
Understanding COVID-19 Pathogenesis: A Drug-Repurposing Effort to Disrupt Nsp-1 Binding to Export Machinery Receptor Complex


Abstract
:1. Introduction
2. Methods
3. Results and Discussions
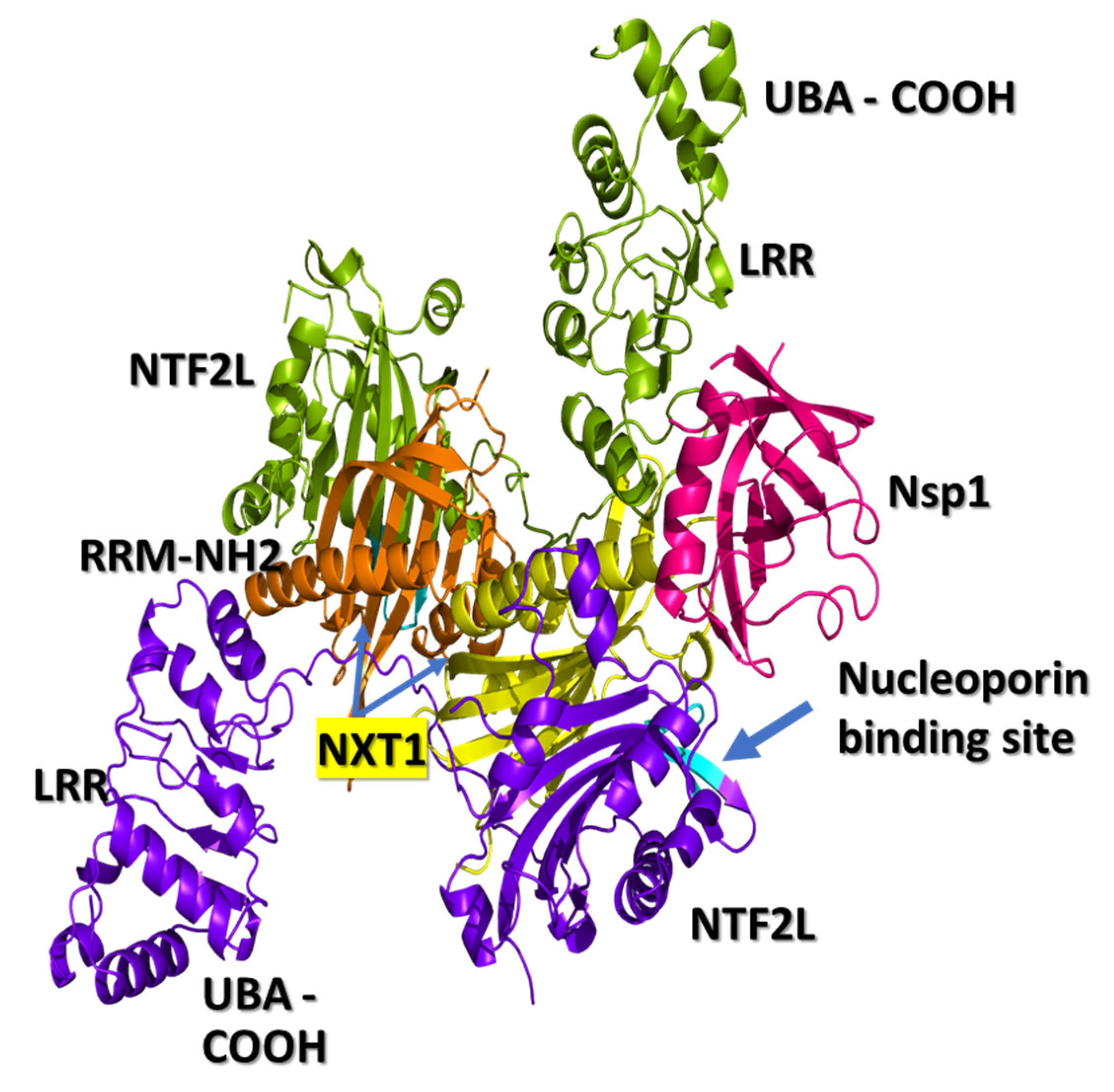
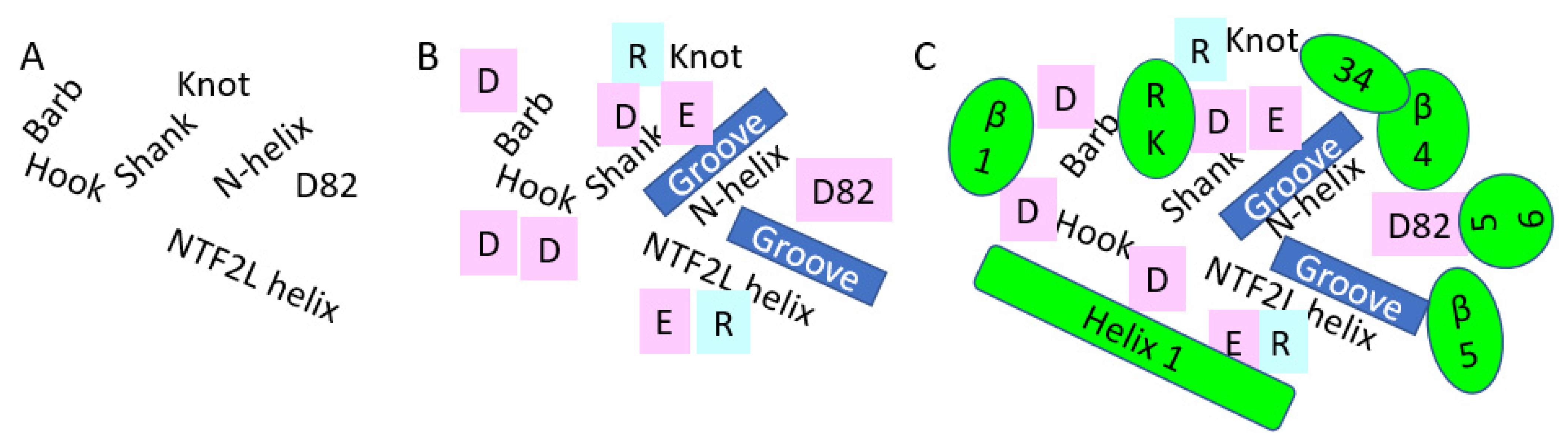
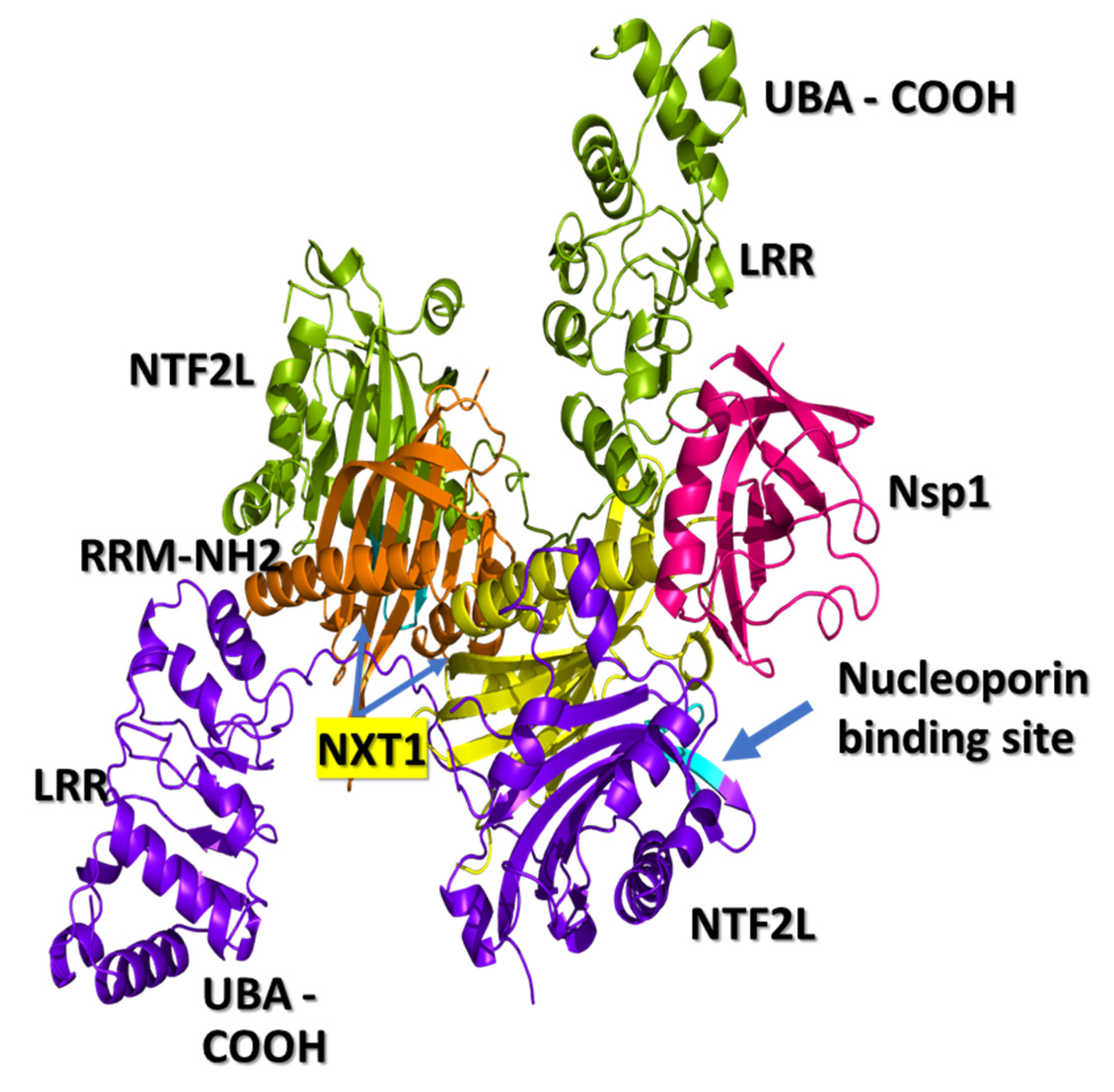
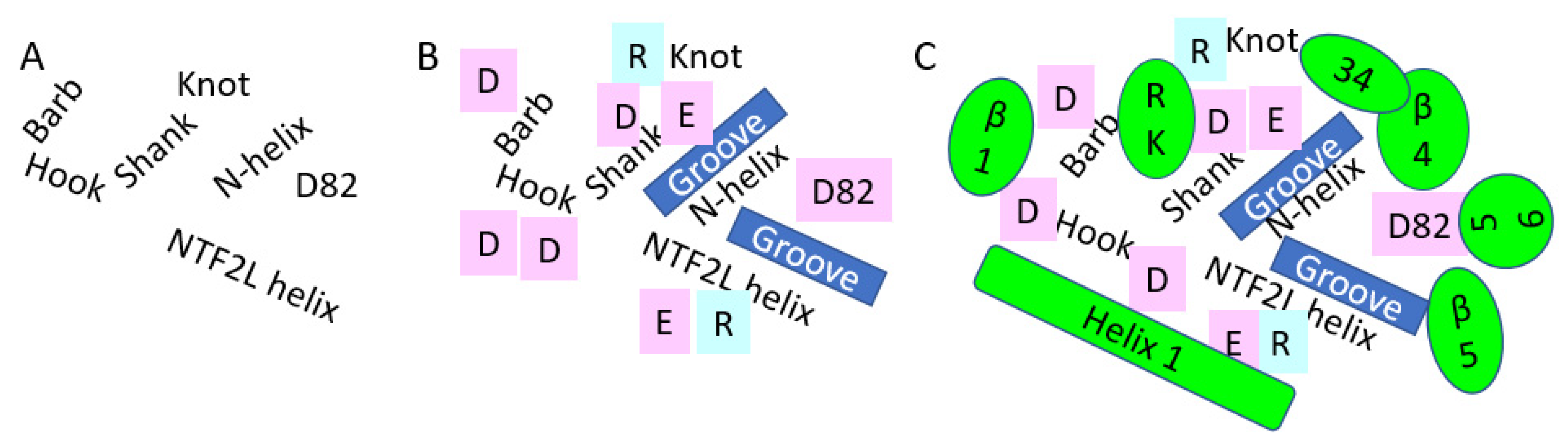
3.1. NXF1–NXT1
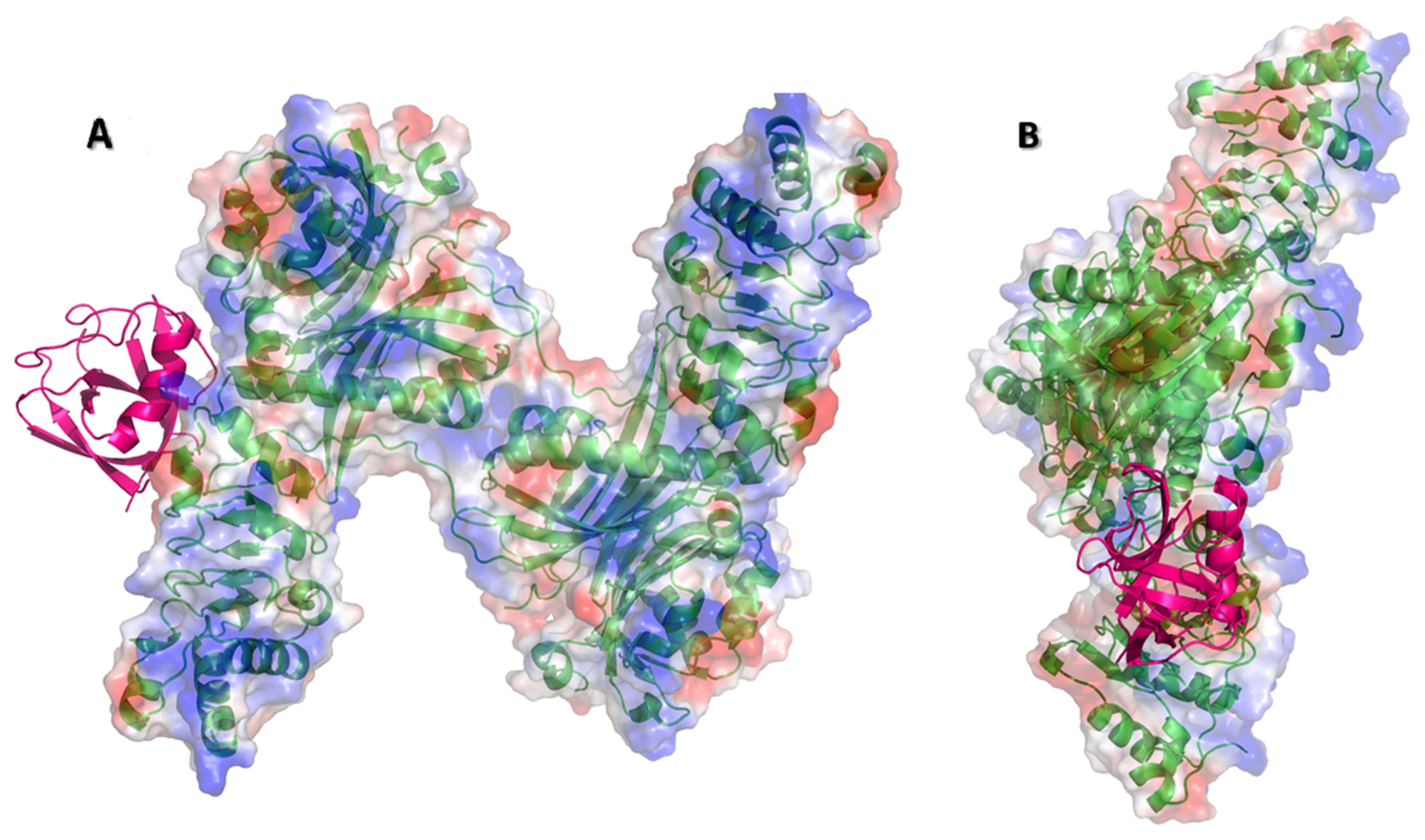
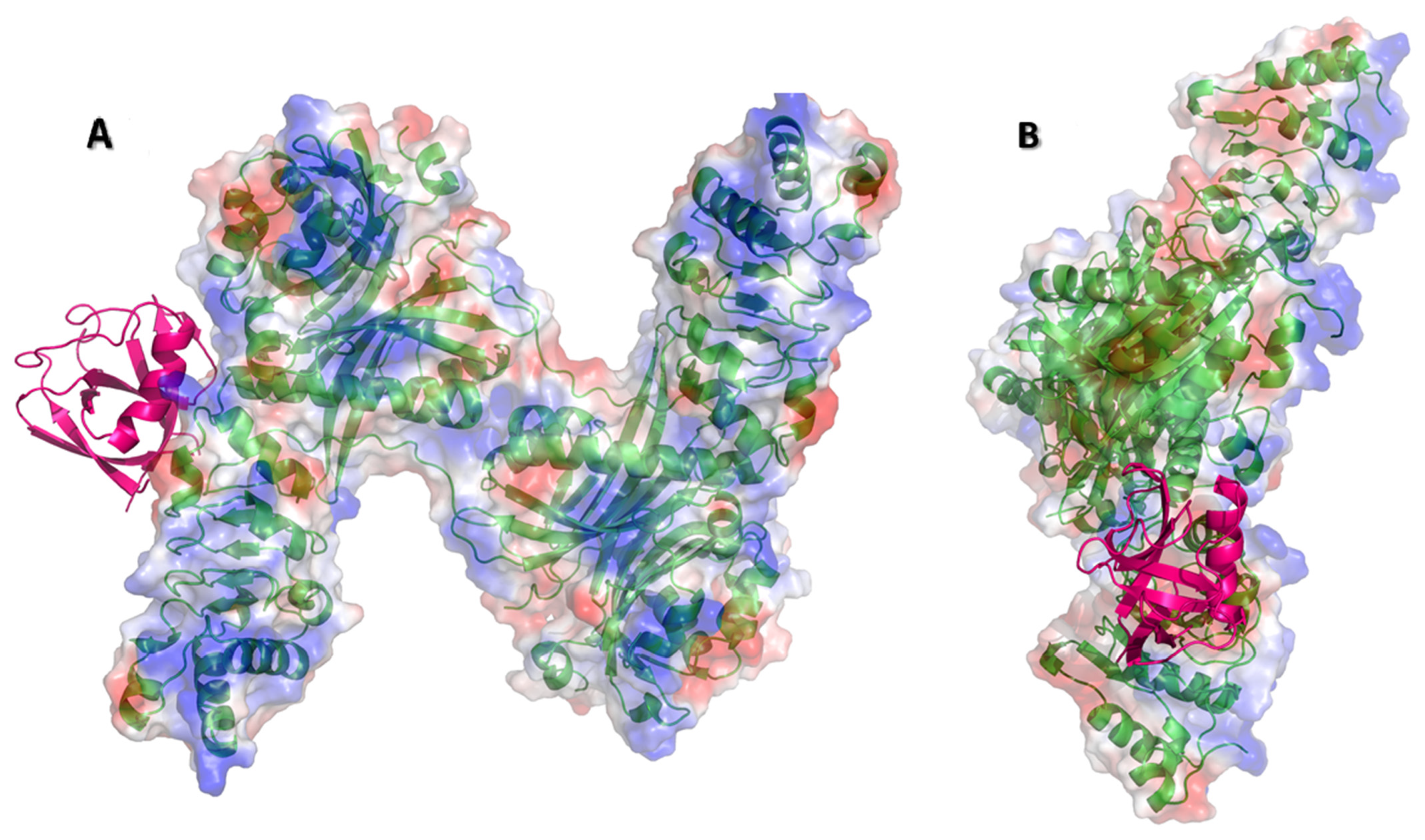
3.2. Nsp1 Docked to NXF1–NXT1
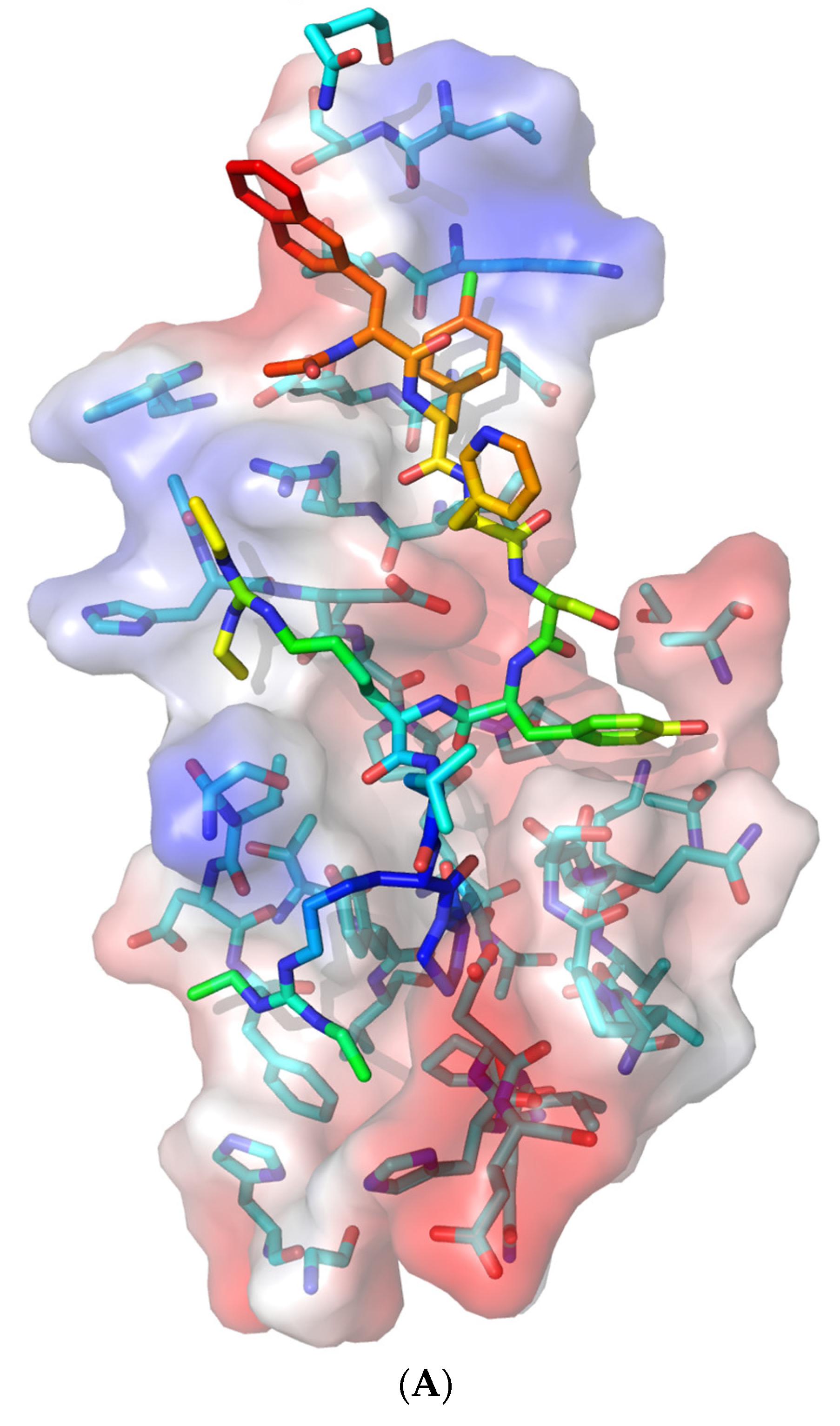
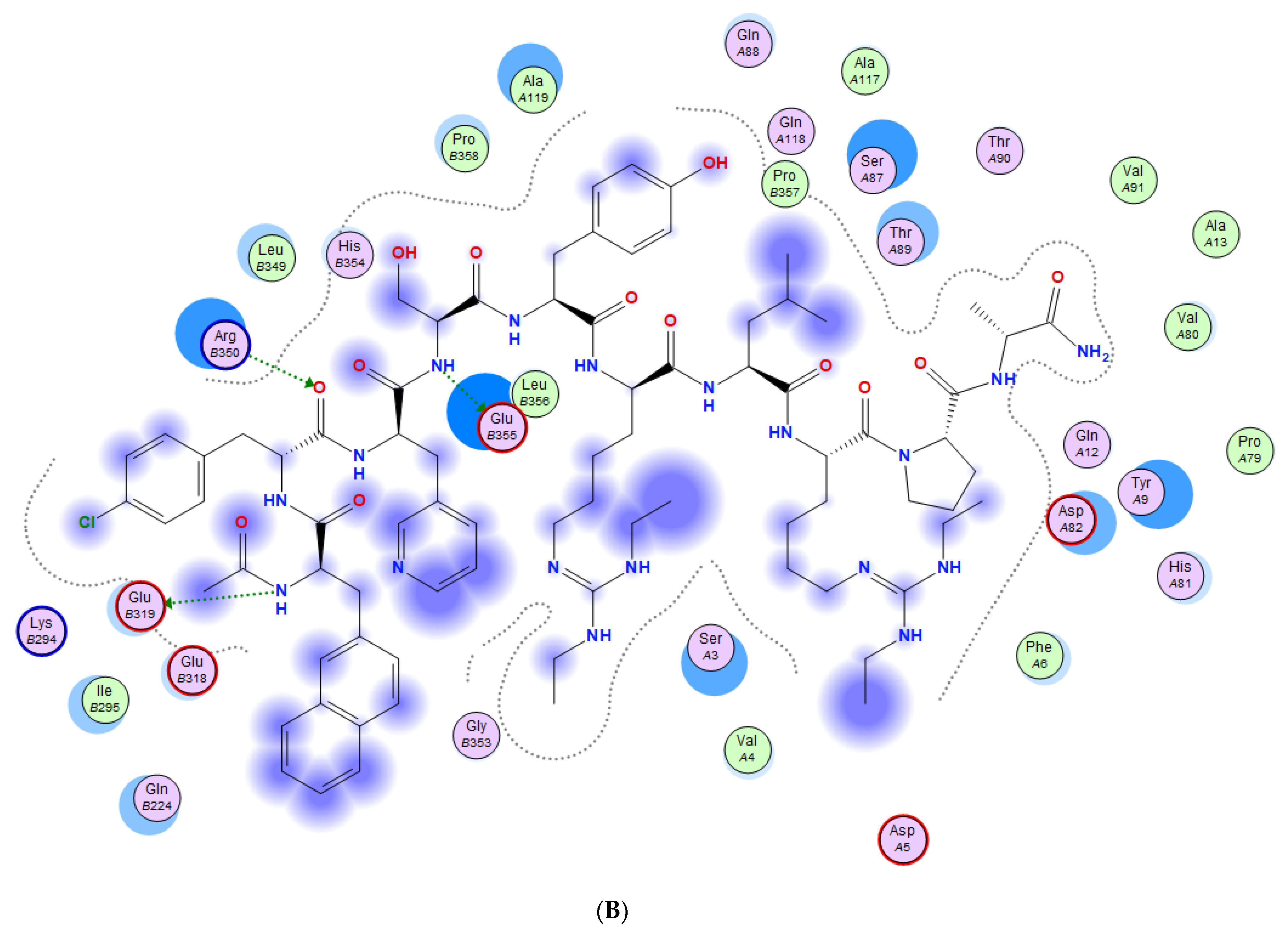
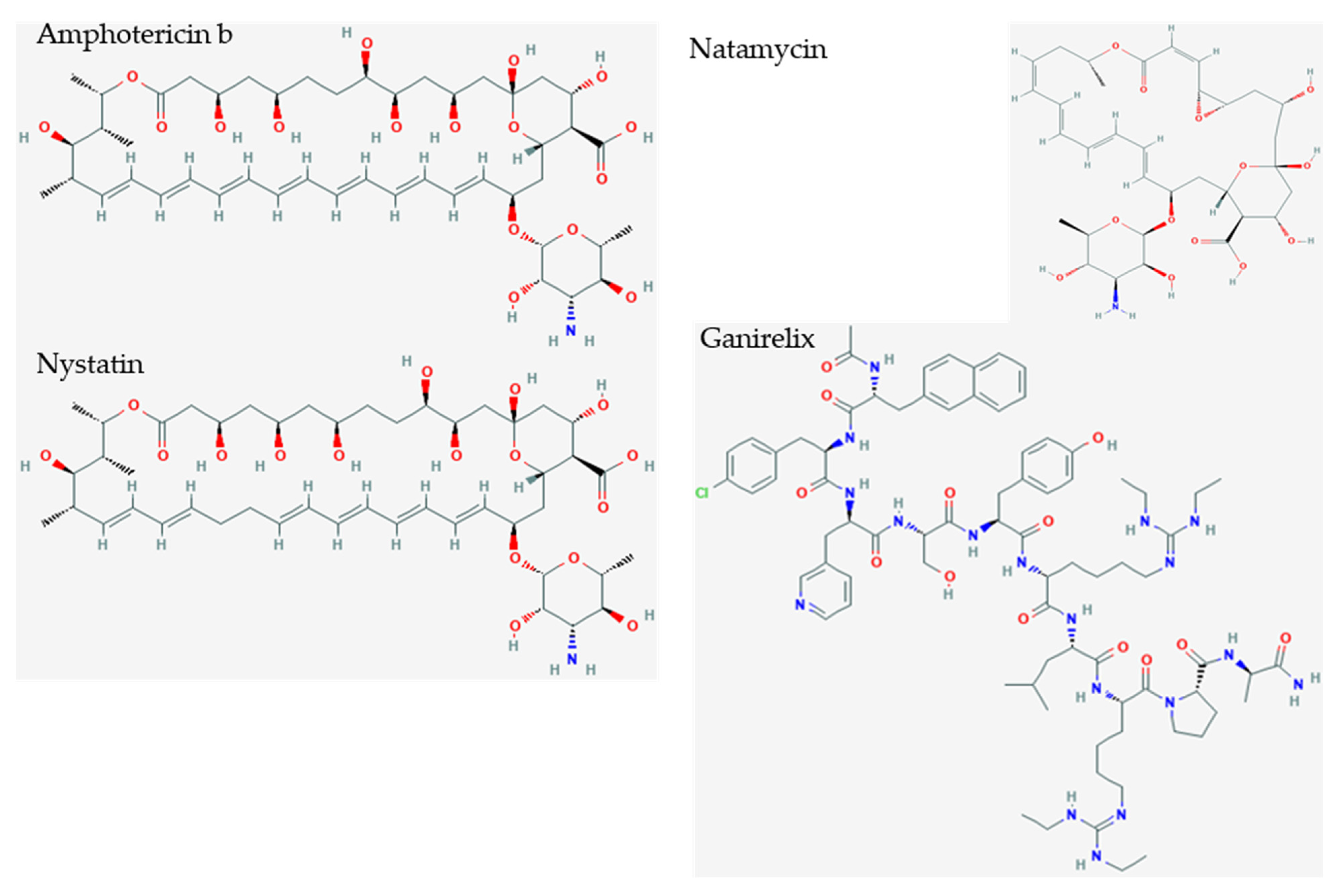
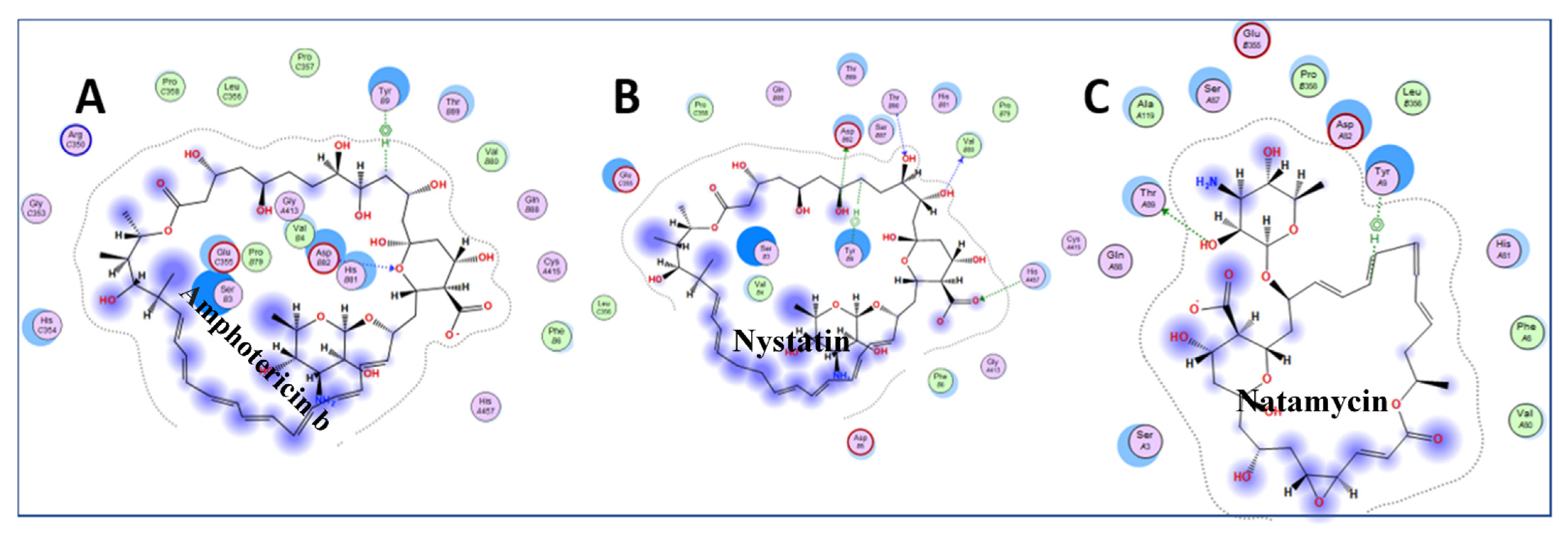
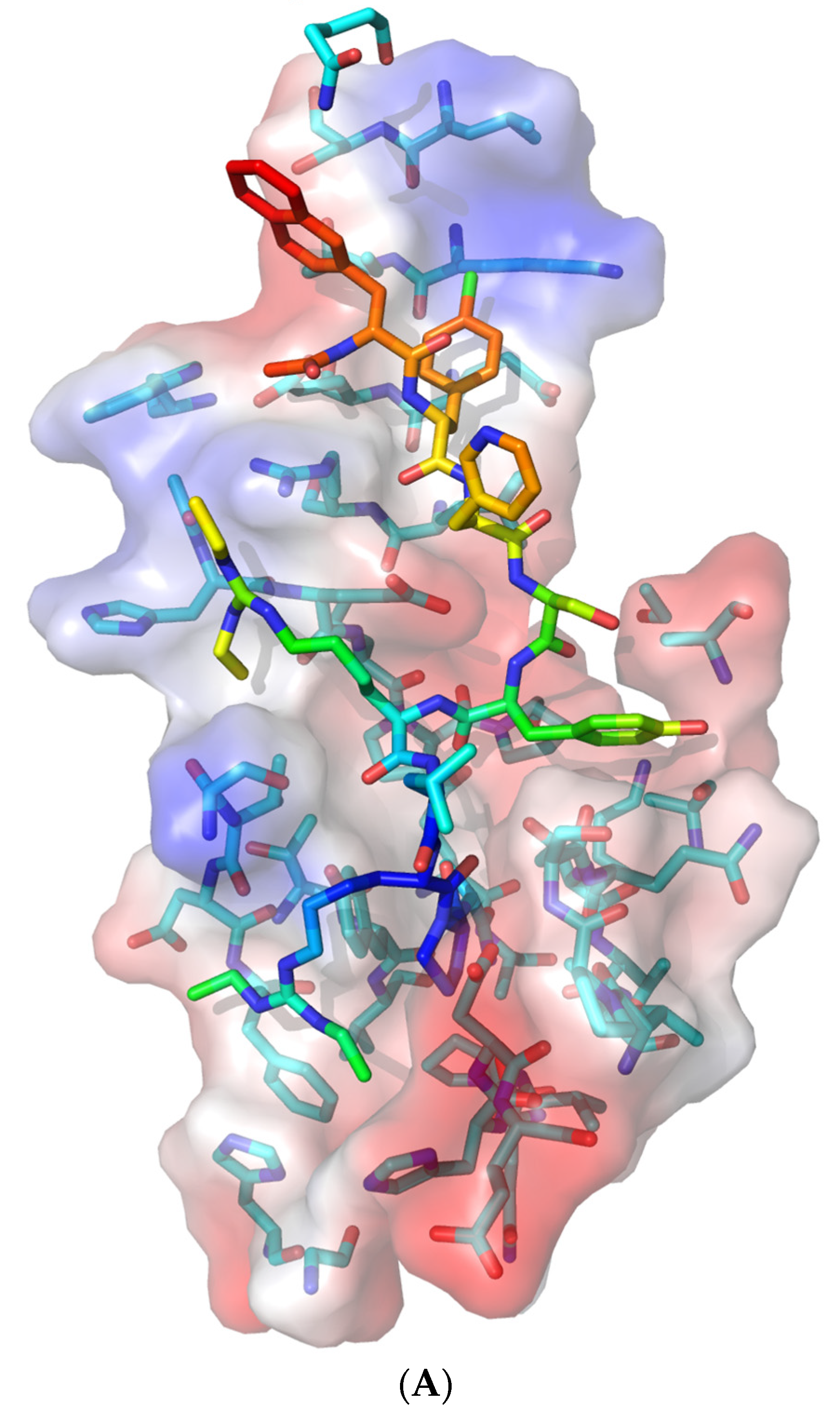
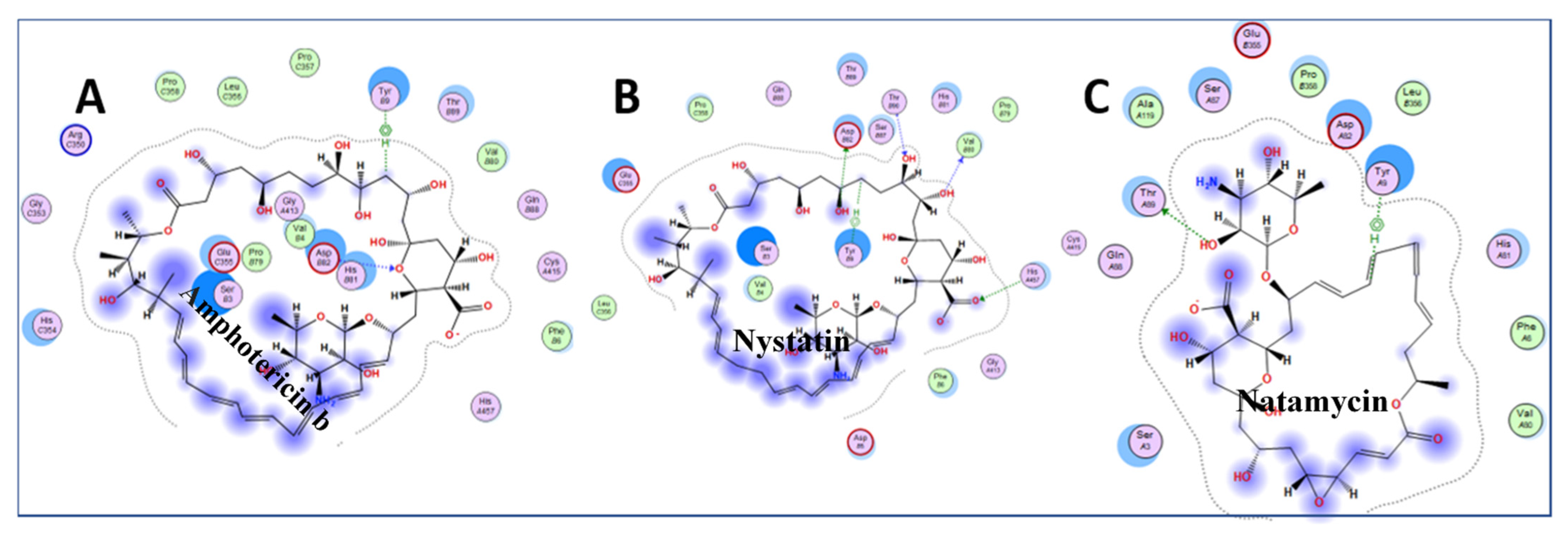
3.3. Molecular Docking of Drugs to Nsp1 Binding Site on NXF1–NXT1
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rotondo, J.C.; Martini, F.; Maritati, M.; Mazziotta, C.; Di Mauro, G.; Lanzillotti, C.; Barp, N.; Gallerani, A.; Tognon, M.; Contini, C. SARS-CoV-2 Infection: New Molecular, Phylogenetic, and Pathogenetic Insights. Efficacy of Current Vaccines and the Potential Risk of Variants. Viruses 2021, 13, 1687. [Google Scholar] [CrossRef]
- Khorsand, B.; Savadi, A.; Naghibzadeh, M. SARS-CoV-2-human protein-protein interaction network. Inform. Med. Unlocked 2020, 20, 100413. [Google Scholar] [CrossRef] [PubMed]
- Shende, P.; Khanolkar, B.; Gaud, R. Drug repurposing: New strategies for addressing COVID-19 outbreak. Expert Rev. Anti-Infect. Ther. 2021, 19, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Sourimant, J.; Aggarwal, M.; Plemper, R.K. Progress and pitfalls of a year of drug repurposing screens against COVID-19. Curr. Opin. Virol. 2021, 49, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339. [Google Scholar] [CrossRef]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Nakagawa, K.; Makino, S. Mechanisms of Coronavirus Nsp1-Mediated Control of Host and Viral Gene Expression. Cells 2021, 10, 300. [Google Scholar] [CrossRef]
- Brown, A.; Baird, M.R.; Yip, M.C.; Murray, J.; Shao, S. Structures of translationally inactive mammalian ribosomes. eLife 2018, 7, 7. [Google Scholar] [CrossRef]
- Ben-Shem, A.; de Loubresse, N.G.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The Structure of the Eukaryotic Ribosome at 3.0 Å Resolution. Science 2011, 334, 1524–1529. [Google Scholar] [CrossRef] [Green Version]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Kamitani, W.; Huang, C.; Narayanan, K.; Lokugamage, K.G.; Makino, S. A two-pronged strategy to suppress host protein synthesis by SARS coronavirus Nsp1 protein. Nat. Struct. Mol. Biol. 2009, 16, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhang, G.; Yang, Y.; Li, M.; Yang, S.; Peng, G. Lysine 164 is critical for SARS-CoV-2 Nsp1 inhibition of host gene expression. J. Gen. Virol. 2021, 102, 001513. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Ishida, R.; Strilets, T.; Cole, J.; Lopez-Orozco, J.; Fayad, N.; Felix-Lopez, A.; Elaish, M.; Evseev, D.; Magor, K.E.; et al. SARS-CoV-2 Nonstructural Protein 1 Inhibits the Interferon Response by Causing Depletion of Key Host Signaling Factors. J. Virol. 2021, 95, e0026621. [Google Scholar] [CrossRef]
- Gomez, G.N.; Abrar, F.; Dodhia, M.P.; Gonzalez, F.G.; Nag, A. SARS coronavirus protein nsp1 disrupts localization of Nup93 from the nuclear pore complex. Biochem. Cell Biol. 2019, 97, 758–766. [Google Scholar] [CrossRef]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. SARS Coronavirus nsp1 Protein Induces Template-Dependent Endonucleolytic Cleavage of mRNAs: Viral mRNAs Are Resistant to nsp1-Induced RNA Cleavage. PLoS Pathog. 2011, 7, e1002433. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kamitani, W.; DeDiego, M.L.; Enjuanes, L.; Matsuura, Y. Severe Acute Respiratory Syndrome Coronavirus nsp1 Facilitates Efficient Propagation in Cells through a Specific Translational Shutoff of Host mRNA. J. Virol. 2012, 86, 11128–11137. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, F.; Snyder, G.A.; Giovanetti, M.; Angeletti, S.; Gallo, R.C.; Ciccozzi, M.; Zella, D. Emerging of a SARS-CoV-2 viral strain with a deletion in nsp1. J. Transl. Med. 2020, 18, 1–6. [Google Scholar] [CrossRef]
- Sajidah, E.; Lim, K.; Wong, R. How SARS-CoV-2 and Other Viruses Build an Invasion Route to Hijack the Host Nucleocytoplasmic Trafficking System. Cells 2021, 10, 1424. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Ren, Y. Mechanisms of nuclear mRNA export: A structural perspective. Traffic 2019, 20, 829–840. [Google Scholar] [CrossRef]
- Gales, J.P.; Kubina, J.; Geldreich, A.; Dimitrova, M. Strength in Diversity: Nuclear Export of Viral RNAs. Viruses 2020, 12, 1014. [Google Scholar] [CrossRef]
- Pühringer, T.; Hohmann, U.; Fin, L.; Pacheco-Fiallos, B.; Schellhaas, U.; Brennecke, J.; Plaschka, C. Structure of the human core transcription-export complex reveals a hub for multivalent interactions. eLife 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Volpon, L.; Culjkovic-Kraljacic, B.; Sohn, H.S.; Blanchet-Cohen, A.; Osborne, M.J.; Borden, K.L. A biochemical framework for eIF4E-dependent mRNA export and nuclear recycling of the export machinery. RNA 2017, 23, 927–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aibara, S.; Katahira, J.; Valkov, E.; Stewart, M. The principal mRNA nuclear export factor NXF1:NXT1 forms a symmetric binding platform that facilitates export of retroviral CTE-RNA. Nucleic Acids Res. 2015, 43, 1883–1893. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-G.; Huang, W.; Lee, H.; van de Leemput, J.; Kane, M.A.; Han, Z. Characterization of SARS-CoV-2 proteins reveals Orf6 pathogenicity, subcellular localization, host interactions and attenuation by Selinexor. Cell Biosci. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- De Lima Menezes, G.; da Silva, R.A. Identification of potential drugs against SARS-CoV-2 non-structural protein 1 (nsp1). J. Biomol. Struct. Dyn. 2021, 39, 5657–5667. [Google Scholar] [CrossRef]
- Sundar, S.; Thangamani, L.; Piramanayagam, S.; Rahul, C.N.; Aiswarya, N.; Sekar, K.; Natarajan, J. Screening of FDA-approved compound library identifies potential small-molecule inhibitors of SARS-CoV-2 non-structural proteins NSP1, NSP4, NSP6 and NSP13: Molecular modeling and molecular dynamics studies. J. Proteins Proteom. 2021, 12, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Alekseenko, A.; Ignatov, M.; Jones, G.; Sabitova, M.; Kozakov, D. Protein–Protein and Protein–Peptide Docking with ClusPro Server. Springer Protoc. Handb. 2020, 2165, 157–174. [Google Scholar] [CrossRef]
- Douguet, D. Data Sets Representative of the Structures and Experimental Properties of FDA-Approved Drugs. ACS Med. Chem. Lett. 2018, 9, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group Inc. Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2019. [Google Scholar]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Fribourg, S.; Braun, I.C.; Izaurralde, E.; Conti, E. Structural Basis for the Recognition of a Nucleoporin FG Repeat by the NTF2-like Domain of the TAP/p15 mRNA Nuclear Export Factor. Mol. Cell 2001, 8, 645–656. [Google Scholar] [CrossRef]
- Lapointe, C.P.; Grosely, R.; Johnson, A.G.; Wang, J.; Fernández, I.S.; Puglisi, J.D. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc. Natl. Acad. Sci. USA 2021, 118, 118. [Google Scholar] [CrossRef]
- Yan, W.; Cheng, L.; Wang, W.; Wu, C.; Yang, X.; Du, X.; Ma, L.; Qi, S.; Wei, Y.; Lu, Z.; et al. Structure of the human gonadotropin-releasing hormone receptor GnRH1R reveals an unusual ligand binding mode. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, C.A.; Manilall, A. Gonadotropin-Releasing Hormone (GnRH) Receptor Structure and GnRH Binding. Front. Endocrinol. 2017, 8, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carolus, H.; Pierson, S.; Lagrou, K.; Van Dijck, P. Amphotericin B and Other Polyenes—Discovery, Clinical Use, Mode of Action and Drug Resistance. J. Fungi 2020, 6, 321. [Google Scholar] [CrossRef]
- Kimura, H.; Kurusu, H.; Sada, M.; Kurai, D.; Murakami, K.; Kamitani, W.; Tomita, H.; Katayama, K.; Ryo, A. Molecular pharmacology of ciclesonide against SARS-CoV-2. J. Allergy Clin. Immunol. 2020, 146, 330–331. [Google Scholar] [CrossRef]
- Tummino, T.A.; Rezelj, V.V.; Fischer, B.; Fischer, A.; O’Meara, M.J.; Monel, B.; Vallet, T.; White, K.M.; Zhang, Z.; Alon, A.; et al. Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2. Science 2021, 373, 541–547. [Google Scholar] [CrossRef]
- Lüllmann, H.; Lüllmann-Rauch, R.; Wassermann, O. Lipidosis induced by amphiphilic cationic drugs. Biochem. Pharmacol. 1978, 27, 1103–1108. [Google Scholar] [CrossRef]
- Breiden, B.; Sandhoff, K. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2019, 401, 31–46. [Google Scholar] [CrossRef]
- Muehlbacher, M.; Tripal, P.; Roas, F.; Kornhuber, J. Identification of Drugs Inducing Phospholipidosis by Novel in vitro Data. ChemMedChem 2012, 7, 1925–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Agback, P.; Agback, T.; Dominguez, F.; Frolova, E.I.; Seisenbaeva, G.; Kessler, V. Site-specific recognition of SARS-CoV-2 nsp1 protein with a tailored titanium dioxide nanoparticle. bioRxiv 2021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function | Official Gene Symbol | Protein Names |
|---|---|---|
| Extracellular | ACE | Angiotensin-converting enzyme (EC 3.4.15.1) |
| ACE2 | Angiotensin-converting enzyme 2 (EC 3.4.17.23) | |
| CRP | C-reactive protein | |
| DPP4 | Dipeptidyl peptidase 4 (EC 3.4.14.5) | |
| F3 | Coagulation factor III | |
| SERPINE1 | Plasminogen activator inhibitor 1 (PAI) | |
| PKP2 | Plakophilin-2 | |
| Immune | TNF | Tumor necrosis factor (Cachectin) |
| IL6 | Interleukin-6 | |
| IFNG | Interferon gamma | |
| CD4 | T-cell surface glycoprotein CD4 | |
| Nuclear transport | NTF2 | Nuclear transport factor 2 |
| NXF1 | Nuclear RNA export factor 1 | |
| NXT1 | NTF2-related export protein 1 | |
| TPR | Nucleoprotein TPR | |
| XPO1 | Exportin-1 | |
| RANBP1 | Ran-specific GTPase-activating protein | |
| RANBP2 | RAN binding protein 2 (Nup358 Nuclear pore complex protein) | |
| Nuclear | POLA2 | DNA polymerase alpha subunit B |
| PRIM1 | DNA primase small subunit (DNA primase 49 kDa subunit) | |
| PRIM2 | DNA primase large subunit (DNA primase 58 kDa subunit) | |
| DDX39B | Spliceosome RNA helicase DEAD box protein | |
| SARS1 | Serine--tRNA ligase, cytoplasmic | |
| Apolipoprotein | APOE | Apolipoprotein E |
| APOL1 | Apolipoprotein L1 | |
| Metabolism | INS | Insulin |
| G6PD | Glucose-6-phosphate 1-dehydrogenase | |
| COLGALT1 | Procollagen galactosyltransferase 1 (EC 2.4.1.50) | |
| LGALS3 | Galectin-3 (Gal-3) (35 kDa lectin) | |
| SLC17A5 | Sialin (H(+)/nitrate cotransporter) | |
| MB | Myoglobin | |
| AQP4 | Aquaporin-4 |
| Drug | Chemical Type | Target | Outcome | S |
|---|---|---|---|---|
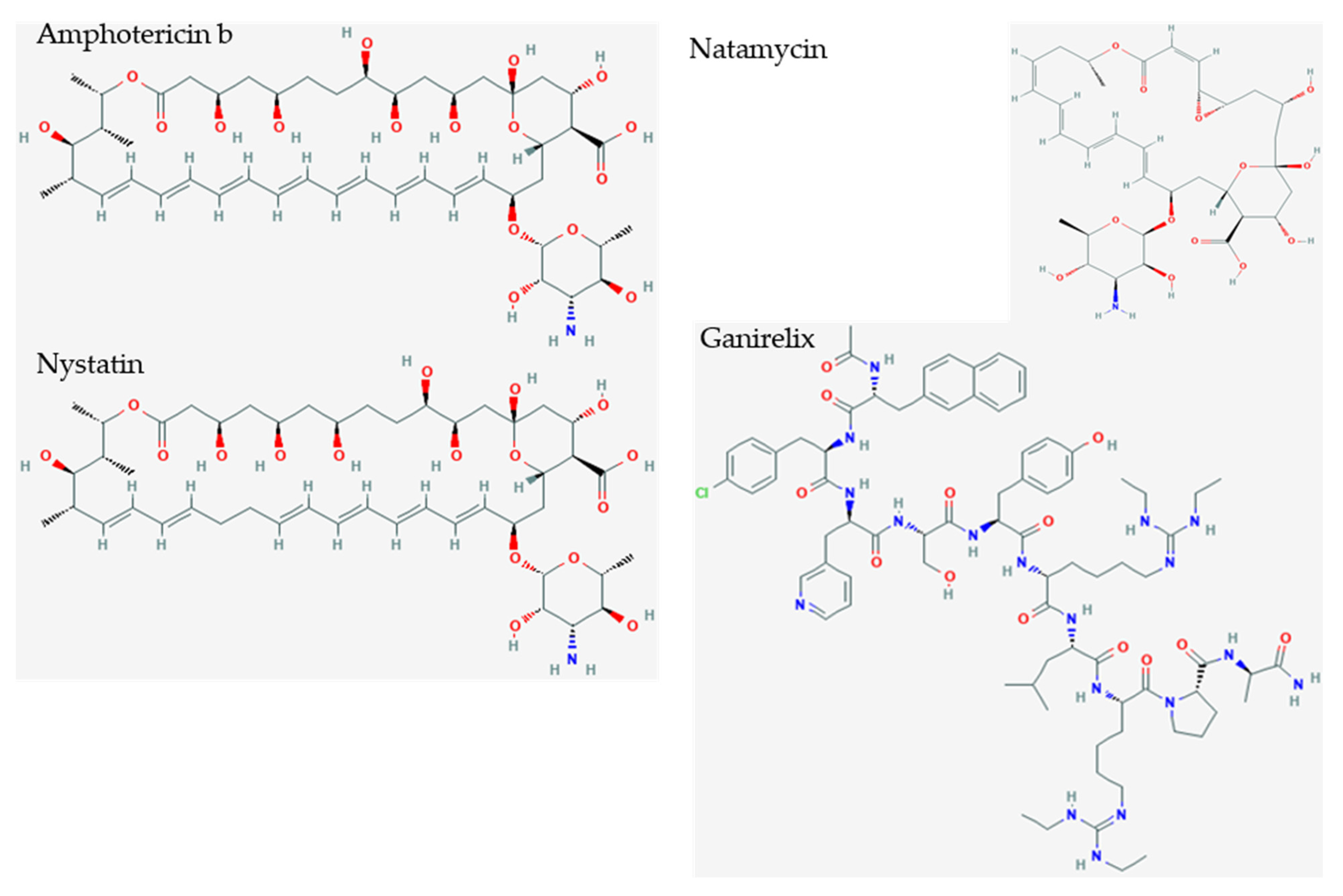
| Ganirelix | decapeptide | GnRH antagonist | competitive gonadotropin-releasing hormone antagonist reduces estrogen and testosterone | −14.49 |
| Triptorelin | decapeptide | luteinizing hormone releasing hormone (LHRH) agonist | reversibly represses gonadotropin-releasing hormone secretion to decrease LH and FSH, and estrogen and testosterone | −13.58 |
| Colistimethate | methanesulfonate derivatives of cyclic polypeptides colistin A and B | surfactant that disrupts bacterial cell membrane | broad-spectrum polymyxin antibiotic against most aerobic Gram-negative bacteria | −13.38 |
| Fondaparinux | synthetic glucopyranoside | activates antithrombin III | neutralizes activated factor X (Factor Xa), and prevents thrombin formation | −13.26 |
| Cetrorelix | decapeptide | gonadotrophin-releasing hormone (GnRH) antagonist | blocking GnRH prevents LH secretion, and reduces estrogen and testosterone | −13.23 |
| Leuprolide | nonapeptide | gonadotropin-releasing hormone (GnRH) receptor agonist | inhibits pituitary FSH and LH secretion, causing decline of testosterone and estradiol | −13.04 |
| Icatibant | decapeptide | bradykinin B2 receptor (B2R) antagonist | prevents B2R-mediated vasodilation, vascular permeability, swelling, inflammation, and pain | −12.76 |
| Afamelanotide | peptide | alpha melanocyte-stimulating hormone (α-MSH) analogue | erythropoietic protoporphyria | −12.70 |
| Nafarelin | peptide | gonadotropin-releasing hormone agonist | −12.50 | |
| Degarelix | peptide | gonadotrophin-releasing hormone (GNRH) antagonist | −12.06 | |
| Sincalide | C-terminal octapeptide of CCK, CCK8 | cholecystokinin agonist | induces gallbladder smooth muscle contraction for bile and pancreatic enzyme secretion | −12.04 |
| Drug | Target | Outcome | S |
|---|---|---|---|
| Amphotericin b | ergosterol binding | membrane pore formation | −10.10 |
| Nystatin | ergosterol binding | membrane pore formation | −9.46 |
| Rifaximin | beta-subunit of the bacterial DNA-dependent RNA polymerase | antibiotic | −8.75 |
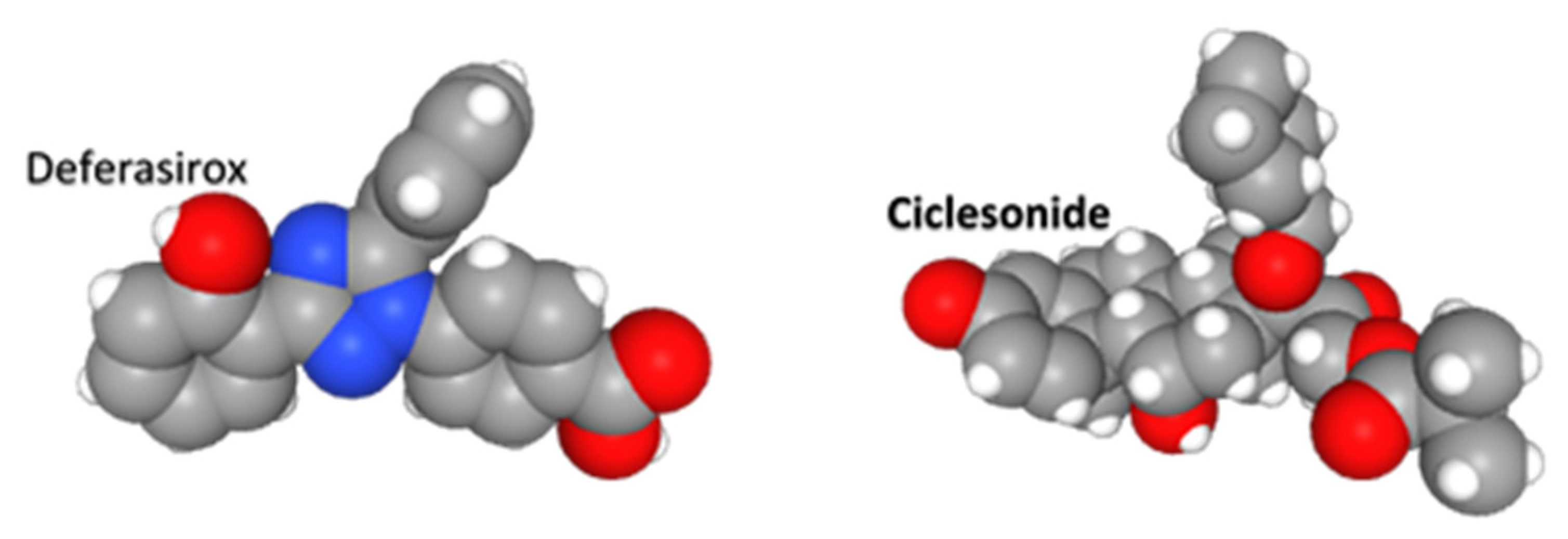
| Deferasirox | binds trivalent (ferric) iron | iron chelation | −8.26 |
| Idarubicin | DNA binding | DNA breaks | −7.77 |
| Natamycin | ergosterol binding | membrane pore formation | −7.74 |
| Loratadine | histamine H1R antagonist | “inverse agonist” for H1R antagonism | −7.73 |
| Ciclesonide | glucocorticoid receptor agonist | inhaled asthma drug | −7.68 |
| Clozapine | 5-HT2A, 5-HT2C, D1-D4 dopamine receptors | D4R antagonist | −7.64 |
| Praziquantel | Schistosome calcium ion channels | anthelmintic | −7.62 |
| Azelastine | histamine H1R antagonist | antihistamine | −7.58 |
| Ixabepilone | microtubules | cell cycle specific antimicrotubule agent | −7.54 |
| Minocycline | aminoacyl-tRNA binding to bacterial 30S ribosome | inhibit protein synthesis | −7.48 |
| Tadalafil | inhibits phosphodiesterase type 5 (PDE5) | increases cGMP to enhance erectile function | −7.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasudevan, S.; Baraniuk, J.N. Understanding COVID-19 Pathogenesis: A Drug-Repurposing Effort to Disrupt Nsp-1 Binding to Export Machinery Receptor Complex. Pathogens 2021, 10, 1634. https://doi.org/10.3390/pathogens10121634
Vasudevan S, Baraniuk JN. Understanding COVID-19 Pathogenesis: A Drug-Repurposing Effort to Disrupt Nsp-1 Binding to Export Machinery Receptor Complex. Pathogens. 2021; 10(12):1634. https://doi.org/10.3390/pathogens10121634
Chicago/Turabian StyleVasudevan, Sona, and James N. Baraniuk. 2021. "Understanding COVID-19 Pathogenesis: A Drug-Repurposing Effort to Disrupt Nsp-1 Binding to Export Machinery Receptor Complex" Pathogens 10, no. 12: 1634. https://doi.org/10.3390/pathogens10121634
APA StyleVasudevan, S., & Baraniuk, J. N. (2021). Understanding COVID-19 Pathogenesis: A Drug-Repurposing Effort to Disrupt Nsp-1 Binding to Export Machinery Receptor Complex. Pathogens, 10(12), 1634. https://doi.org/10.3390/pathogens10121634