
PIAS1 Regulates Hepatitis C Virus-Induced Lipid Droplet Accumulation by Controlling Septin 9 and Microtubule Filament Assembly

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Result
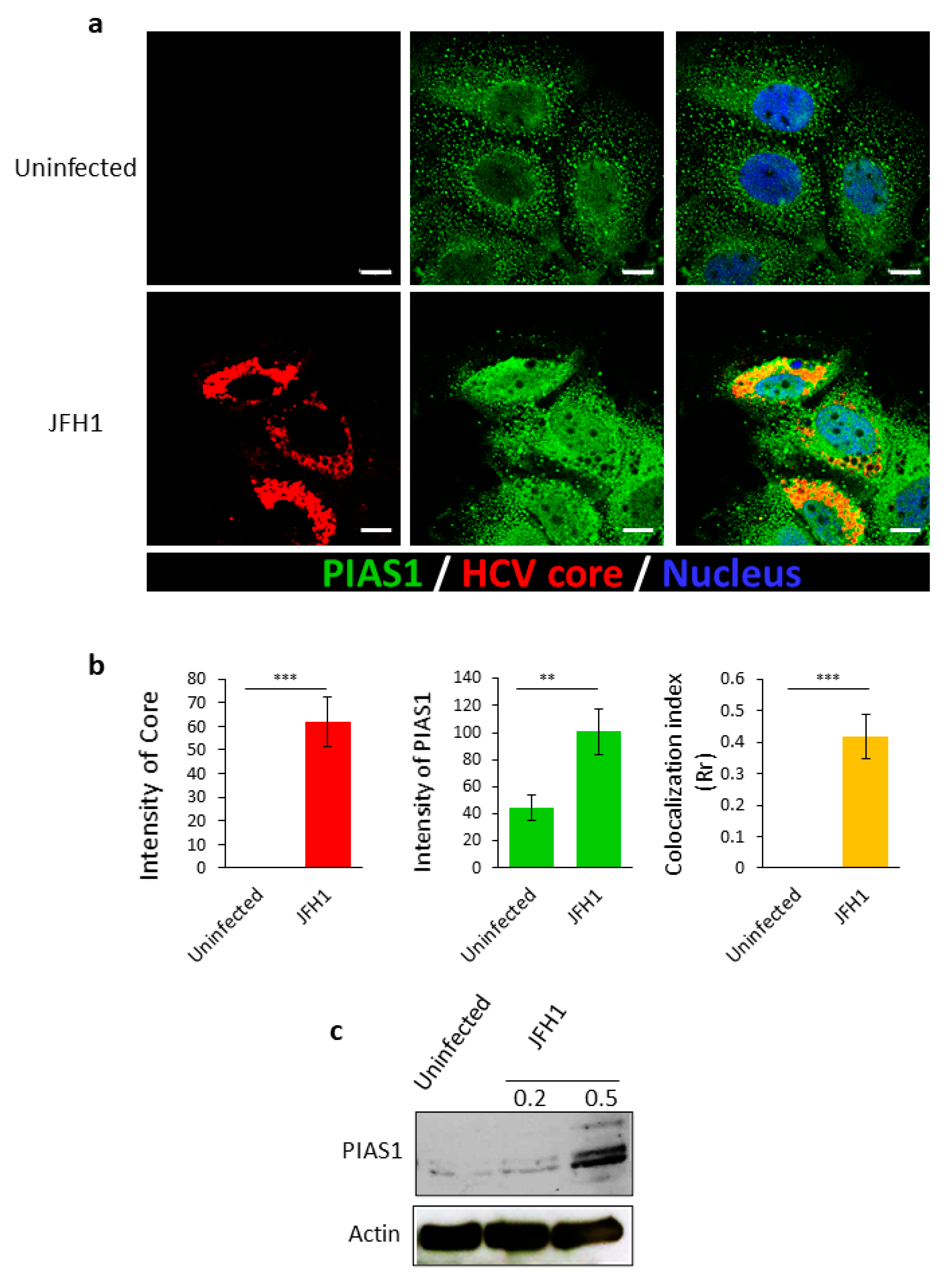
2.1. HCV Infection Increases the Expression of PIAS1, Which Co-Localizes with Its Core Protein
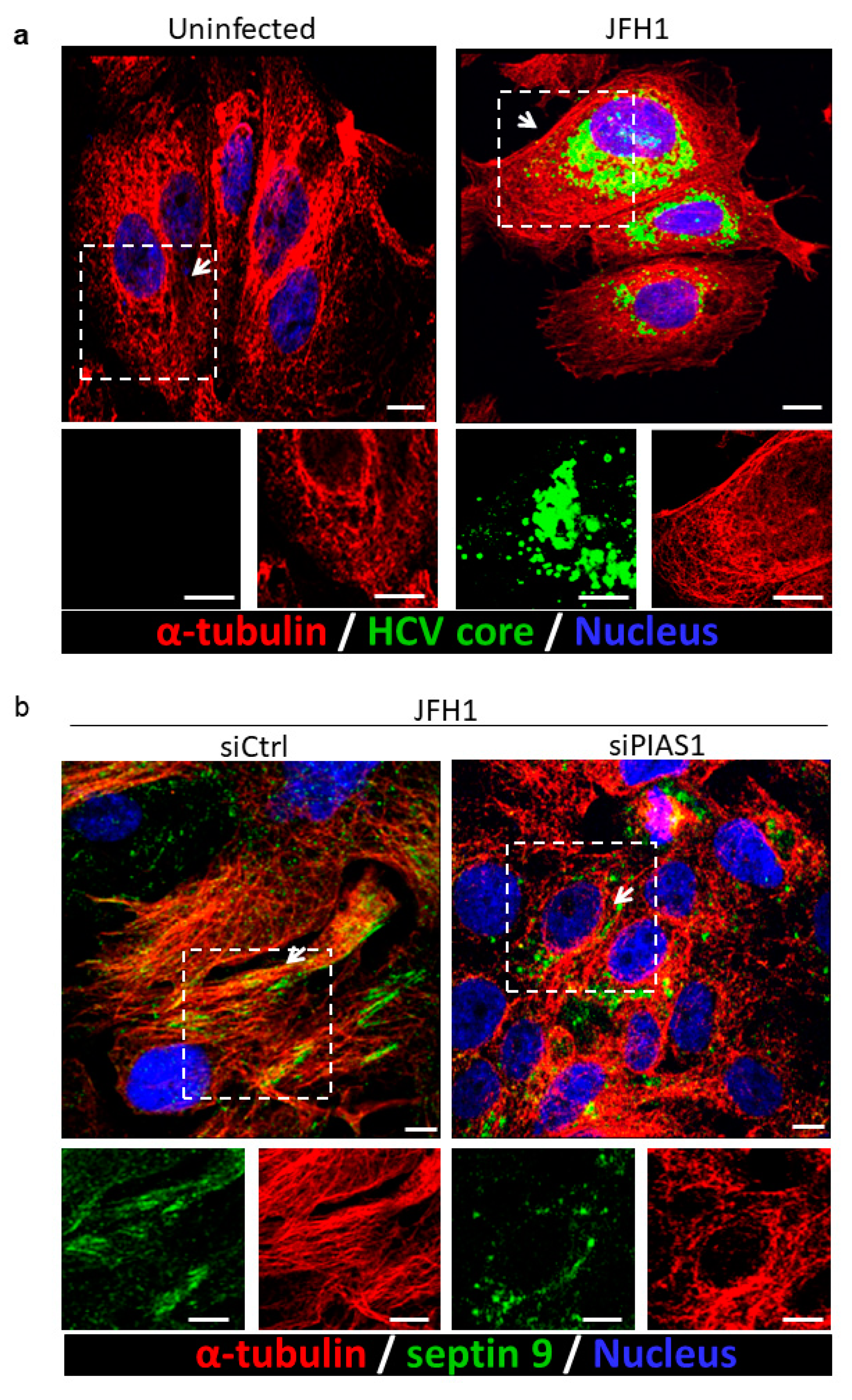
2.2. PIAS1 Regulates the HCV-Induced Stabilization of Microtubules and Endogenous Septin 9 Filaments
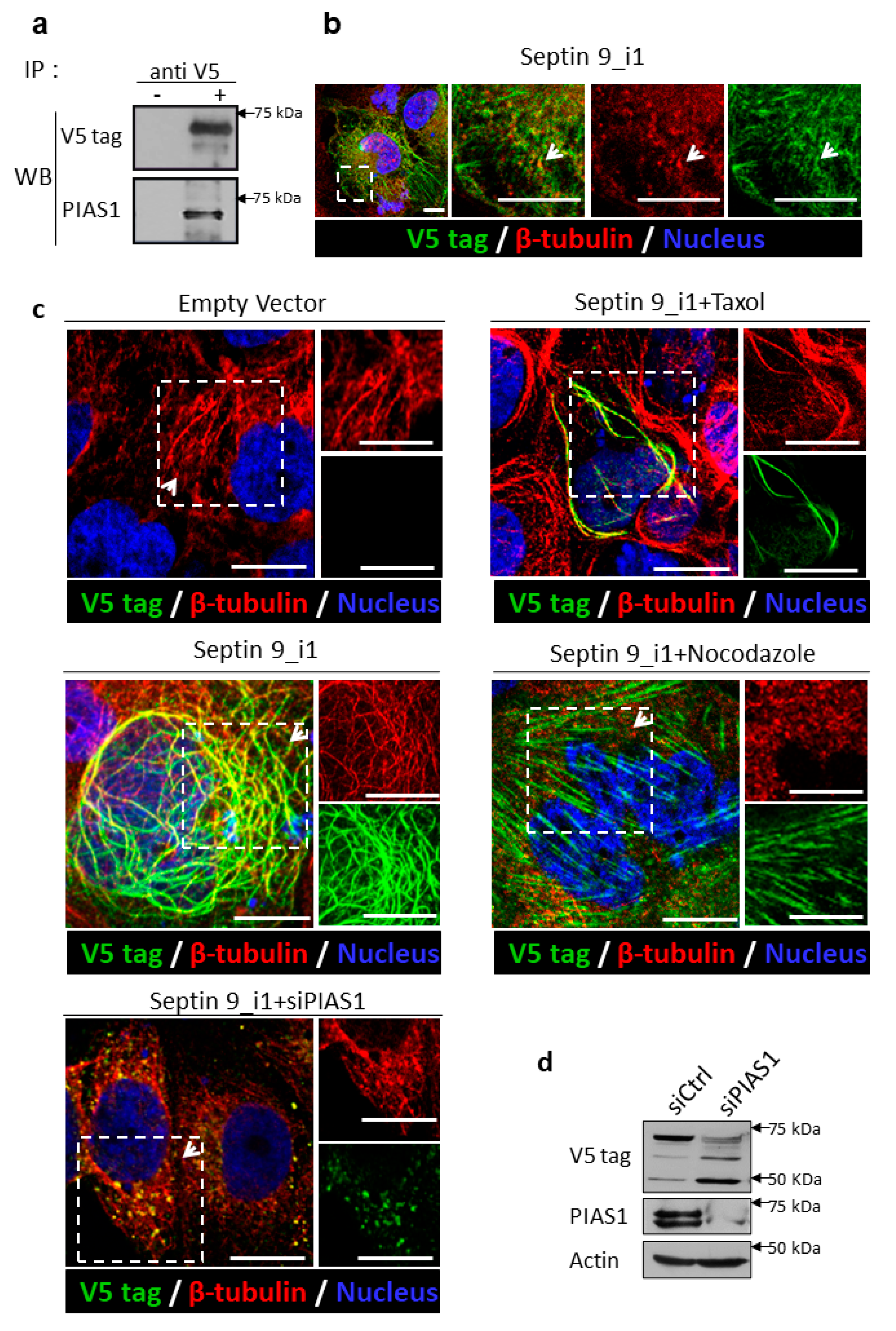
2.3. PIAS1 Interacts with Septin 9_i1 and Controls Its Filamentous Structure Formation and MT Organization in the Presence of the HCV Genomic Replicon
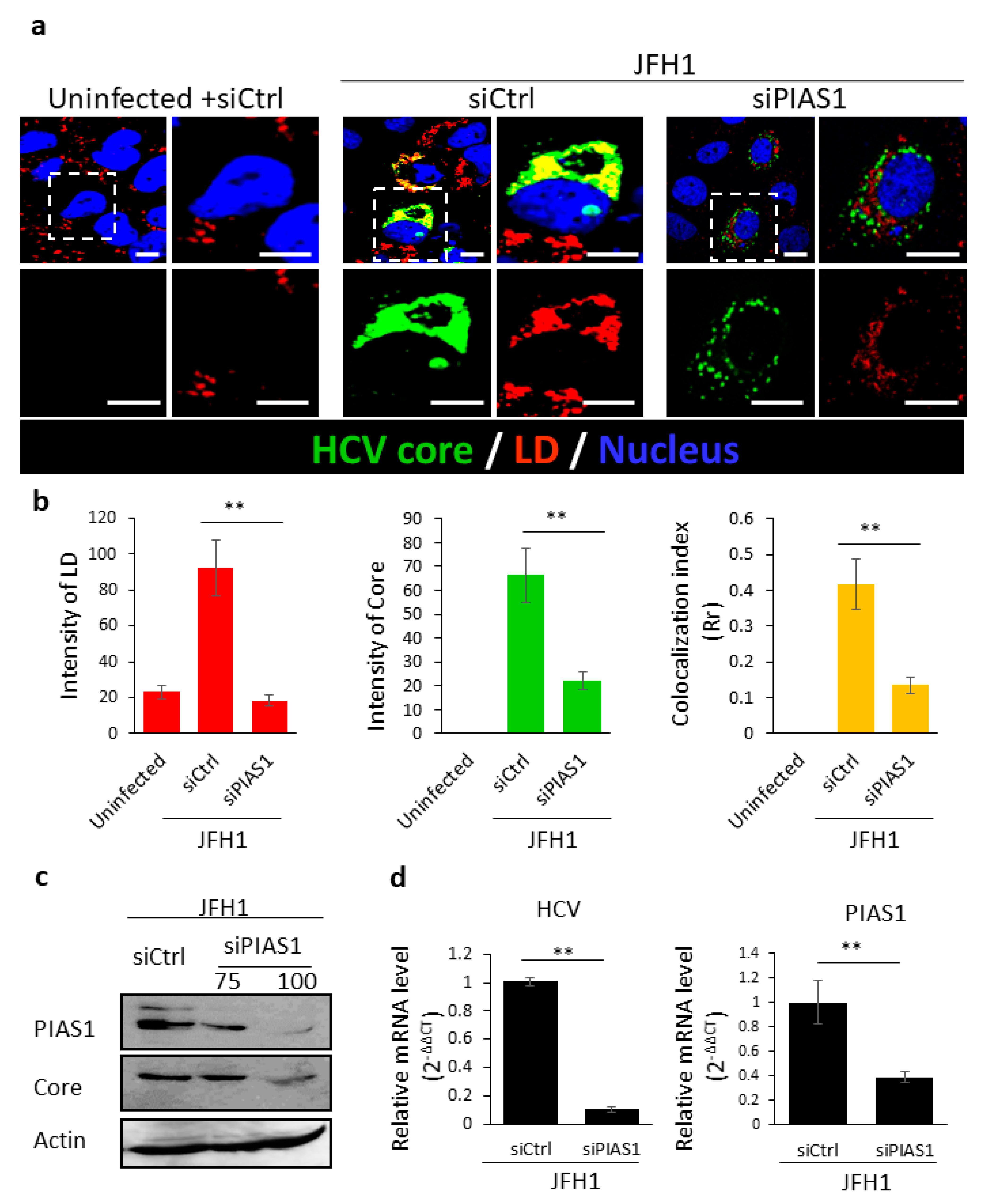
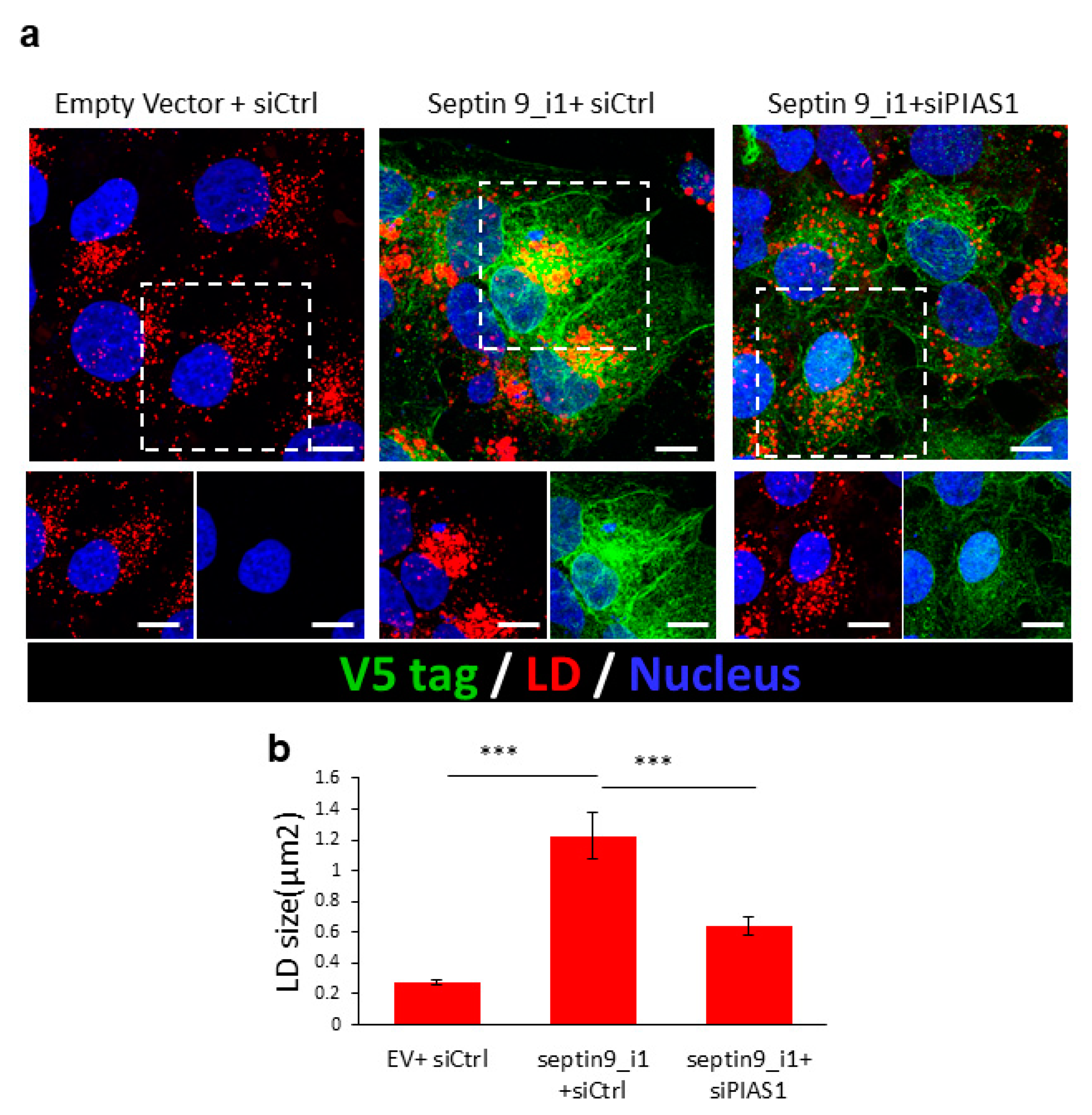
2.4. PIAS1 Regulates Septin 9-Induced LD Accumulation
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture
4.3. siRNAs and Cell Transfection
4.4. Immunoblotting
4.5. Immunofluorescence Staining
4.6. JFH1/HCVcc Inoculation
4.7. Determination of Infectious HCV Titer
4.8. RNA Extraction and RT-PCR Analysis
4.9. Image Acquisition and Analysis
4.10. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alqahtani, S.A.; Colombo, M. Viral Hepatitis as a Risk Factor for the Development of Hepatocellular Carcinoma. Hepatoma Res. 2020, 6, 58. [Google Scholar] [CrossRef]
- Wu, P.-S.; Chang, T.-S.; Lu, S.-N.; Su, H.-J.; Chang, S.-Z.; Hsu, C.-W.; Chen, M.-Y. An Investigation of the Side Effects, Patient Feedback, and Physiological Changes Associated with Direct-Acting Antiviral Therapy for Hepatitis C. Int. J. Environ. Res. Public Health 2019, 16, 4981. [Google Scholar] [CrossRef] [Green Version]
- Asselah, T.; Rubbia-Brandt, L.; Marcellin, P.; Negro, F. Steatosis in Chronic Hepatitis C: Why Does It Really Matter? Gut 2006, 55, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Koike, K. Steatosis, Liver Injury, and Hepatocarcinogenesis in Hepatitis C Viral Infection. J. Gastroenterol. 2009, 44 (Suppl. 19), 82–88. [Google Scholar] [CrossRef]
- Leandro, G.; Mangia, A.; Hui, J.; Fabris, P.; Rubbia-Brandt, L.; Colloredo, G.; Adinolfi, L.E.; Asselah, T.; Jonsson, J.R.; Smedile, A.; et al. Relationship between Steatosis, Inflammation, and Fibrosis in Chronic Hepatitis C: A Meta-Analysis of Individual Patient Data. Gastroenterology 2006, 130, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Herms, A.; Bosch, M.; Reddy, B.J.N.; Schieber, N.L.; Fajardo, A.; Rupérez, C.; Fernández-Vidal, A.; Ferguson, C.; Rentero, C.; Tebar, F.; et al. AMPK Activation Promotes Lipid Droplet Dispersion on Detyrosinated Microtubules to Increase Mitochondrial Fatty Acid Oxidation. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty Acid Trafficking in Starved Cells: Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurat, C.F.; Wolinski, H.; Petschnigg, J.; Kaluarachchi, S.; Andrews, B.; Natter, K.; Kohlwein, S.D. Cdk1/Cdc28-Dependent Activation of the Major Triacylglycerol Lipase Tgl4 in Yeast Links Lipolysis to Cell-Cycle Progression. Mol. Cell 2009, 33, 53–63. [Google Scholar] [CrossRef]
- Chauhan, N.; Visram, M.; Cristobal-Sarramian, A.; Sarkleti, F.; Kohlwein, S.D. Morphogenesis Checkpoint Kinase Swe1 Is the Executor of Lipolysis-Dependent Cell-Cycle Progression. Proc. Natl. Acad. Sci. USA 2015, 112, E1077–E1085. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-Mediated Fat Catabolism Regulates Cardiac Mitochondrial Function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.; Abbott, M.J.; Ahmadian, M.; Lopes, A.B.; Wang, Y.; Sul, H.S. Desnutrin/ATGL Activates PPARδ to Promote Mitochondrial Function for Insulin Secretion in Islet β Cells. Cell Metab. 2013, 18, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Schuldiner, M.; Bohnert, M. A Different Kind of Love—Lipid Droplet Contact Sites. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1188–1196. [Google Scholar] [CrossRef]
- Barbosa, A.D.; Siniossoglou, S. Function of Lipid Droplet-Organelle Interactions in Lipid Homeostasis. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.D.; Savage, D.B.; Siniossoglou, S. Lipid Droplet-Organelle Interactions: Emerging Roles in Lipid Metabolism. Curr. Opin. Cell Biol. 2015, 35, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Akil, A.; Peng, J.; Omrane, M.; Gondeau, C.; Desterke, C.; Marin, M.; Tronchère, H.; Taveneau, C.; Sar, S.; Briolotti, P.; et al. Septin 9 Induces Lipid Droplets Growth by a Phosphatidylinositol-5- Phosphate and Microtubule-Dependent Mechanism Hijacked by HCV. Nat. Commun. 2016, 7, 12203. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Froese, C.D.; Estey, M.P.; Trimble, W.S. SEPT9 Occupies the Terminal Positions in Septin Octamers and Mediates Polymerization-Dependent Functions in Abscission. J. Cell Biol. 2011, 195, 815–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, E.A.; Tokhtaeva, E.; Turdikulova, S.; Capri, J.; Whitelegge, J.P.; Scott, D.R.; Sachs, G.; Berditchevski, F.; Vagin, O. Septin Oligomerization Regulates Persistent Expression of ErbB2/HER2 in Gastric Cancer Cells. Biochem. J. 2016, 473, 1703–1718. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, M.; Field, C.M.; Coughlin, M.L.; Straight, A.F.; Mitchison, T.J. Self- and Actin-Templated Assembly of Mammalian Septins. Dev. Cell 2002, 3, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Mostowy, S.; Cossart, P. Septins: The Fourth Component of the Cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Sellin, M.E.; Sandblad, L.; Stenmark, S.; Gullberg, M. Deciphering the Rules Governing Assembly Order of Mammalian Septin Complexes. Mol. Biol. Cell 2011, 22, 3152–3164. [Google Scholar] [CrossRef]
- Barral, Y.; Kinoshita, M. Structural Insights Shed Light onto Septin Assemblies and Function. Curr. Opin. Cell Biol. 2008, 20, 12–18. [Google Scholar] [CrossRef]
- Ihara, M.; Tomimoto, H.; Kitayama, H.; Morioka, Y.; Akiguchi, I.; Shibasaki, H.; Noda, M.; Kinoshita, M. Association of the Cytoskeletal GTP-Binding Protein Sept4/H5 with Cytoplasmic Inclusions Found in Parkinson’s Disease and Other Synucleinopathies. J. Biol. Chem. 2003, 278, 24095–24102. [Google Scholar] [CrossRef] [Green Version]
- Shehadeh, L.; Mitsi, G.; Adi, N.; Bishopric, N.; Papapetropoulos, S. Expression of Lewy Body Protein Septin 4 in Postmortem Brain of Parkinson’s Disease and Control Subjects. Mov. Disord. 2009, 24, 204–210. [Google Scholar] [CrossRef]
- Pennington, K.; Beasley, C.L.; Dicker, P.; Fagan, A.; English, J.; Pariante, C.M.; Wait, R.; Dunn, M.J.; Cotter, D.R. Prominent Synaptic and Metabolic Abnormalities Revealed by Proteomic Analysis of the Dorsolateral Prefrontal Cortex in Schizophrenia and Bipolar Disorder. Mol. Psychiatry 2008, 13, 1102–1117. [Google Scholar] [CrossRef]
- Tada, T.; Simonetta, A.; Batterton, M.; Kinoshita, M.; Edbauer, D.; Sheng, M. Role of Septin Cytoskeleton in Spine Morphogenesis and Dendrite Development in Neurons. Curr. Biol. 2007, 17, 1752–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro-Cerdá, J.; Jonquières, R.; Gouin, E.; Vandekerckhove, J.; Garin, J.; Cossart, P. Distinct Protein Patterns Associated with Listeria Monocytogenes InlA- or InlB-Phagosomes. Cell Microbiol. 2002, 4, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Boddy, K.C.; Gao, A.D.; Truong, D.; Kim, M.S.; Froese, C.D.; Trimble, W.S.; Brumell, J.H. Septinregulated Actin Dynamics Promote Salmonella Invasion of Host Cells. Cell Microbiol. 2018, 20, e12866. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.P.; Lobato-Márquez, D.; Pramanik, N.; Sirianni, A.; Daza-Cajigal, V.; Rivers, E.; Cavazza, A.; Bouma, G.; Moulding, D.; Hultenby, K.; et al. Wiskott-Aldrich Syndrome Protein Regulates Autophagy and Inflammasome Activity in Innate Immune Cells. Nat. Commun. 2017, 8, 1576. [Google Scholar] [CrossRef]
- Mostowy, S.; Cossart, P. Cytoskeleton Rearrangements during Listeria Infection: Clathrin and Septins as New Players in the Game. Cell Motil. 2009, 66, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S.; Janel, S.; Forestier, C.; Roduit, C.; Kasas, S.; Pizarro-Cerdá, J.; Cossart, P.; Lafont, F. A Role for Septins in the Interaction between the Listeria Monocytogenes INVASION PROTEIN InlB and the Met Receptor. Biophys. J. 2011, 100, 1949–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfanzelter, J.; Mostowy, S.; Way, M. Septins Suppress the Release of Vaccinia Virus from Infected Cells. J. Cell Biol. 2018, 217, 2911–2929. [Google Scholar] [CrossRef] [PubMed]
- Dolat, L.; Hu, Q.; Spiliotis, E.T. Septin Functions in Organ System Physiology and Pathology. Biol. Chem. 2014, 395, 123–141. [Google Scholar] [CrossRef] [Green Version]
- Montagna, C.; Sagie, M.; Zechmeister, J. Mammalian Septins in Health and Disease. Res. Rep. Biochem. 2015, 2015, 59–72. [Google Scholar] [CrossRef] [Green Version]
- Marcus, J.; Bejerano-Sagie, M.; Patterson, N.; Bagchi, S.; Verkhusha, V.V.; Connolly, D.; Goldberg, G.L.; Golden, A.; Sharma, V.P.; Condeelis, J.; et al. Septin 9 Isoforms Promote.Tumorigenesis in Mammary Epithelial Cells by Increasing Migration and ECM Degradation through Metalloproteinase Secretion at Focal Adhesions. Oncogene 2019, 38, 5839–5859. [Google Scholar] [CrossRef]
- Diesenberg, K.; Beerbaum, M.; Fink, U.; Schmieder, P.; Krauss, M. SEPT9 Negatively Regulates Ubiquitin-Dependent Downregulation of EGFR. J. Cell Sci. 2015, 128, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Liu, A.; Wang, X.; Chen, Y.; Shen, Y.; Tan, Z.; Qiu, M. Repression of Septin9 and Septin2 Suppresses Tumor Growth of Human Glioblastoma Cells. Cell Death Dis. 2018, 9, 514. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Liu, L.; Fan, N.; Zhang, Q.; Wang, W.; Zheng, M.; Ma, L.; Li, Y.; Shi, L. The Requirement of SEPT2 and SEPT7 for Migration and Invasion in Human Breast Cancer via MEK/ERK Activation. Oncotarget 2016, 7, 61587–61600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, K.; Asano, T.; Nozawa, Y.; Inagaki, M. Biochemical and Cell Biological Analyses of a Mammalian Septin Complex, Sept7/9b/11. J. Biol. Chem. 2004, 279, 55895–55904. [Google Scholar] [CrossRef] [Green Version]
- Sirajuddin, M.; Farkasovsky, M.; Hauer, F.; Kühlmann, D.; Macara, I.G.; Weyand, M.; Stark, H.; Wittinghofer, A. Structural Insight into Filament Formation by Mammalian Septins. Nature 2007, 449, 311–315. [Google Scholar] [CrossRef]
- Spiliotis, E.T.; Nelson, W.J. Here Come the Septins: Novel Polymers That Coordinate Intracellular Functions and Organization. J. Cell Sci. 2006, 119, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.O.; Nielsen, M.L. Site-Specific Mapping of the Human SUMO Proteome Reveals Co-Modification with Phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Boscaini, S.; Cauvin, C.; Siguier, M.; Mostowy, S.; Echard, A.; Cossart, P. SUMOylation of Human Septins Is Critical for Septin Filament Bundling and Cytokinesis. J. Cell Biol. 2017, 216, 4041–4052. [Google Scholar] [CrossRef] [Green Version]
- Omrane, M.; Camara, A.S.; Taveneau, C.; Benzoubir, N.; Tubiana, T.; Yu, J.; Guérois, R.; Samuel, D.; Goud, B.; Poüs, C.; et al. Septin 9 Has Two Polybasic Domains Critical to Septin Filament Assembly and Golgi Integrity. iScience 2019, 13, 138–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Rodríguez, Y.; Momany, M. Posttranslational Modifications and Assembly of Septin Heteropolymers and Higher-Order Structures. Curr. Opin. Microbiol. 2012, 15, 660–668. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Iwase, M.; Konishi, M.; Tanaka, M.; Toh-e, A.; Kikuchi, Y. Smt3, a SUMO-1 Homolog, Is Conjugated to Cdc3, a Component of Septin Rings at the Mother-Bud Neck in Budding Yeast. Biochem. Biophys. Res. Commun. 1999, 259, 582–587. [Google Scholar] [CrossRef]
- Garcia, G.; Bertin, A.; Li, Z.; Song, Y.; McMurray, M.A.; Thorner, J.; Nogales, E. Subunit-Dependent Modulation of Septin Assembly: Budding Yeast Septin Shs1 Promotes Ring and Gauze Formation. J. Cell Biol. 2011, 195, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.S.; Blobel, G. Cell Cycle-Regulated Attachment of the Ubiquitin-Related Protein SUMO to the Yeast Septins. J. Cell Biol. 1999, 147, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.S.; Gupta, A.A. An E3-like Factor That Promotes SUMO Conjugation to the Yeast Septins. Cell 2001, 106, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Hoefer, J.; Schäfer, G.; Klocker, H.; Erb, H.H.H.; Mills, I.G.; Hengst, L.; Puhr, M.; Culig, Z. PIAS1 Is Increased in Human Prostate Cancer and Enhances Proliferation through Inhibition of P21. Am. J. Pathol. 2012, 180, 2097–2107. [Google Scholar] [CrossRef] [Green Version]
- Morozko, E.L.; Smith-Geater, C.; Monteys, A.M.; Pradhan, S.; Lim, R.G.; Langfelder, P.; Kachemov, M.; Hill, A.; Stocksdale, J.T.; Cullis, P.R.; et al. PIAS1 Modulates Striatal Transcription, DNA Damage Repair, and SUMOylation with Relevance to Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2021836118. [Google Scholar] [CrossRef]
- Zlatanou, A.; Stewart, G.S. A PIAS-Ed View of DNA Double Strand Break Repair Focuses on SUMO. DNA Repair 2010, 9, 588–592. [Google Scholar] [CrossRef]
- Liu, B.; Shuai, K. Targeting the PIAS1 SUMO Ligase Pathway to Control Inflammation. Trends Pharm. Sci. 2008, 29, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Shuai, K. Regulation of Cytokine Signaling Pathways by PIAS Proteins. Cell Res. 2006, 16, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jääskeläinen, T.; Palvimo, J.J. PIAS Proteins: Pleiotropic Interactors Associated with SUMO. Cell Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The Lipid Droplet Is an Important Organelle for Hepatitis C Virus Production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Roohvand, F.; Maillard, P.; Lavergne, J.-P.; Boulant, S.; Walic, M.; Andréo, U.; Goueslain, L.; Helle, F.; Mallet, A.; McLauchlan, J.; et al. Initiation of Hepatitis C Virus Infection Requires the Dynamic Microtubule Network. J. Biol. Chem. 2009, 284, 13778–13791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulant, S.; Douglas, M.W.; Moody, L.; Budkowska, A.; Targett-Adams, P.; McLauchlan, J. Hepatitis C Virus Core Protein Induces Lipid Droplet Redistribution in a Microtubule- and Dynein-Dependent Manner. Traffic 2008, 9, 1268–1282. [Google Scholar] [CrossRef]
- Blight, K.J.; Kolykhalov, A.A.; Rice, C.M. Efficient Initiation of HCV RNA Replication in Cell Culture. Science 2000, 290, 1972–1974. [Google Scholar] [CrossRef]
- Surka, M.C.; Tsang, C.W.; Trimble, W.S. The Mammalian Septin MSF Localizes with Microtubules and Is Required for Completion of Cytokinesis. Mol. Biol. Cell 2002, 13, 3532–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA Replication and Assembly: Living on the Fat of the Land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-C.; Yang, C.-H.; Lo, S.-Y. Cellular Factors Involved in the Hepatitis C Virus Life Cycle. World J. Gastroenterol. 2021, 27, 4555–4581. [Google Scholar] [CrossRef] [PubMed]
- El-Saadany, S.; Ziada, D.H.; El Bassat, H.; Farrag, W.; El-Serogy, H.; Eid, M.; Abdallah, M.; Ghazy, M.; Salem, H.A. The Role of Hepatic Expression of STAT1, SOCS3 and PIAS1 in the Response of Chronic Hepatitis C Patients to Therapy. Can. J. Gastroenterol. 2013, 27, e13–e17. [Google Scholar] [CrossRef] [PubMed]
- Duong, F.H.T.; Filipowicz, M.; Tripodi, M.; La Monica, N.; Heim, M.H. Hepatitis C Virus Inhibits Interferon Signaling through Up-Regulation of Protein Phosphatase 2A. Gastroenterology 2004, 126, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.; Bermúdez-Silva, F.J.; Lasarte, J.J.; Rodriguez-Fonseca, F.; Baixeras, E. Liver Expression of Proteins Controlling Interferon-Mediated Signalling as Predictive Factors in the Response to Therapy in Patients with Hepatitis C Virus Infection. J. Pathol. 2007, 213, 347–355. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akil, A.; Song, P.; Peng, J.; Gondeau, C.; Samuel, D.; Gassama-Diagne, A. PIAS1 Regulates Hepatitis C Virus-Induced Lipid Droplet Accumulation by Controlling Septin 9 and Microtubule Filament Assembly. Pathogens 2021, 10, 1327. https://doi.org/10.3390/pathogens10101327
Akil A, Song P, Peng J, Gondeau C, Samuel D, Gassama-Diagne A. PIAS1 Regulates Hepatitis C Virus-Induced Lipid Droplet Accumulation by Controlling Septin 9 and Microtubule Filament Assembly. Pathogens. 2021; 10(10):1327. https://doi.org/10.3390/pathogens10101327
Chicago/Turabian StyleAkil, Abdellah, Peixuan Song, Juan Peng, Claire Gondeau, Didier Samuel, and Ama Gassama-Diagne. 2021. "PIAS1 Regulates Hepatitis C Virus-Induced Lipid Droplet Accumulation by Controlling Septin 9 and Microtubule Filament Assembly" Pathogens 10, no. 10: 1327. https://doi.org/10.3390/pathogens10101327
APA StyleAkil, A., Song, P., Peng, J., Gondeau, C., Samuel, D., & Gassama-Diagne, A. (2021). PIAS1 Regulates Hepatitis C Virus-Induced Lipid Droplet Accumulation by Controlling Septin 9 and Microtubule Filament Assembly. Pathogens, 10(10), 1327. https://doi.org/10.3390/pathogens10101327