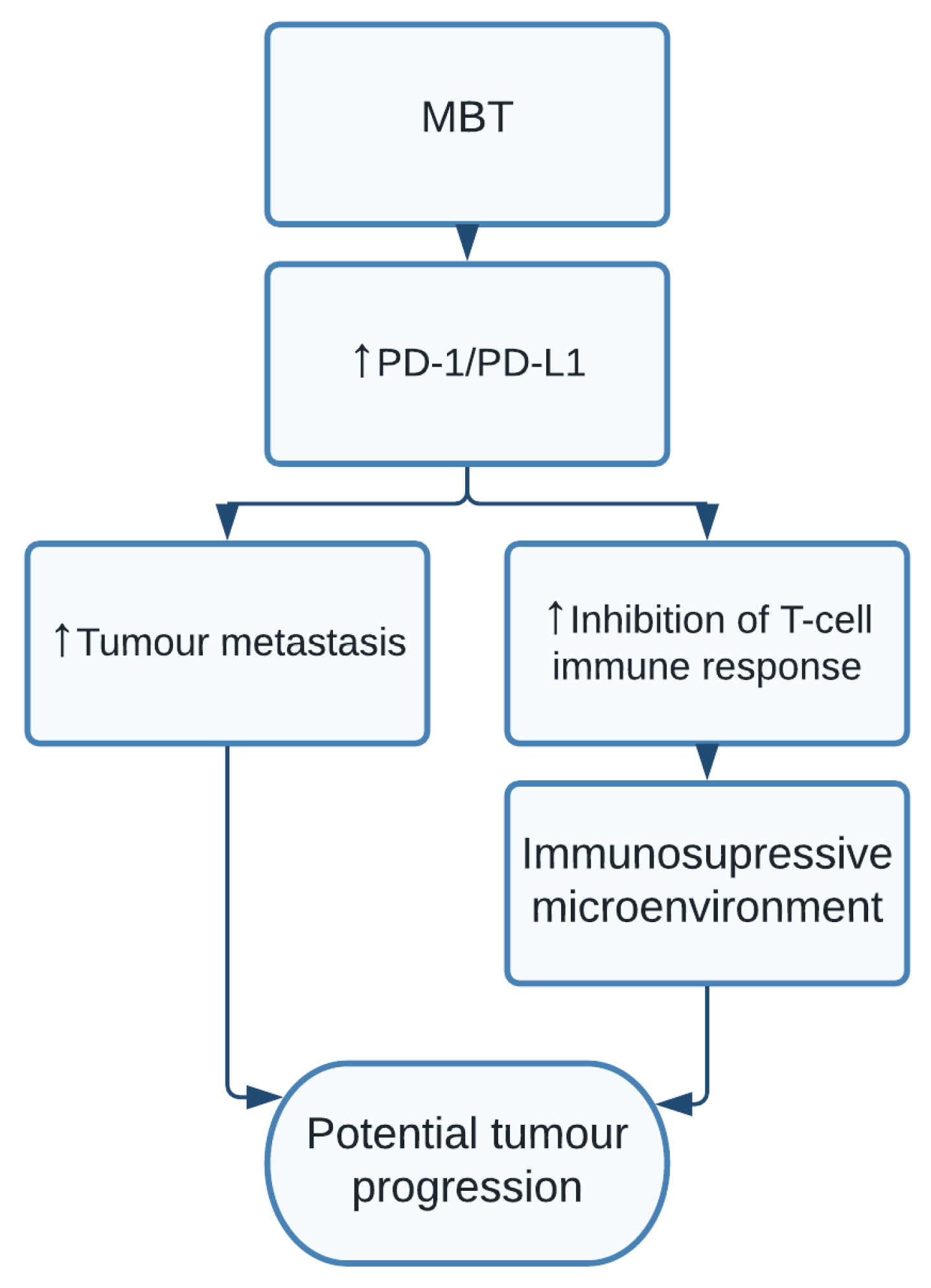
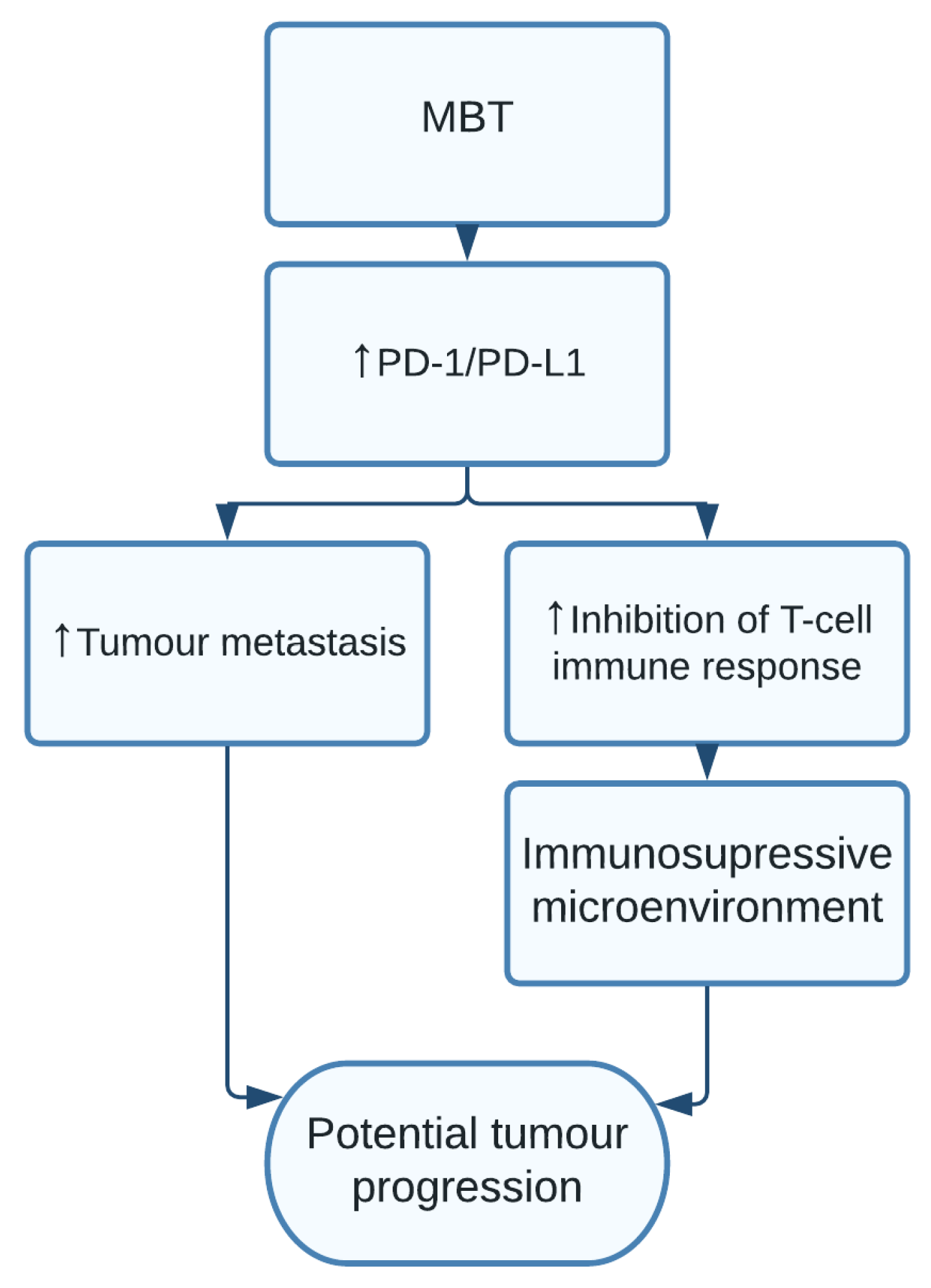
1.12. Mycobacterium tuberculosis
2. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tavares, R.; Pathak, S.K. Helicobacter pylori Protein JHP0290 Exhibits Proliferative and Anti-Apoptotic Effects in Gastric Epithelial Cells. PLoS ONE 2015, 10, e0124407. [Google Scholar] [CrossRef]
- Wang, H.; Sun, Y.; Liu, S.; Yu, H.; Li, W.; Zeng, J.; Chen, C.; Jia, J. Upregulation of progranulin byHelicobacter pyloriin human gastric epithelial cells via p38MAPK and MEK1/2 signaling pathway: Role in epithelial cell proliferation and migration. FEMS Immunol. Med. Microbiol. 2011, 63, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C.; Wang, Y.; Li, J.-Y.; Xu, W.-R.; Zhang, Y.-L. H pyloristimulates proliferation of gastric cancer cells through activating mitogen-activated protein kinase cascade. World J. Gastroenterol. 2006, 12, 5972–5977. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziubańska-Kusibab, P.J.; Berger, H.; Battistini, F.; Bouwman, B.A.M.; Iftekhar, A.; Katainen, R.; Cajuso, T.; Crosetto, N.; Orozco, M.; Aaltonen, L.A.; et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat. Med. 2020, 26, 1063–1069. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- Abdulamir, A.S.; Hafidh, R.R.; Abu Bakar, F. The association of Streptococcus bovis/gallolyticus with colorectal tumors: The nature and the underlying mechanisms of its etiological role. J. Exp. Clin. Cancer Res. 2011, 30, 11. [Google Scholar] [CrossRef] [Green Version]
- Ellmerich, S.; Schöller, M.; Duranton, B.; Gossé, F.; Galluser, M.; Klein, J.-P.; Raul, F. Promotion of intestinal carcinogenesis by Streptococcus bovis. Carcinogenesis 2000, 21, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Cano, D.A.; Martínez-Moya, M.; Pucciarelli, M.G.; Groisman, E.A.; Casadesús, J.; Portillo, F.G.-D. Salmonella enterica Serovar Typhimurium Response Involved in Attenuation of Pathogen Intracellular Proliferation. Infect. Immun. 2001, 69, 6463–6474. [Google Scholar] [CrossRef] [Green Version]
- Scanu, T.; Spaapen, R.; Bakker, J.M.; Pratap, C.B.; Wu, L.-E.; Hofland, I.; Broeks, A.; Shukla, V.; Kumar, M.; Janssen, H.; et al. Salmonella Manipulation of Host Signaling Pathways Provokes Cellular Transformation Associated with Gallbladder Carcinoma. Cell Host Microbe 2015, 17, 763–774. [Google Scholar] [CrossRef] [Green Version]
- Mughini-Gras, L.; Schaapveld, M.; Kramers, J.; Mooij, S.; Neefjes-Borst, A.; Van Pelt, W.; Neefjes, J. Increased colon cancer risk after severe Salmonella infection. PLoS ONE 2018, 13, e0189721. [Google Scholar] [CrossRef] [Green Version]
- Abed, J.; Maalouf, N.; Manson, A.L.; Earl, A.M.; Parhi, L.; Emgård, J.E.M.; Klutstein, M.; Tayeb, S.; Almogy, G.; Atlan, K.A.; et al. Colon Cancer-Associated Fusobacterium nucleatum May Originate from the Oral Cavity and Reach Colon Tumors via the Circulatory System. Front. Cell. Infect. Microbiol. 2020, 10, 400. [Google Scholar] [CrossRef]
- Wu, J.; Li, Q.; Fu, X. Fusobacterium nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Haghi, F.; Goli, E.; Mirzaei, B.; Zeighami, H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer 2019, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Wang, Z.; Yan, Y.; Ji, L.; He, J.; Xuan, B.; Shen, C.; Ma, Y.; Jiang, S.; Ma, D.; et al. Enterotoxigenic Bacteroides fragilis Promotes Intestinal Inflammation and Malignancy by Inhibiting Exosome-Packaged miR-149-3p. Gastroenterology 2021. [Google Scholar] [CrossRef]
- Xie, X.; Jiang, D.; Zhou, X.; Ye, X.; Yang, P.; He, Y. Recombinant Bacteroides fragilis enterotoxin-1 (rBFT-1) promotes proliferation of colorectal cancer via CCL3-related molecular pathways. Open Life Sci. 2021, 16, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Khan, Z.; Siddiqui, N.; Saif, M.W. Enterococcus Faecalis Infective Endocarditis and Colorectal Carcinoma: Case of New Association Gaining Ground. Gastroenterol. Res. 2018, 11, 238–240. [Google Scholar] [CrossRef] [Green Version]
- Zurmeyer, S.; Fotopoulou, C.; Braicu, E.; Schlichting, U.; Sehouli, J. Clostridium septicum can cause distant myonecrosis in patients with ovarian cancer. Anticancer. Res. 2013, 33, 1585–1589. [Google Scholar]
- Sidhu, J.S.; Mandal, A.; Virk, J.; Gayam, V. Early Detection of Colon Cancer Following Incidental Finding of Clostridium septicum Bacteremia. J. Investig. Med. High Impact Case Rep. 2019, 7, 2324709619832050. [Google Scholar] [CrossRef] [Green Version]
- Vielfort, K.; Söderholm, N.; Weyler, L.; Vare, D.; Löfmark, S.; Aro, H. Neisseria gonorrhoeae infection causes DNA damage and affects the expression of p21, p27, and p53 in non-tumor epithelial cells. J. Cell Sci. 2013, 126, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Lian, W.-Q.; Luo, F.; Song, X.-L.; Lu, Y.-J.; Zhao, S.-C. Gonorrhea and Prostate Cancer Incidence: An Updated Meta-Analysis of 21 Epidemiologic Studies. Med. Sci. Monit. 2015, 21, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Chung, C.-H.; Chen, J.-H.; Chiang, M.-H.; Yin, T.-; Tsao, C.-H.; Lin, F.-H.; Chien, W.-C.; Shang, S.-T.; Chang, F.-Y. Gonorrhea infection increases the risk of prostate cancer in Asian population: A nationwide population-based cohort study. Eur. J. Clin. Microbiol. Infect. Dis. 2016, 36, 813–821. [Google Scholar] [CrossRef]
- Michaud, D.S.; Platz, E.A.; Giovannucci, E. Gonorrhoea and male bladder cancer in a prospective study. Br. J. Cancer 2006, 96, 169–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.; Li, J.; Lu, J.; Zhong, R.; Zhong, H. Mycobacterium tuberculosis antigens repress Th1 immune response suppression and promotes lung cancer metastasis through PD-1/PDl-1 signaling pathway. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-Y.; Hu, H.-Y.; Pu, C.-Y.; Huang, N.; Shen, H.-C.; Li, C.-P.; Chou, Y.-J. Pulmonary tuberculosis increases the risk of lung cancer. Cancer 2010, 117, 618–624. [Google Scholar] [CrossRef]
- Rajeeve, K.; Vollmuth, N.; Janaki-Raman, S.; Wulff, T.F.; Baluapuri, A.; Dejure, F.R.; Huber, C.; Fink, J.; Schmalhofer, M.; Schmitz, W.; et al. Author Correction: Reprogramming of host glutamine metabolism during Chlamydia trachomatis infection and its key role in peptidoglycan synthesis. Nat. Microbiol. 2021, 6, 1. [Google Scholar] [CrossRef]
- Siegl, C.; Prusty, B.K.; Karunakaran, K.; Wischhusen, J.; Rudel, T. Tumor Suppressor p53 Alters Host Cell Metabolism to Limit Chlamydia trachomatis Infection. Cell Rep. 2014, 9, 918–929. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Machuy, N.; Böhme, L.; Karunakaran, K.; Mäurer, A.P.; Meyer, T.F.; Rudel, T. HIF-1α is involved in mediating apoptosis resistance to Chlamydia trachomatis-infected cells. Cell. Microbiol. 2011, 13, 1573–1585. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Hoffmann, K.; Fritsche, K.; Brinkmann, V.; Mollenkopf, H.-J.; Thieck, O.; Da Costa, A.R.T.; Braicu, E.I.; Sehouli, J.; Mangler, M.; et al. Chronic Chlamydia infection in human organoids increases stemness and promotes age-dependent CpG methylation. Nat. Commun. 2019, 10, 1194. [Google Scholar] [CrossRef]
- Chumduri, C.; Gurumurthy, R.K.; Zadora, P.K.; Mi, Y.; Meyer, T.F. Chlamydia Infection Promotes Host DNA Damage and Proliferation but Impairs the DNA Damage Response. Cell Host Microbe 2013, 13, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Lei, W.; Su, S.; Bu, J.; Zhu, S.; Huang, Q.; Li, Z. Chlamydia trachomatis plasmid-encoded protein Pgp3 inhibits apoptosis via the PI3K-AKT-mediated MDM2-p53 axis. Mol. Cell. Biochem. 2018, 452, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Rajeeve, K.; Das, S.; Prusty, B.K.; Rudel, T. Chlamydia trachomatis paralyses neutrophils to evade the host innate immune response. Nat. Microbiol. 2018, 3, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, H.; Chen, W.; Yao, X.; Xing, Y.; Wang, X.; Zhong, J.; Meng, G. Mycoplasma hyorhinis Activates the NLRP3 Inflammasome and Promotes Migration and Invasion of Gastric Cancer Cells. PLoS ONE 2013, 8, e77955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Rong, Z.; Shou, C. Mycoplasma hyorhinis infection promotes gastric cancer cell motility via β-catenin signaling. Cancer Med. 2019, 8, 5301–5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.; Wu, J.C.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef] [Green Version]
- Dorer, M.S.; Talarico, S.; Salama, N.R. Helicobacter pylori’s Unconventional Role in Health and Disease. PLoS Pathog. 2009, 5, e1000544. [Google Scholar] [CrossRef]
- McClain, M.S.; Beckett, A.C.; Cover, T.L. Helicobacter pylori Vacuolating Toxin and Gastric Cancer. Toxins 2017, 9, 316. [Google Scholar] [CrossRef] [Green Version]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Schütte, K.; Mayerle, J.; Malfertheiner, P. The role of the gastric bacterial microbiome in gastric cancer: Helicobacter pylori and beyond. Ther. Adv. Gastroenterol. 2019, 12, 1756284819894062. [Google Scholar] [CrossRef] [Green Version]
- Ohnishi, N.; Yuasa, H.; Tanaka, S.; Sawa, H.; Miura, M.; Matsui, A.; Higashi, H.; Musashi, M.; Iwabuchi, K.; Suzuki, M.; et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc. Natl. Acad. Sci. USA 2008, 105, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Hisatsune, J.; Yamasaki, E.; Isomoto, H.; Kurazono, H.; Hatakeyama, M.; Azuma, T.; Yamaoka, Y.; Yahiro, K.; Moss, J.; et al. Helicobacter pylori VacA-induced Inhibition of GSK3 through the PI3K/Akt Signaling Pathway. J. Biol. Chem. 2009, 284, 1612–1619. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, M. Helicobacter pylori CagA and Gastric Cancer: A Paradigm for Hit-and-Run Carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Umehara, S.; Higashi, H.; Ohnishi, N.; Asaka, M.; Hatakeyama, M. Effects of Helicobacter pylori CagA protein on the growth and survival of B lymphocytes, the origin of MALT lymphoma. Oncogene 2003, 22, 8337–8342. [Google Scholar] [CrossRef] [Green Version]
- Lamb, A.; Yang, X.-D.; Tsang, Y.N.; Li, J.-D.; Higashi, H.; Hatakeyama, M.; Peek, R.M.; Blanke, S.R.; Chen, L. Helicobacter pylori CagA activates NF-κB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 2009, 10, 1242–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 Tyrosine Phosphatase as an Intracellular Target of Helicobacter pylori CagA Protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharafutdinov, I.; Backert, S.; Tegtmeyer, N. Cortactin: A Major Cellular Target of the Gastric Carcinogen Helicobacter pylori. Cancers 2020, 12, 159. [Google Scholar] [CrossRef] [Green Version]
- Selbach, M.; Moese, S.; Hurwitz, R.; Hauck, C.R.; Meyer, T.F.; Backert, S. The Helicobacter pylori CagA protein induces cortactin dephosphorylation and actin rearrangement by c-Src inactivation. EMBO J. 2003, 22, 515–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Zhao, Z.-X.; Li, Y.; Zhou, Z.-Q.; You, T.-G. Cortactin expression confers a more malignant phenotype to gastric cancer SGC-7901 cells. World J. Gastroenterol. 2014, 20, 3287–3300. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Wang, Y.; Xie, B.; Ma, J.; Wang, Y. Cortactin and HER2 as potential markers for dural-targeted therapy in advanced gastric cancer. Clin. Exp. Med. 2021. [Google Scholar] [CrossRef]
- Sharafutdinov, I.; Backert, S.; Tegtmeyer, N. The Helicobacter pylori type IV secretion system upregulates epithelial cortactin expression by a CagA - and JNK -dependent pathway. Cell. Microbiol. 2021, 23, e13376. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Nakaya, A.; Tsutsumi, R.; Yokoyama, K.; Fujii, Y.; Ishikawa, S.; Higuchi, M.; Takahashi, A.; Kurashima, Y.; Teishikata, Y.; et al. Helicobacter pylori CagA Induces Ras-independent Morphogenetic Response through SHP-2 Recruitment and Activation. J. Biol. Chem. 2004, 279, 17205–17216. [Google Scholar] [CrossRef] [Green Version]
- Muratakamiya, N.; Kurashima, Y.; Teishikata, Y.; Yamahashi, Y.; Saito, Y.; Higashi, H.; Aburatani, H.; Akiyama, T.; Peek, R.M.; Azuma, T.; et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene 2007, 26, 4617–4626. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.-G.; Kim, H.S.; Lee, Y.S.; Kim, S.; Cha, S.; Ota, I.; Kim, N.H.; Cha, Y.H.; Yang, D.H.; Lee, Y.; et al. Helicobacter pylori CagA promotes Snail-mediated epithelial–mesenchymal transition by reducing GSK-3 activity. Nat. Commun. 2014, 5, 4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buti, L.; Spooner, E.; Van der Veen, A.G.; Rappuoli, R.; Covacci, A.; Ploegh, H.L. Helicobacter pylori cytotoxin-associated gene A (CagA) subverts the apoptosis-stimulating protein of p53 (ASPP2) tumor suppressor pathway of the host. Proc. Natl. Acad. Sci. USA 2011, 108, 9238–9243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassam, F.H.; Graham, D.Y.; Yamaoka, Y. Helicobacter pyloriactivate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3β phosphorylation. Cell. Microbiol. 2009, 11, 70–82. [Google Scholar] [CrossRef] [Green Version]
- Franco, A.; Johnston, E.; Krishna, U.; Yamaoka, Y.; Israel, D.A.; Nagy, T.A.; Wroblewski, L.E.; Piazuelo, M.B.; Correa, P.; Peek, R.M. Regulation of Gastric Carcinogenesis by Helicobacter pylori Virulence Factors. Cancer Res. 2008, 68, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.C.; Yuan, Y.; Moayyedi, P. Helicobacter pylori eradication therapy to prevent gastric cancer: Systematic review and meta-analysis. Gut 2020, 69, 2113–2121. [Google Scholar] [CrossRef]
- Boren, T.; Falk, P.; Roth, K.; Larson, G.; Normark, S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science 1993, 262, 1892–1895. [Google Scholar] [CrossRef]
- Ilver, D.; Arnqvist, A.; Ögren, J.; Frick, I.-M.; Kersulyte, D.; Incecik, E.T.; Berg, D.E.; Covacci, A.; Engstrand, L.; Borén, T. Helicobacter pylori Adhesin Binding Fucosylated Histo-Blood Group Antigens Revealed by Retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef]
- Ishijima, N.; Suzuki, M.; Ashida, H.; Ichikawa, Y.; Kanegae, Y.; Saito, I.; Borén, T.; Haas, R.; Sasakawa, C.; Mimuro, H. BabA-mediated Adherence Is a Potentiator of the Helicobacter pylori Type IV Secretion System Activity. J. Biol. Chem. 2011, 286, 25256–25264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolitho, C.; Hahn, M.A.; Baxter, R.; Marsh, D.J. The chemokine CXCL1 induces proliferation in epithelial ovarian cancer cells by transactivation of the epidermal growth factor receptor. Endocr.-Relat. Cancer 2010, 17, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Wu, S.; Zhang, Y.-G.; Xia, Y.; Liu, X.; Zheng, Y.; Chen, H.; Schaefer, K.L.; Zhou, Z.; Bissonnette, M.; et al. Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta-catenin signaling pathway. Oncogenesis 2014, 3, e105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, V.; Singh, H.; Pandey, M.; Upadhyay, S.; Nath, G. Carcinoma of the Gallbladder—Is It a Sequel of Typhoid? Dig. Dis. Sci. 2000, 45, 900–903. [Google Scholar] [CrossRef]
- LaRock, D.; Chaudhary, A.; Miller, S.I. Salmonellae interactions with host processes. Nat. Rev. Genet. 2015, 13, 191–205. [Google Scholar] [CrossRef]
- Kuper, H.; Adami, H.-O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Intern. Med. 2000, 248, 171–183. [Google Scholar] [CrossRef]
- Belluz, L.D.B.; Guidi, R.; Pateras, I.S.; Levi, L.; Mihaljevic, B.; Rouf, S.F.; Wrande, M.; Candela, M.; Turroni, S.; Nastasi, C.; et al. The Typhoid Toxin Promotes Host Survival and the Establishment of a Persistent Asymptomatic Infection. PLoS Pathog. 2016, 12, e1005528. [Google Scholar] [CrossRef]
- Kang, M.; Martin, A. Microbiome and colorectal cancer: Unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin. Immunol. 2017, 32, 3–13. [Google Scholar] [CrossRef]
- Lu, R.; Bosland, M.; Xia, Y.; Zhang, Y.-G.; Kato, I.; Sun, J. Presence of Salmonella AvrA in colorectal tumor and its precursor lesions in mouse intestine and human specimens. Oncotarget 2017, 8, 55104–55115. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Vogelstein, B.; Kinzler, K.W. Phosphorylation of beta-catenin at S33, S37, or T41 can occur in the absence of phosphorylation at T45 in colon cancer cells. Cancer Res. 2003, 63, 5234–5235. [Google Scholar]
- Wu, S.; Ye, Z.; Liu, X.; Zhao, Y.; Xia, Y.; Steiner, A.; Petrof, E.O.; Claud, E.C.; Sun, J. Salmonella typhimurium infection increases p53 acetylation in intestinal epithelial cells. Am. J. Physiol. Liver Physiol. 2010, 298, G784–G794. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Woods, N.T.; Piluso, L.G.; Lee, H.-H.; Chen, J.; Bhalla, K.N.; Monteiro, A.; Liu, X.; Hung, M.-C.; Wang, H.-G. p53 Acetylation Is Crucial for Its Transcription-independent Proapoptotic Functions. J. Biol. Chem. 2009, 284, 11171–11183. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Wu, S.; Zhang, Y.-G.; Xia, Y.; Zhou, Z.; Kato, I.; Dong, H.; Bissonnette, M.; Sun, J. Salmonella Protein AvrA Activates the STAT3 Signaling Pathway in Colon Cancer. Neoplasia 2016, 18, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Parry, C.M.; Hien, T.T.; Dougan, G.; White, N.J.; Farrar, J.J. Typhoid Fever. N. Engl. J. Med. 2002, 347, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Iyer, P.; Barreto, S.G.; Sahoo, B.; Chandrani, P.; Ramadwar, M.R.; Shrikhande, S.V.; Dutt, A. Non-typhoidal Salmonella DNA traces in gallbladder cancer. Infect. Agents Cancer 2016, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Dongol, S.; Thompson, C.N.; Clare, S.; Nga, T.V.T.; Duy, P.T.; Karkey, A.; Arjyal, A.; Koirala, S.; Khatri, N.S.; Maskey, P.; et al. The Microbiological and Clinical Characteristics of Invasive Salmonella in Gallbladders from Cholecystectomy Patients in Kathmandu, Nepal. PLoS ONE 2012, 7, e47342. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Ning, S.; Guo, Q.; Liu, M.; Yuan, H.; Wang, Y.; Chen, Z.; Ji, R.; Li, Q.; Zhou, Y. COX-2 regulates Snail expression in gastric cancer via the Notch1 signaling pathway. Int. J. Mol. Med. 2017, 40, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Sorski, L.; Melamed, R.; Matzner, P.; Lavon, H.; Shaashua, L.; Rosenne, E.; Ben-Eliyahu, S. Reducing liver metastases of colon cancer in the context of extensive and minor surgeries through β-adrenoceptors blockade and COX2 inhibition. Brain Behav. Immun. 2016, 58, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swidsinski, A.; Khilkin, M.; Kerjaschki‡, D.; Schreiber, S.; Ortner, M.; Weber, J.; Lochs, H. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology 1998, 115, 281–286. [Google Scholar] [CrossRef]
- Raisch, J. Colon cancer-associated B2Escherichia colicolonize gut mucosa and promote cell proliferation. World J. Gastroenterol. 2014, 20, 6560–6572. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Scanlon, K.M.; Donnenberg, M.S. An Escherichia coli Effector Protein Promotes Host Mutation via Depletion of DNA Mismatch Repair Proteins. mBio 2013, 4, e00152-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef]
- Abd-El-Raouf, R.; Ouf, S.A.; Gabr, M.M.; Zakaria, M.M.; El-Yasergy, K.F.; Ali-El-Dein, B. Escherichia coli foster bladder cancer cell line progression via epithelial mesenchymal transition, stemness and metabolic reprogramming. Sci. Rep. 2020, 10, 18024. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Huber, A.R.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Laurila, A.L.; Anttila, T.; Läärä, E.; Bloigu, A.; Virtamo, J.; Albanes, D.; Leinonen, M.; Saikku, P. Serological evidence of an association betweenChlamydia pneumoniae infection and lung cancer. Int. J. Cancer 1997, 74, 31–34. [Google Scholar] [CrossRef]
- Littman, A.J.; Jackson, L.A.; Vaughan, T.L. Chlamydia pneumoniae and Lung Cancer: Epidemiologic Evidence. Cancer Epidemiol. Biomark. Prev. 2005, 14, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, A.K.; Gaydos, C.A.; Agreda, P.; Holden, J.P.; Chatterjee, N.; Goedert, J.J.; Caporaso, N.E.; Engels, E.A. Chlamydia pneumoniae Infection and Risk for Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1498–1505. [Google Scholar] [CrossRef] [Green Version]
- Alshamsan, A.; Khan, S.; Imran, A.; Aljuffali, I.A.; Alsaleh, K. Prediction of Chlamydia pneumoniae protein localization in host mitochondria and cytoplasm and possible involvements in lung cancer etiology: A computational approach. Saudi Pharm. J. 2017, 25, 1151–1157. [Google Scholar] [CrossRef]
- Rizzo, A.; Carratelli, C.R. Transforming activities of Chlamydia pneumoniae in human mesothelial cells. Int. Microbiol. 2014, 17, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Shen, Z.; Luo, H.; Zhang, W.; Zhu, X. Chlamydia Trachomatis Infection-Associated Risk of Cervical Cancer. Medicine 2016, 95, e3077. [Google Scholar] [CrossRef] [PubMed]
- Koskela, P.; Anttila, T.; Bjørge, T.; Brunsvig, A.; Dillner, J.; Hakama, M.; Hakulinen, T.; Jellum, E.; Lehtinen, M.; Lenner, P.; et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int. J. Cancer 2000, 85, 35–39. [Google Scholar] [CrossRef]
- Smith, J.S.; Bosetti, C.; Muñoz, N.; Herrero, R.; Bosch, F.X.; Eluf-Neto, J.; Meijer, C.J.; Brule, A.J.V.D.; Franceschi, S.; Peeling, R.W. Chlamydia trachomatisand invasive cervical cancer: A pooled analysis of the IARC multicentric case-control study. Int. J. Cancer 2004, 111, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Paavonen, J.; Karunakaran, K.P.; Noguchi, Y.; Anttila, T.; Bloigu, A.; Dillner, J.; Hallmans, G.; Hakulinen, T.; Jellum, E.; Koskela, P.; et al. Serum antibody response to the heat shock protein 60 of Chlamydia trachomatis in women with developing cervical cancer. Am. J. Obstet. Gynecol. 2003, 189, 1287–1292. [Google Scholar] [CrossRef]
- Trabert, B.; Waterboer, T.; Idahl, A.; Brenner, N.; Brinton, L.A.; Butt, J.; Coburn, S.B.; Hartge, P.; Hufnagel, K.; Inturrisi, F.; et al. Antibodies Against Chlamydia trachomatis and Ovarian Cancer Risk in Two Independent Populations. J. Natl. Cancer Inst. 2018, 111, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Sater, A.A.; Saïd-Sadier, N.; Lam, V.M.; Singh, B.; Pettengill, M.A.; Soares, F.; Tattoli, I.; Lipinski, S.; Girardin, S.E.; Rosenstiel, P.; et al. Enhancement of Reactive Oxygen Species Production and Chlamydial Infection by the Mitochondrial Nod-like Family Member NLRX. J. Biol. Chem. 2010, 285, 41637–41645. [Google Scholar] [CrossRef] [Green Version]
- Mi, Y.; Gurumurthy, R.K.; Zadora, P.K.; Meyer, T.F.; Chumduri, C. Chlamydia trachomatis Inhibits Homologous Recombination Repair of DNA Breaks by Interfering with PP2A Signaling. mBio 2018, 9, e01465-18. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Lam, J.L.; Dooley, C.A.; Noriea, N.F.; Hansen, B.T.; Hoyt, F.H.; Carmody, A.B.; Sturdevant, G.L.; Hackstadt, T. Absence of Specific Chlamydia trachomatis Inclusion Membrane Proteins Triggers Premature Inclusion Membrane Lysis and Host Cell Death. Cell Rep. 2017, 19, 1406–1417. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Fischer, S.F.; Pettengill, M.; Conte, D.; Paschen, S.A.; Ojcius, D.; Häcker, G. Characterization of Host Cell Death Induced by Chlamydia trachomatis. Infect. Immun. 2006, 74, 6057–6066. [Google Scholar] [CrossRef] [Green Version]
- Rajalingam, K.; Sharma, M.; Lohmann, C.; Oswald, M.; Thieck, O.; Froelich, C.J.; Rudel, T. Mcl-1 Is a Key Regulator of Apoptosis Resistance in Chlamydia trachomatis-Infected Cells. PLoS ONE 2008, 3, e3102. [Google Scholar] [CrossRef]
- Fischer, A.; Harrison, K.S.; Ramirez, Y.; Auer, D.; Chowdhury, S.R.; Prusty, B.K.; Sauer, F.; Dimond, Z.; Kisker, C.; Hefty, P.S.; et al. Chlamydia trachomatis-containing vacuole serves as deubiquitination platform to stabilize Mcl-1 and to interfere with host defense. eLife 2017, 6, 6192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum—Symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Shang, F.-M.; Liu, H.-L. Fusobacterium nucleatum and colorectal cancer: A review. World J. Gastrointest. Oncol. 2018, 10, 71–81. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Chen, J.; Chen, F.; Zeng, Q.; Liu, W.-L.; Zhang, G. Exosomes derived from Fusobacterium nucleatum-infected colorectal cancer cells facilitate tumour metastasis by selectively carrying miR-1246/92b-3p/27a-3p and CXCL. Gut 2021, 70, 1507–1519. [Google Scholar] [CrossRef]
- Kong, C.; Yan, X.; Zhu, Y.; Zhu, H.; Luo, Y.; Liu, P.; Ferrandon, S.; Kalady, M.F.; Gao, R.; He, J.; et al. Fusobacterium Nucleatum Promotes the Development of Colorectal Cancer by Activating a Cytochrome P450/Epoxyoctadecenoic Acid Axis via TLR4/Keap1/NRF2 Signaling. Cancer Res. 2021, 81, 4485–4498. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Recco, R.A.; Catalano, M.T.; Edberg, S.C.; Casey, J.I.; Steigbigel, N. Association ofStreptococcus boviswith Carcinoma of the Colon. N. Engl. J. Med. 1977, 297, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Madani, R.; Mukhtar, H. Streptococcus bovisendocarditis, a silent sign for colonic tumour. Color. Dis. 2010, 12, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Aymeric, L.; Donnadieu, F.; Mulet, C.; du Merle, L.; Nigro, G.; Saffarian, A.; Bérard, M.; Poyart, C.; Robine, S.; Regnault, B.; et al. Colorectal cancer specific conditions promote Streptococcus gallolyticus gut colonization. Proc. Natl. Acad. Sci. USA 2018, 115, E283–E291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Herold, J.L.; Schady, D.; Davis, J.; Kopetz, S.; Martinez-Moczygemba, M.; Murray, B.E.; Han, F.; Li, Y.; Callaway, E.; et al. Streptococcus gallolyticus subsp. gallolyticus promotes colorectal tumor development. PLoS Pathog. 2017, 13, e1006440. [Google Scholar] [CrossRef]
- Huang, S.; Li, J.-Y.; Wu, J.; Meng, L.; Shou, C.-C. Mycoplasma infections and different human carcinomas. World J. Gastroenterol. 2001, 7, 266–269. [Google Scholar] [CrossRef]
- Barykova, Y.A.; Logunov, D.Y.; Shmarov, M.; Vinarov, A.; Fiev, D.N.; Vinarova, N.A.; Rakovskaya, I.V.; Baker, P.S.; Shyshynova, I.; Stephenson, A.J.; et al. Association of Mycoplasma hominis infection with prostate cancer. Oncotarget 2011, 2, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Miyake, M.; Ohnishi, K.; Hori, S.; Nakano, A.; Nakano, R.; Yano, H.; Ohnishi, S.; Owari, T.; Morizawa, Y.; Itami, Y.; et al. Mycoplasma genitalium Infection and Chronic Inflammation in Human Prostate Cancer: Detection Using Prostatectomy and Needle Biopsy Specimens. Cells 2019, 8, 212. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Ren, T.; Jiang, B.; Gong, M.; Shou, C. Mycoplasmal membrane protein p37 promotes malignant changes in mammalian cells. Can. J. Microbiol. 2007, 53, 270–276. [Google Scholar] [CrossRef]
- Gong, M.; Meng, L.; Jiang, B.; Zhang, J.; Yang, H.; Wu, J.; Shou, C. p37 fromMycoplasma hyorhinispromotes cancer cell invasiveness and metastasis through activation of MMP-2 and followed by phosphorylation of EGFR. Mol. Cancer Ther. 2008, 7, 530–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.-A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C.; et al. The Bacteroides fragilis Toxin Gene Is Prevalent in the Colon Mucosa of Colorectal Cancer Patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef]
- Chung, L.; Orberg, E.T.; Geis, A.L.; Chan, J.L.; Fu, K.; Shields, C.E.D.; Dejea, C.M.; Fathi, P.; Chen, J.; Finard, B.B.; et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe 2018, 23, 421. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, S.; Giovannucci, E.; De Marzo, A.M.; Leitzmann, M.F.; Willett, W.C.; Platz, E.A. Gonorrhea, Syphilis, Clinical Prostatitis, and the Risk of Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2160–2166. [Google Scholar] [CrossRef] [Green Version]
- Balamurugan, R.; Rajendiran, E.; George, S.; Samuel, G.V.; Ramakrishna, B.S. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, DesulfovibrioandEnterococcus faecalisin the feces of patients with colorectal cancer. J. Gastroenterol. Hepatol. 2008, 23, 1298–1303. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Y.; Huycke, M.M. Commensal-infected macrophages induce dedifferentiation and reprogramming of epithelial cells during colorectal carcinogenesis. Oncotarget 2017, 8, 102176–102190. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Komiya, M.; Fujii, G.; Hamoya, T.; Nakanishi, R.; Fujimoto, K.; Tamura, S.; Kurokawa, Y.; Takahashi, M.; Ijichi, T.; et al. Preventive Effects of Heat-Killed Enterococcus faecalis Strain EC-12 on Mouse Intestinal Tumor Development. Int. J. Mol. Sci. 2017, 18, 826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Almeida, C.V.; Taddei, A.; Amedei, A. The controversial role of Enterococcus faecalis in colorectal cancer. Ther. Adv. Gastroenterol. 2018, 11, 1756284818783606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickertsson, J.A.B.; Desler, C.; Martin-Bertelsen, T.; Machado, A.M.D.; Wadstrøm, T.; Winther, O.; Rasmussen, L.J.; Friis-Hansen, L. Enterococcus faecalis Infection Causes Inflammation, Intracellular Oxphos-Independent ROS Production, and DNA Damage in Human Gastric Cancer Cells. PLoS ONE 2013, 8, e63147. [Google Scholar] [CrossRef] [Green Version]
- Hermsen, J.L.; Schurr, M.J.; Kudsk, K.A.; Faucher, L.D. Phenotyping Clostridium septicum Infection: A Surgeon’s Infectious Disease. J. Surg. Res. 2008, 148, 67–76. [Google Scholar] [CrossRef]
- Chew, S.S.B.; Lubowski, D.Z. Clostridium septicumand malignancy. ANZ J. Surg. 2001, 71, 647–649. [Google Scholar] [CrossRef]
- Dylewski, J.; Luterman, L. Septic arthritis and Clostridium septicum: A clue to colon cancer. Can. Med. Assoc. J. 2010, 182, 1446–1447. [Google Scholar] [CrossRef]
- Engels, E.A.; Shen, M.; Chapman, R.S.; Pfeiffer, R.M.; Yu, Y.-Y.; He, X.; Lan, Q. Tuberculosis and subsequent risk of lung cancer in Xuanwei, China. Int. J. Cancer 2009, 124, 1183–1187. [Google Scholar] [CrossRef]
- Tian, Y.; Hao, T.; Cao, B.; Zhang, W.; Ma, Y.; Lin, Q.; Li, X. Clinical End-Points Associated withMycobacterium tuberculosisand Lung Cancer: Implications into Host-Pathogen Interaction and Coevolution. BioMed Res. Int. 2015, 2015, 827829. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Potential Hallmarks of Cancer and Other Associated Pathologies | Type of Carcinoma Associated |
|---|---|---|
| Helicobacter pylori | Increase proliferation signalling [1,2,3] | Gastric cancer [1,2,3] |
| Escherichia coli(pks+ve strain) | Genomic instability (DNA damage), tumour-promoting inflammation, and increased cell proliferation [4,5,6] | Colorectal cancer [4,5,6] |
| Streptococcus bovis | Increase proliferation signalling | Colorectal cancer [7,8] |
| Salmonella enterica (serovar Enteritidis and Typhi) | Increased cell proliferation, transformation, and tumour-promoting inflammation [9,10,11] | Colorectal cancer and gallbladder carcinoma [9,10,11] |
| Fusobacterium nucleatum | Increased cell proliferation, avoid immune destruction, and tumour-promoting inflammation [12,13] | Colorectal cancer (potentially oral, head, neck, oesophageal, cervical, and gastric cancer tissues) [12,13] |
| Enterotoxigenic Bacteroides fragilis (ETBF) | Increased cell proliferation and tumour-promoting inflammation [14,15,16] | Colorectal cancer [14,15,16] |
| Enterococcus faecalis | Genomic instability, tumour-promoting inflammation, and increased cell proliferation [17] | Colorectal cancer [17] |
| Clostridium septicum | Myonecrosis [18,19] | Potential involvement in colorectal cancer [19] |
| Neisseria gonorrhoeae | DNA damage [20], Genomic instability | Prostate cancer [21,22] and bladder cancer [23] |
| Mycobacterium tuberculosis | Tumour metastasis and avoiding immune destruction [24,25] | Lung cancer [24,25] |
| Chlamydia trachomatis and Chlamydia pneumoniae | Increased cell proliferation, tumour-promoting [26,27,28] inflammation [29], genomic instability [30], and evasion of apoptosis [31], evade immune system [32] | Cervical cancer [26,27,28] and lung cancer |
| Mycoplasma hyorhinis | Cell immortality, invasion, and increased cell proliferation [33,34] | Prostate cancer, gastric carcinoma, oesophageal cancer, lung cancer, and breast cancer [33,34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, J.P.; Ali, W.M.; Sivadasan, R.; Rajeeve, K. Bacteria–Cancer Interface: Awaiting the Perfect Storm. Pathogens 2021, 10, 1321. https://doi.org/10.3390/pathogens10101321
Hansen JP, Ali WM, Sivadasan R, Rajeeve K. Bacteria–Cancer Interface: Awaiting the Perfect Storm. Pathogens. 2021; 10(10):1321. https://doi.org/10.3390/pathogens10101321
Chicago/Turabian StyleHansen, Jonathan Pommer, Waled Mohammed Ali, Rajeeve Sivadasan, and Karthika Rajeeve. 2021. "Bacteria–Cancer Interface: Awaiting the Perfect Storm" Pathogens 10, no. 10: 1321. https://doi.org/10.3390/pathogens10101321
APA StyleHansen, J. P., Ali, W. M., Sivadasan, R., & Rajeeve, K. (2021). Bacteria–Cancer Interface: Awaiting the Perfect Storm. Pathogens, 10(10), 1321. https://doi.org/10.3390/pathogens10101321