
Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
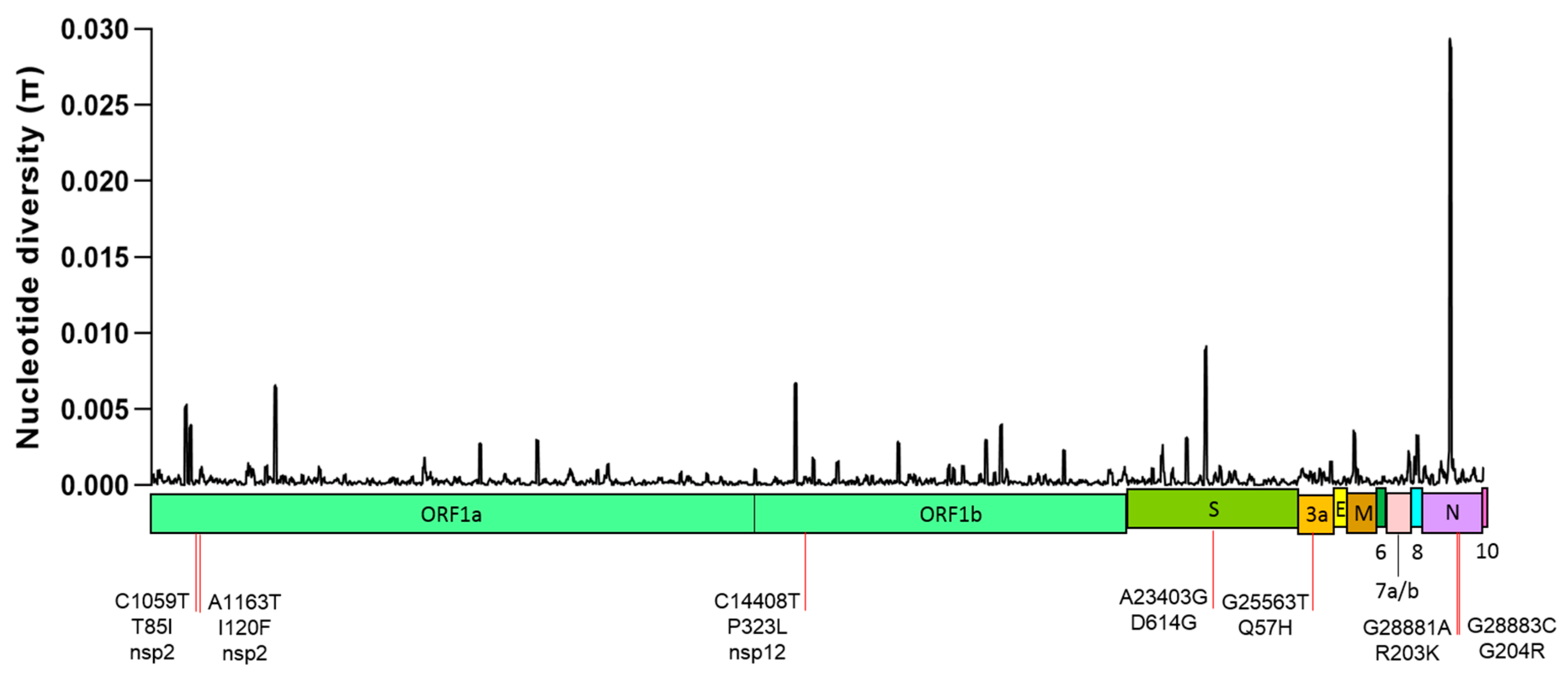
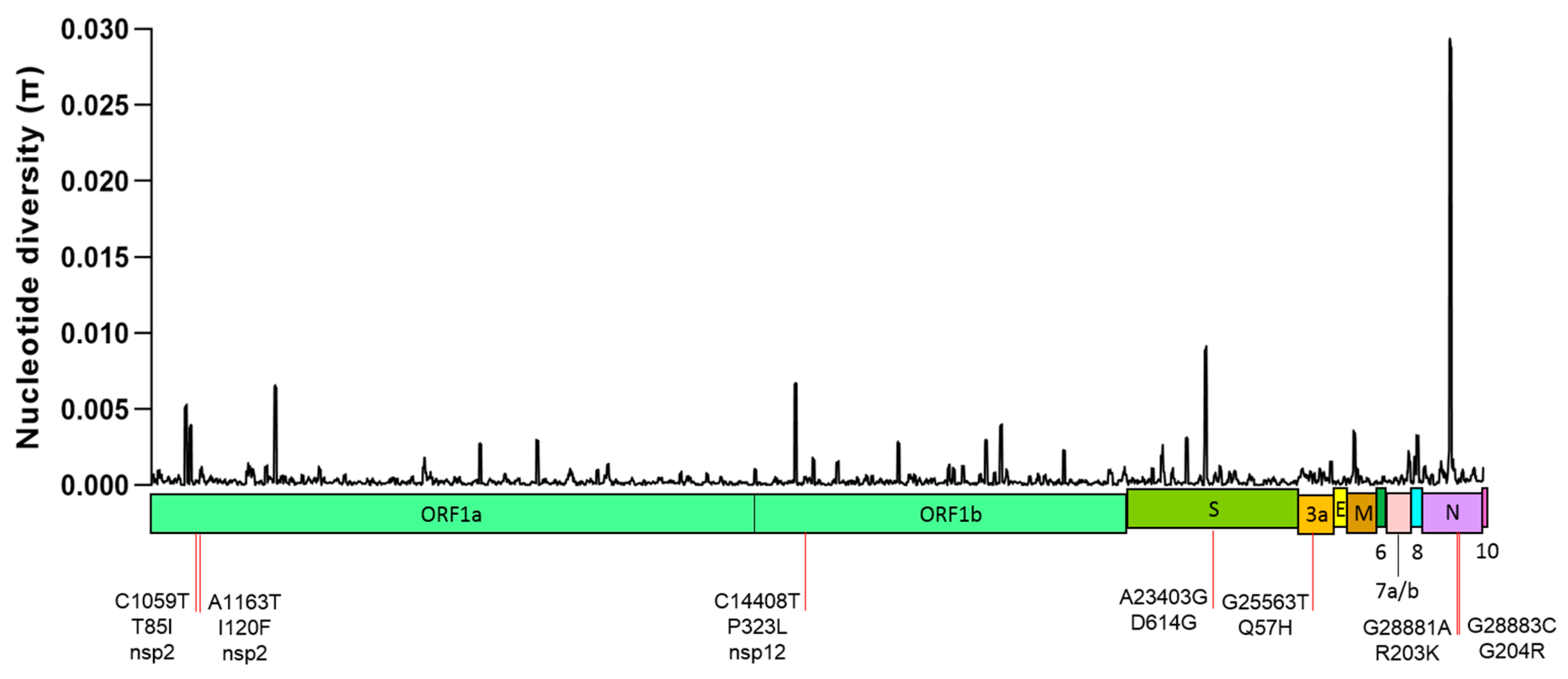
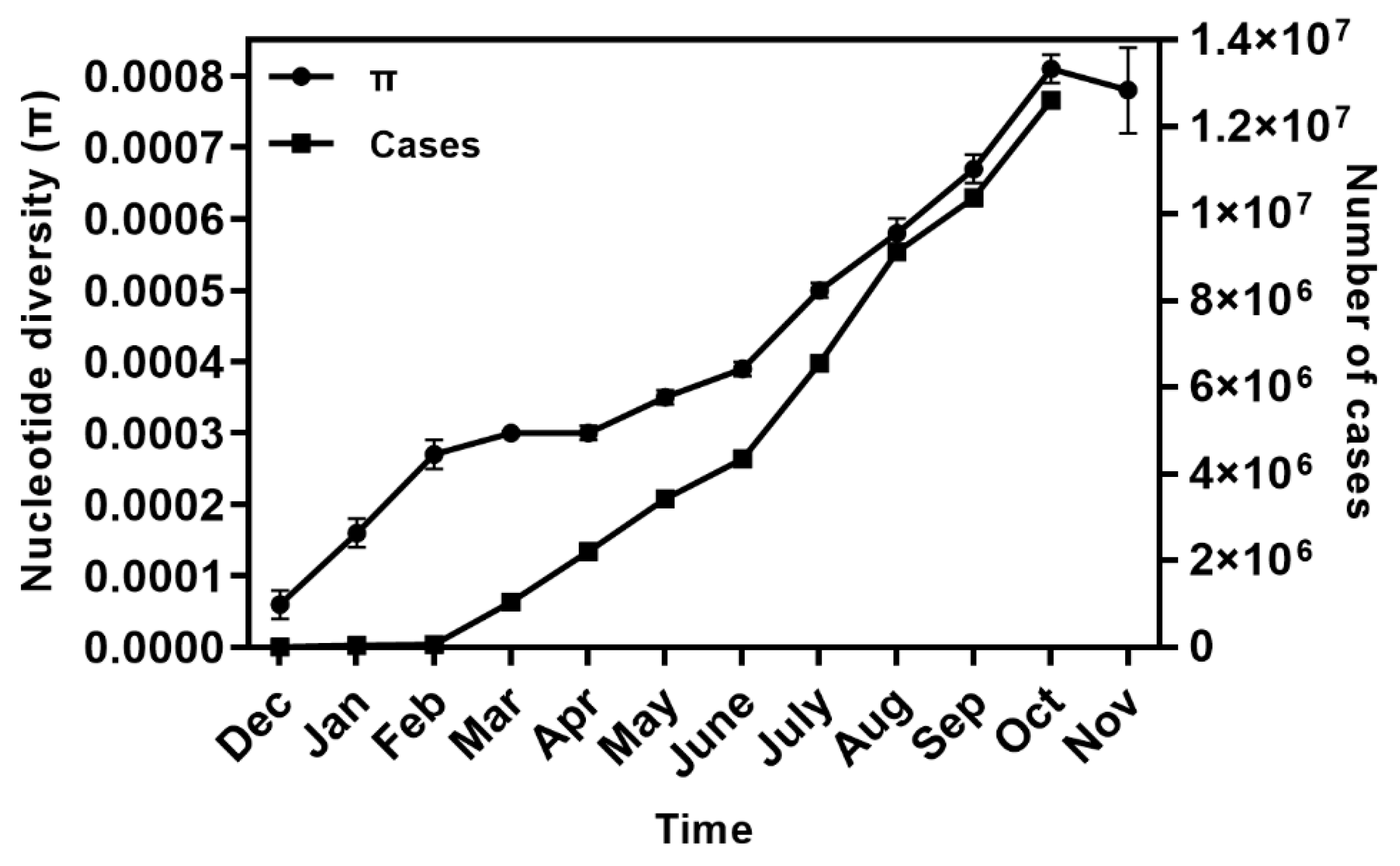
2.1. Global Genetic Diversity of SARS-CoV-2
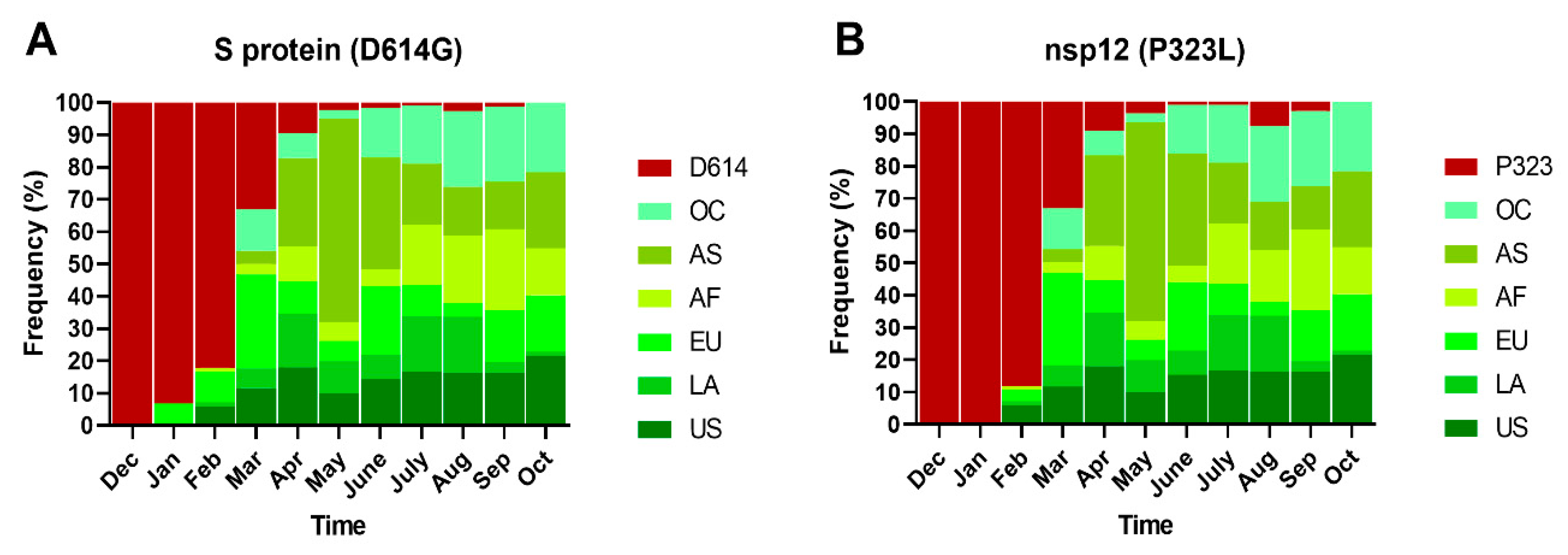
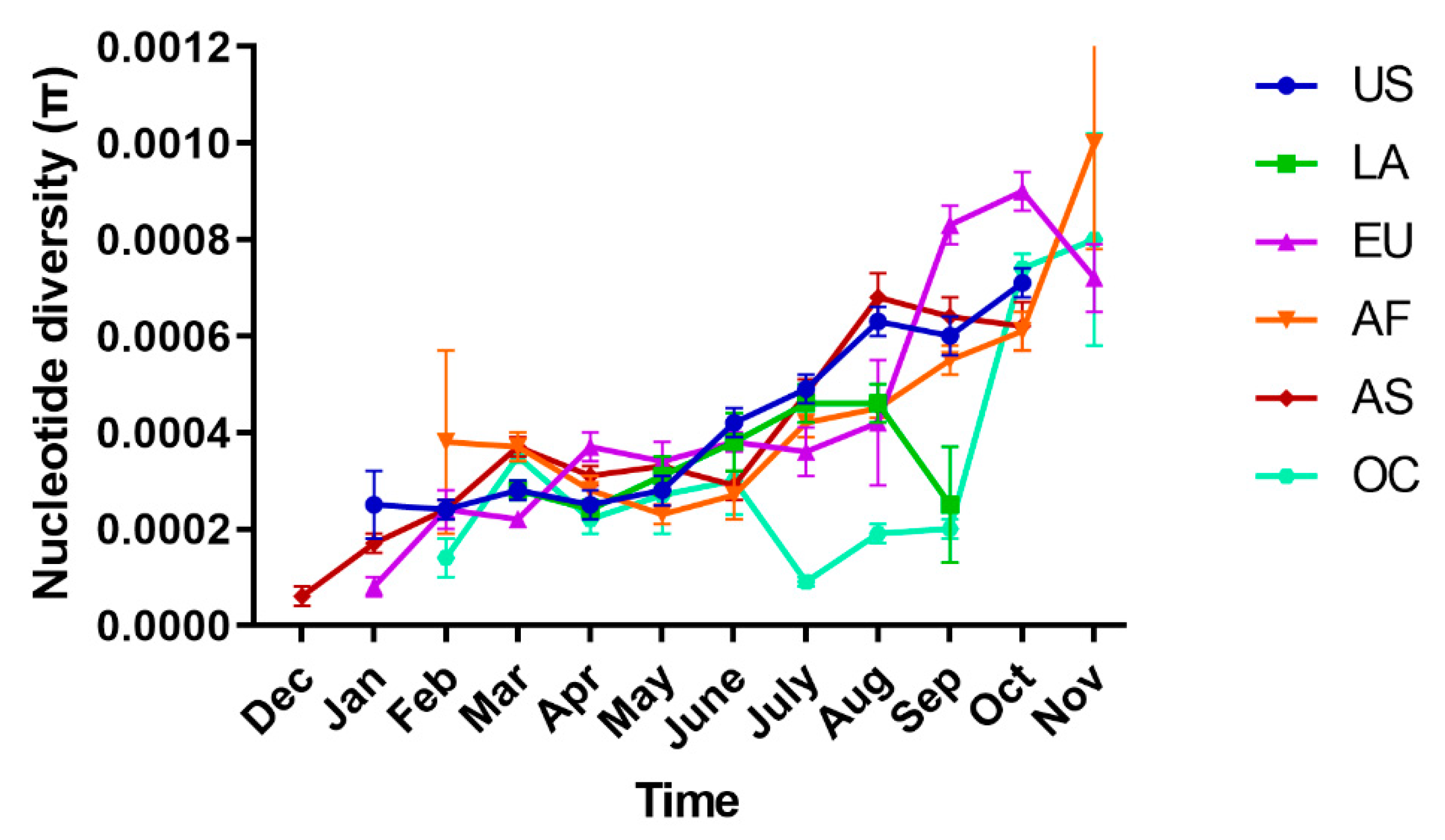
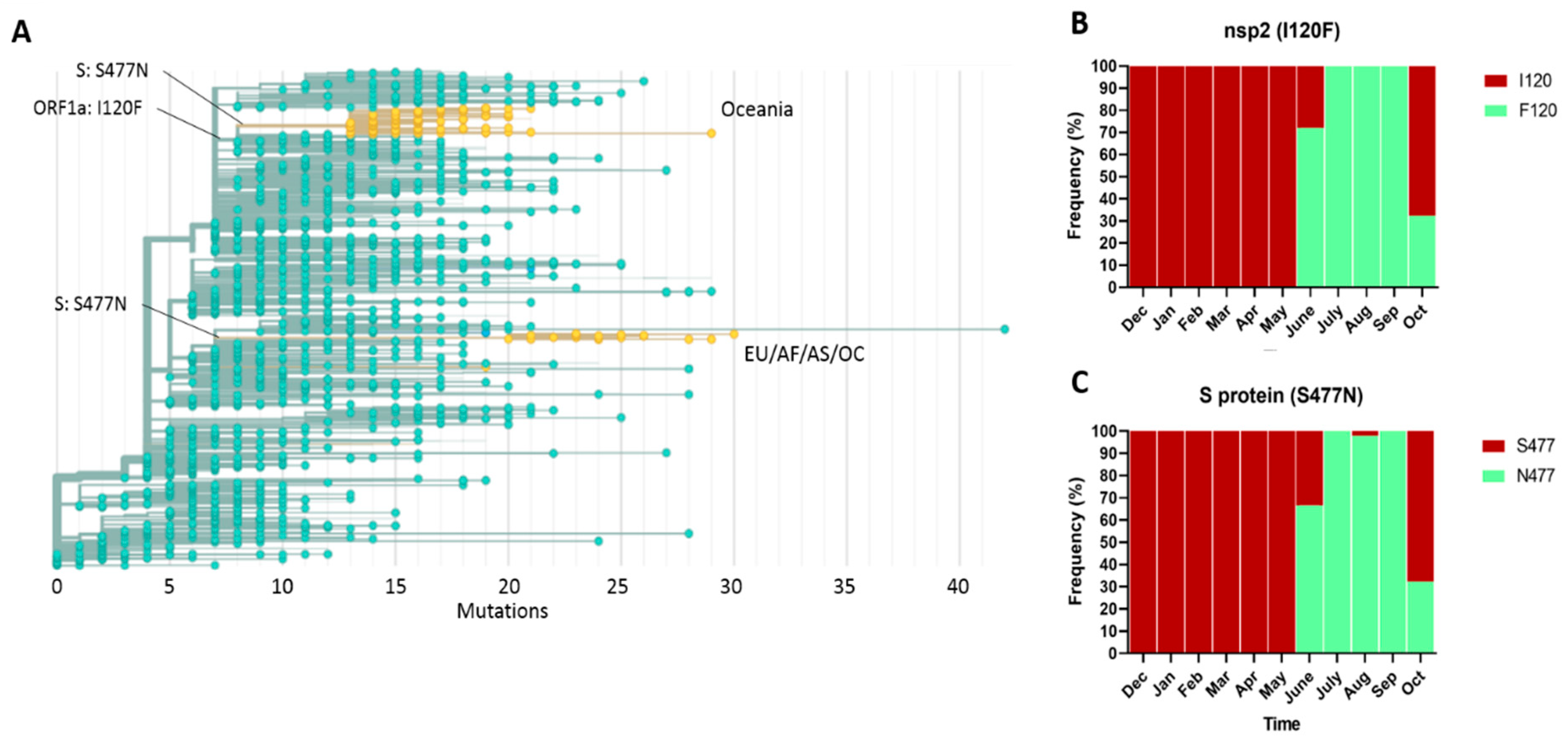
2.2. Spatial–Temporal Genetic Diversity of SARS-CoV-2
2.3. Non-Synonymous Substitutions and Natural Selection
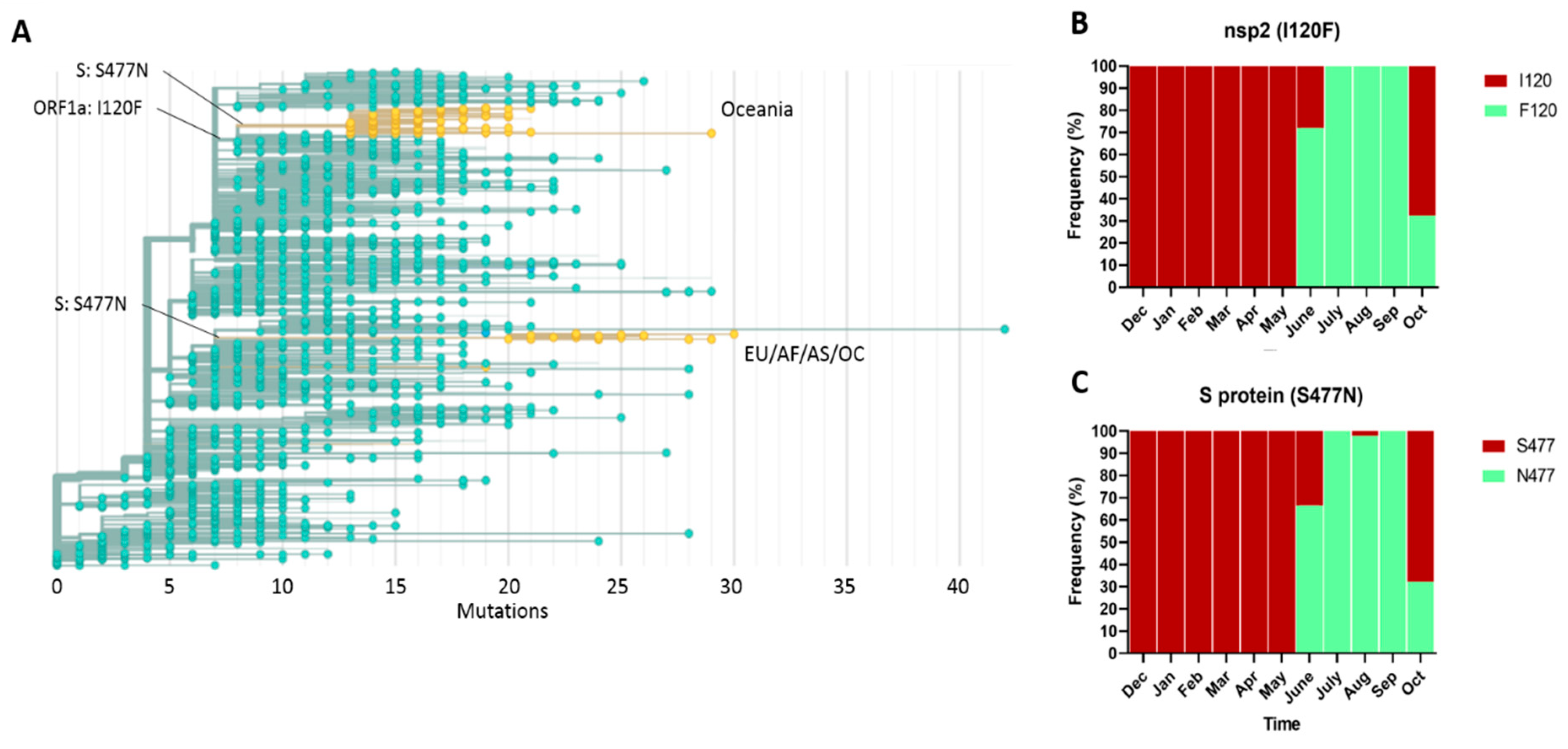
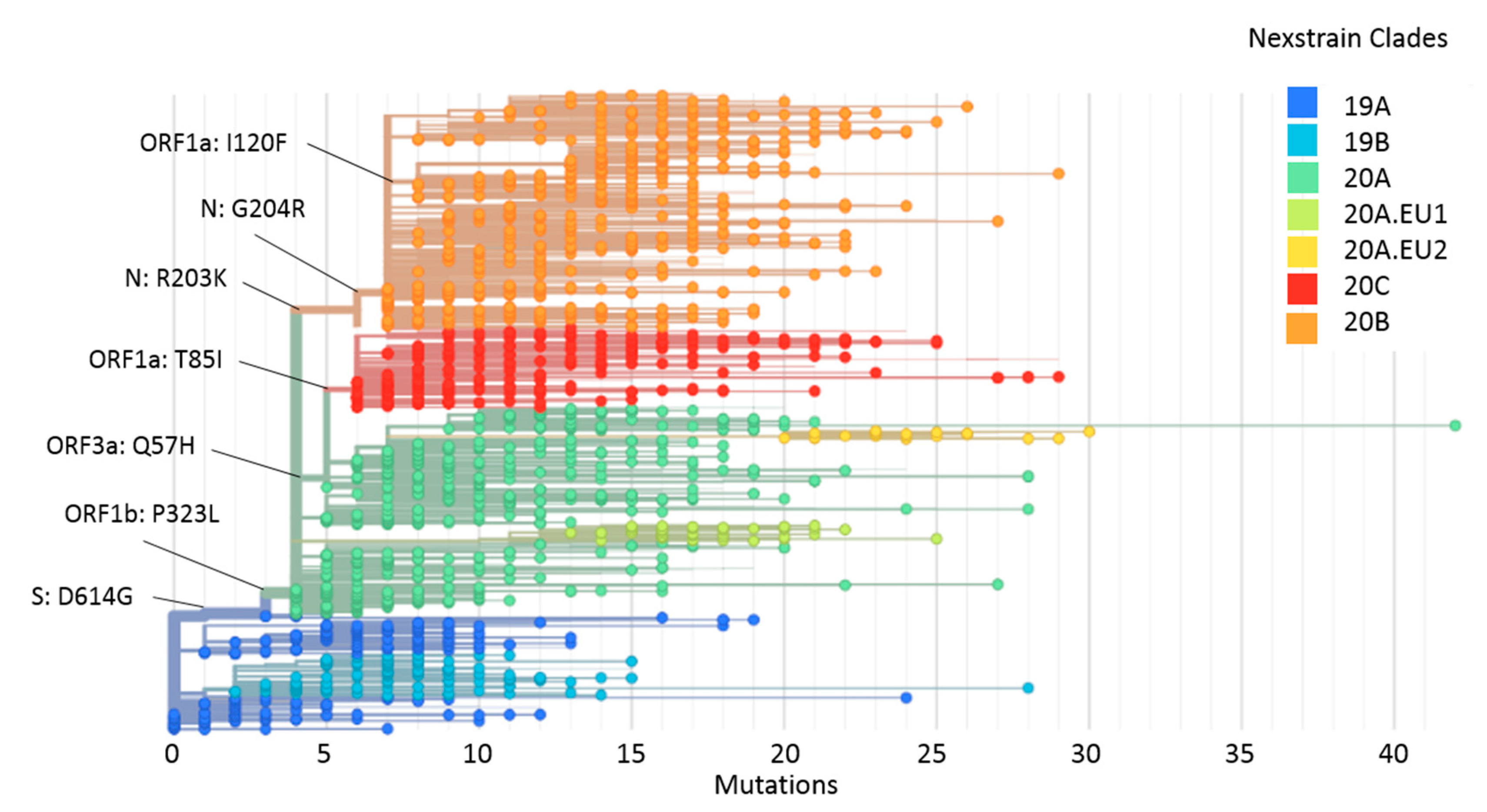
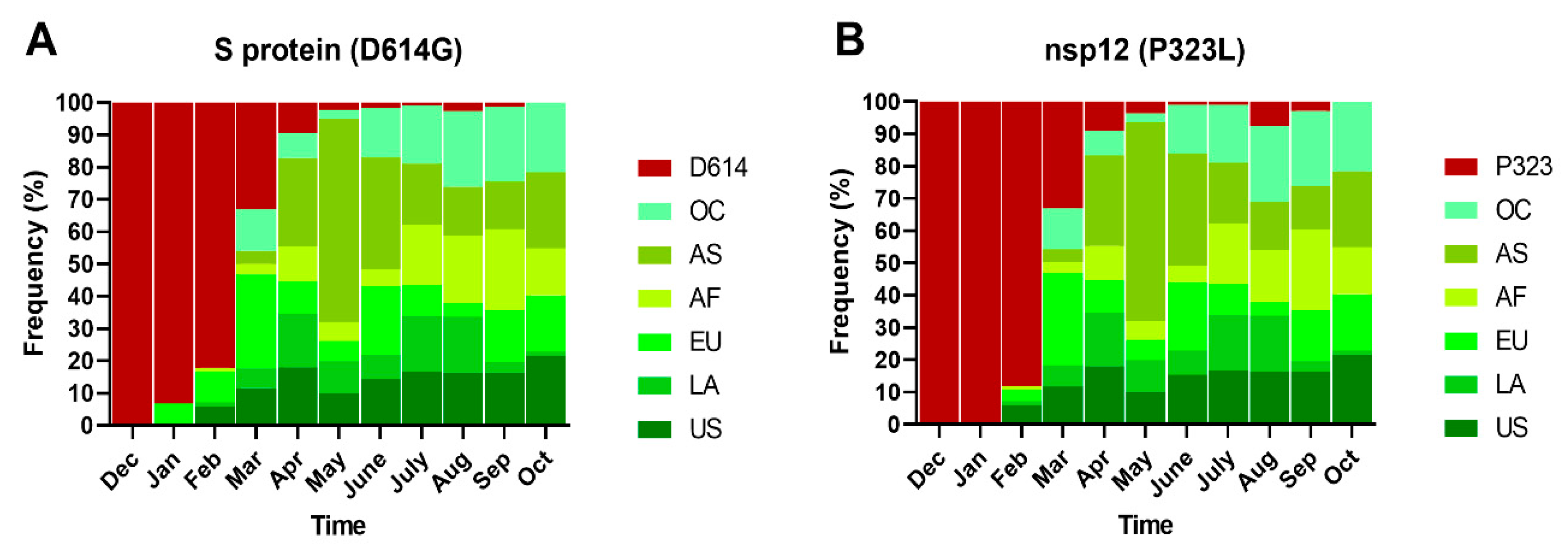
2.4. Phylogeny and Dynamics of the Highly Frequent Global dN Substitutions
2.5. Emergence and Transmission of New Variants of SARS-CoV-2
2.6. Association between Amino acid Variation and Disease Severity
3. Discussion
4. Materials and Methods
4.1. Sequences, Alignments and Quality Control
4.2. Genetic Analyses
4.3. Phylogenetic Analysis
4.4. Clinical Classification and Genetic–Phenotype Association Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2020. Available online: https://covid19.who.int/ (accessed on 29 September 2020).
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Von Brunn, A.; Teepe, C.; Simpson, J.C.; Pepperkok, R.; Friedel, C.C.; Zimmer, R.; Roberts, R.; Baric, R.; Haas, J. Analysis of Intraviral Protein-Protein Interactions of the SARS Coronavirus ORFeome. PLoS ONE 2007, 2, e459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ogando, N.S.; Ferron, F.; Decroly, E.; Canard, B.; Posthuma, C.C.; Snijder, E.J. The Curious Case of the Nidovirus Exoribonuclease: Its Role in RNA Synthesis and Replication Fidelity. Front. Microbiol. 2019, 10, 1813. [Google Scholar] [CrossRef]
- Bedford, T.; Riley, S.; Barr, I.G.; Broor, S.; Chadha, M.; Cox, N.J.; Daniels, R.S.; Gunasekaran, C.P.; Hurt, A.C.; Kelso, A.; et al. Global circulation patterns of seasonal influenza viruses vary with antigenic drift Europe PMC Funders Group. Nature 2015, 523, 217–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, E.C.; Dudas, G.; Rambaut, A.; Andersen, K.G. The evolution of Ebola virus: Insights from the 2013–2016 epidemic. Nature 2016, 538, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhao, S.; Shi, C.-M.; Song, S.; Zhu, S.; Su, Y.; Zhao, W.; Li, M.; Bao, Y.; Xue, Y.; et al. Population Genetics of SARS-CoV-2: Disentangling Effects of Sampling Bias and Infection Clusters. Genom. Proteom. Bioinform. 2020, 4–11. [Google Scholar] [CrossRef]
- Vitti, J.J.; Grossman, S.R.; Sabeti, P.C. Detecting Natural Selection in Genomic Data. Annu. Rev. Genet. 2013, 47, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Nextclade. Available online: https://clades.nextstrain.org/ (accessed on 16 December 2020).
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Day, T.; Gandon, S.; Lion, S.; Otto, S.P. On the evolutionary epidemiology of SARS-CoV-2. Curr. Biol. 2020, 30, R849–R857. [Google Scholar] [CrossRef]
- Ramírez, J.D.; Florez, C.; Muñoz, M.; Hernández, C.; Castillo, A.; Gomez, S.; Rico, A.; Pardo, L.; Barros, E.C.; Castañeda, S.; et al. The arrival and spread of SARS-CoV-2 in Colombia. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Lakdawala, S.; Cooper, V. SARS-CoV-2 genome evolution exposes early human adaptations. bioRxiv 2020. [Google Scholar] [CrossRef]
- Van Dorp, L.; Acman, M.; Richard, D.; Shaw, L.P.; Ford, C.E.; Ormond, L.; Owen, C.J.; Pang, J.; Tan, C.C.; Boshier, F.A.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020, 83, 104351. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. The effect of linkage on directional selection. Genetics 1965, 52, 349–363. [Google Scholar] [CrossRef]
- Karlin, S.; Feldman, M.W. Linkage and selection: Two locus symmetric viability model. Theor. Popul. Biol. 1970, 1, 39–71. [Google Scholar] [CrossRef]
- Bedford, T.; Greninger, A.L.; Roychoudhury, P.; Starita, L.M.; Famulare, M.; Huang, M.-L.; Nalla, A.; Pepper, G.; Reinhardt, A.; Xie, H.; et al. Cryptic transmission of SARS-CoV-2 in Washington state. Science 2020, 370, 571–575. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Crawford, K.H.D.; Bloom, J.D.; Stadler, T.; Neher, R.A. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020 SeqCOVID-SPAIN consortium, 14. medRxiv 2020. [Google Scholar] [CrossRef]
- GISAID–Initiative. Available online: https://www.gisaid.org/ (accessed on 30 November 2020).
- Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations—SARS-CoV-2 Coronavirus/nCoV-2019 Genomic Epidemiology—Virological. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 23 December 2020).
- Wise, J. Covid-19: New coronavirus variant is identified in UK. BMJ 2020, 371, m4857. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Hopkins, S.; Gandy, A.; Rambaut, A.; Ferguson, N.M. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2021. [Google Scholar] [CrossRef]
- Davies, N.G.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Abbott, S.; Gimma, A.; et al. Preliminary-not peer reviewed Estimat-ed transmissibility and severity of novel SARS-CoV-2 Variant of Concern 202012/01 in England. medRxiv 2020. [Google Scholar] [CrossRef]
- Gu, H.; Chen, Q.; Yang, G.; He, L.; Fan, H.; Deng, Y.-Q.; Wang, Y.; Teng, Y.; Zhao, Z.; Cui, Y.; et al. Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science 2020, 369, 1603–1607. [Google Scholar] [CrossRef]
- What Do We Know about the Two New Covid-19 Variants in the UK?|World News|The Guardian. Available online: https://www.theguardian.com/world/2020/dec/23/what-do-we-know-about-the-two-new-covid-19-variants-in-the-uk (accessed on 23 December 2020).
- Puenpa, J.; Suwannakarn, K.; Chansaenroj, J.; Nilyanimit, P.; Yorsaeng, R.; Auphimai, C.; Kitphati, R.; Mungaomklang, A.; Kongklieng, A.; Chirathaworn, C.; et al. Molecular epidemiology of the first wave of severe acute respiratory syndrome coronavirus 2 infection in Thailand in 2020. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Laiton-Donato, K.; Villabona Arenas, C.J.; Usme Ciro, J.; Franco Munoz, C.; Alvarez-Diaz, D.A.; Villabona-Arenas, L.; Echeverria-Londono, S.; Franco-Sierra, N.; Cucunuba, Z.; Florez-Sanchez, A.C.; et al. Genomic epidemiology of SARS-CoV-2 in Colombia. medRxiv 2020. [Google Scholar] [CrossRef]
- Lemieux, J.E.; Siddle, K.J.; Shaw, B.M.; Loreth, C.; Schaffner, S.F.; Gladden-Young, A.; Adams, G.; Fink, T.; Tomkins-Tinch, C.H.; Krasilnikova, L.A.; et al. Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science 2021, 371, eabe3261. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.M.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular Tracing of SARS-CoV-2 in Italy in the First Three Months of the Epidemic. Viruses 2020, 12, 798. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.W. Characterizing SARS-CoV-2 mutations in the united states. arXiv 2020. [Google Scholar] [CrossRef]
- Chand, G.B.; Banerjee, A.; Azad, G.K. Identification of novel mutations in RNA-dependent RNA polymerases of SARS-CoV-2 and their implications on its protein structure. PeerJ 2020, 8, e9492. [Google Scholar] [CrossRef]
- Ruan, Z.; Liu, C.; Guo, Y.; He, Z.; Huang, X.; Jia, X.; Yang, T. SARS-CoV-2 and SARS-CoV: Virtual screening of potential inhibitors targeting RNA-dependent RNA polymerase activity (NSP12). J. Med. Virol. 2020, 93, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2021, 370, 1464–1468. [Google Scholar] [CrossRef]
- Brufsky, A. Distinct viral clades of SARS-CoV-2: Implications for modeling of viral spread. J. Med. Virol. 2020, 92, 1386–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brufsky, A.; Lotze, M.T. DC/L-SIGNs of hope in the COVID-19 pandemic. J. Med. Virol. 2020, 92, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; He, C.L.; Gao, Q.; Zhang, G.J.; Cao, X.X.; Long, Q.X.; Deng, H.J.; Huang, L.Y.; Chen, J.; Wang, K.; et al. The D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Yao, H.; Lu, X.; Chen, Q.; Xu, K.; Chen, Y.; Cheng, M.; Chen, K.; Cheng, L.; Weng, T.; Shi, D.; et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Lorenzo-Redondo, R.; Nam, H.H.; Roberts, S.C.; Simons, L.M.; Jennings, L.J.; Qi, C.; Achenbach, C.J.; Hauser, A.R.; Ison, M.G.; Hultquist, J.F.; et al. A clade of SARS-CoV-2 viruses associated with lower viral loads in patient upper airways. EBioMedicine 2020, 62, 103112. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 1–6. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recogni-tion by polyclonal human serum antibodies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; Yang, Z.; et al. mRNA vaccineelicited antibodies to SARS-CoV-2 and circulating variants 2 3. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Lambson, B.E.; Vermeulen, M.; Van Den Berg, K.; Rossouw, T.; Boswell, M.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. bioRxiv 2021. [Google Scholar] [CrossRef]
- Novavax Vaccine Delivers 89% Efficacy against COVID-19 in UK—but Is Less Potent in South Africa|Science|AAAS. Available online: https://www.sciencemag.org/news/2021/01/novavax-vaccine-delivers-89-efficacy-against-covid-19-uk-less-potent-south-africa (accessed on 29 January 2021).
- Davies, J.; Almasy, K.; McDonald, E.; Plate, L. Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus non-structural proteins identifies unique and shared host-cell dependencies. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.; Yee, R.; Shen, L.; Judkins, A.R.; Bootwalla, M.; Ryutov, A.; Maglinte, D.T.; Ostrow, D.; Precit, M.; Biegel, J.A.; et al. High Prevalence of SARS-CoV-2 Genetic Variation and D614G Mutation in Pediatric Patients with COVID-19. Open Forum Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- SARS-CoV-2 Resources—NCBI. Available online: https://www.ncbi.nlm.nih.gov/sars-cov-2/ (accessed on 30 November 2020).
- Katoh, K.; Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequences alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinforma. Appl. NOTE 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Muse, S.V.; Gaut, B.S. A likelihood approach for comparing synonymous and nonsynonymous nucleotide substitution rates, with application to the chloroplast genome. Mol. Biol. Evol. 1994, 11, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nextstrain/Ncov/Global. Available online: https://nextstrain.org/ncov/global (accessed on 30 November 2020).
- RStudio|Open Source & Professional Software for Data Science Teams—RStudio. Available online: https://rstudio.com/ (accessed on 29 January 2021).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | Amino Acid Change | Genomic Location | dN/dS (p Value) | Distribution and Frequency (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| US | LA | EU | AF | AS | OC | Global | ||||
| C1059T | T85I | ORF1a (nsp2) | 5.89 (0.009) | 49.2 | 12.5 | 9.80 | 5.80 | 2.40 | 11.4 | 14.41 |
| A1163T | I120F | ORF1a (nsp2) | 4.79 (0.052) | 0.00 | 0.00 | 0.20 | 0.00 | 11.4 | 43.2 | 10.08 |
| C14408T | P323L | ORF1b (nsp12) | 7.49 (0.002) | 80.6 | 92.3 | 81.8 | 88.4 | 68.2 | 81.1 | 79.58 |
| A23403G | D614G | S gene | 2.42 (0.153) | 80.3 | 90.9 | 85.3 | 92.6 | 69.0 | 81.1 | 80.80 |
| G25563T | Q57H | ORF3 | 7.13 (0.105) | 59.8 | 18.7 | 21.1 | 18.6 | 25.9 | 16.2 | 27.47 |
| G28881A | R203K | N gene | −0.43 (0.805) | 7.90 | 41.8 | 34.2 | 48.8 | 26.9 | 54.0 | 33.44 |
| G28883C | G204R | N gene | 1.79 (0.285) | 7.90 | 41.8 | 34.0 | 48.3 | 26.7 | 54.0 | 33.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Alanis, A.; Cruz-Rangel, A.; Rodríguez-Gómez, F.; González, J.; Torres-Guerrero, C.A.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging. Pathogens 2021, 10, 184. https://doi.org/10.3390/pathogens10020184
Flores-Alanis A, Cruz-Rangel A, Rodríguez-Gómez F, González J, Torres-Guerrero CA, Delgado G, Cravioto A, Morales-Espinosa R. Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging. Pathogens. 2021; 10(2):184. https://doi.org/10.3390/pathogens10020184
Chicago/Turabian StyleFlores-Alanis, Alejandro, Armando Cruz-Rangel, Flor Rodríguez-Gómez, James González, Carlos Alberto Torres-Guerrero, Gabriela Delgado, Alejandro Cravioto, and Rosario Morales-Espinosa. 2021. "Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging" Pathogens 10, no. 2: 184. https://doi.org/10.3390/pathogens10020184
APA StyleFlores-Alanis, A., Cruz-Rangel, A., Rodríguez-Gómez, F., González, J., Torres-Guerrero, C. A., Delgado, G., Cravioto, A., & Morales-Espinosa, R. (2021). Molecular Epidemiology Surveillance of SARS-CoV-2: Mutations and Genetic Diversity One Year after Emerging. Pathogens, 10(2), 184. https://doi.org/10.3390/pathogens10020184