
The Role of Lipid Metabolism in Influenza A Virus Infection


Abstract
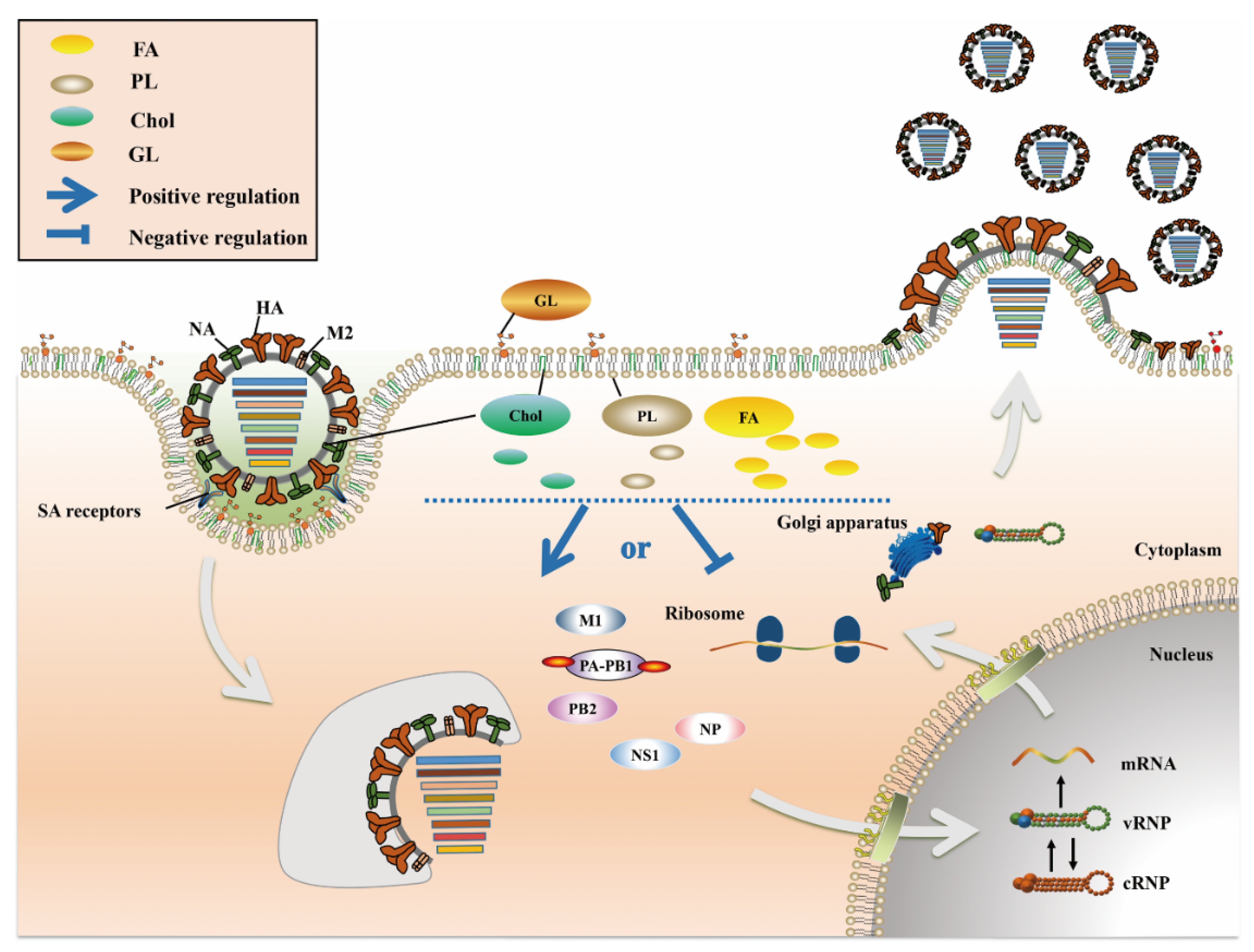
:1. Introduction
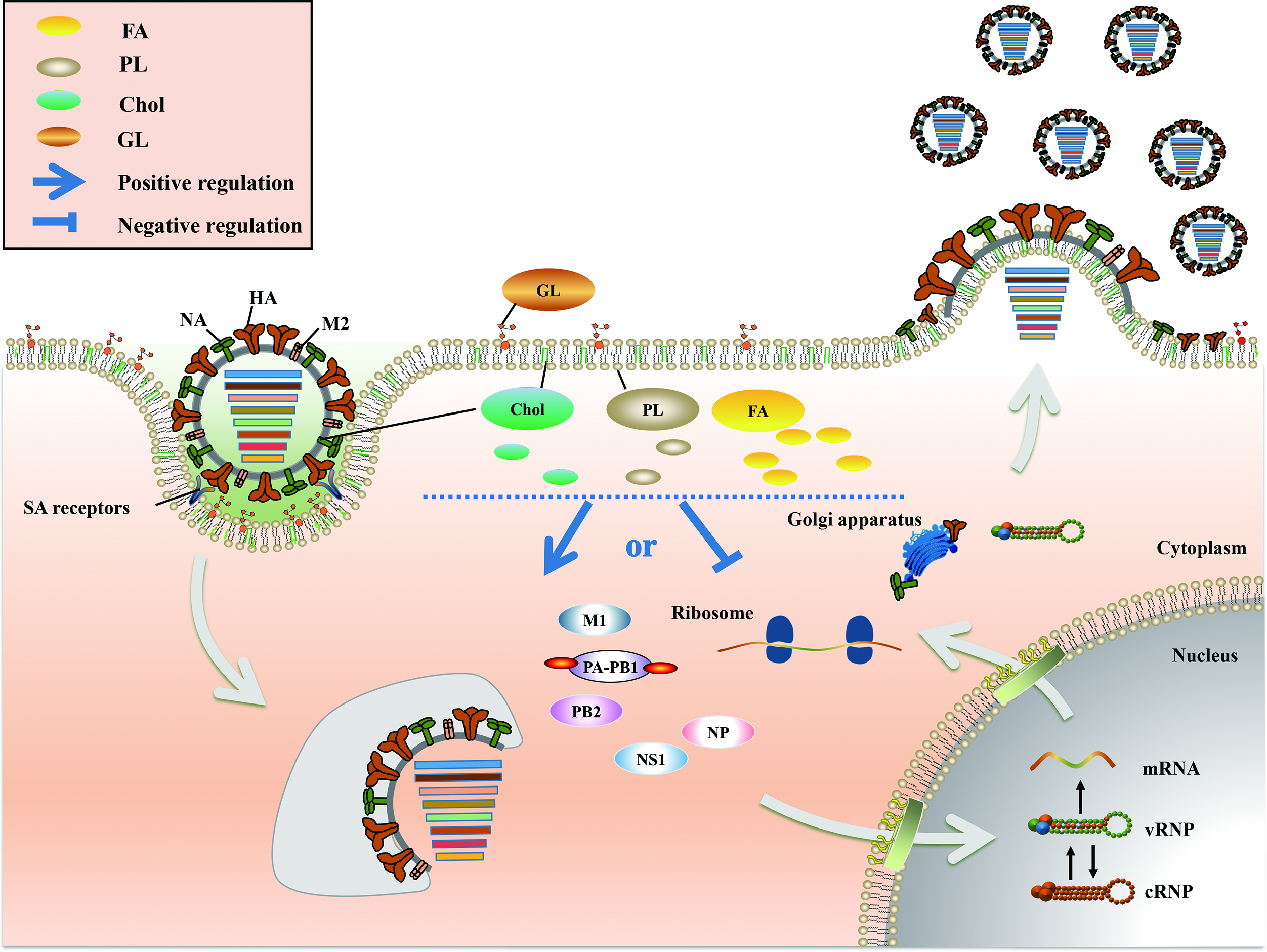
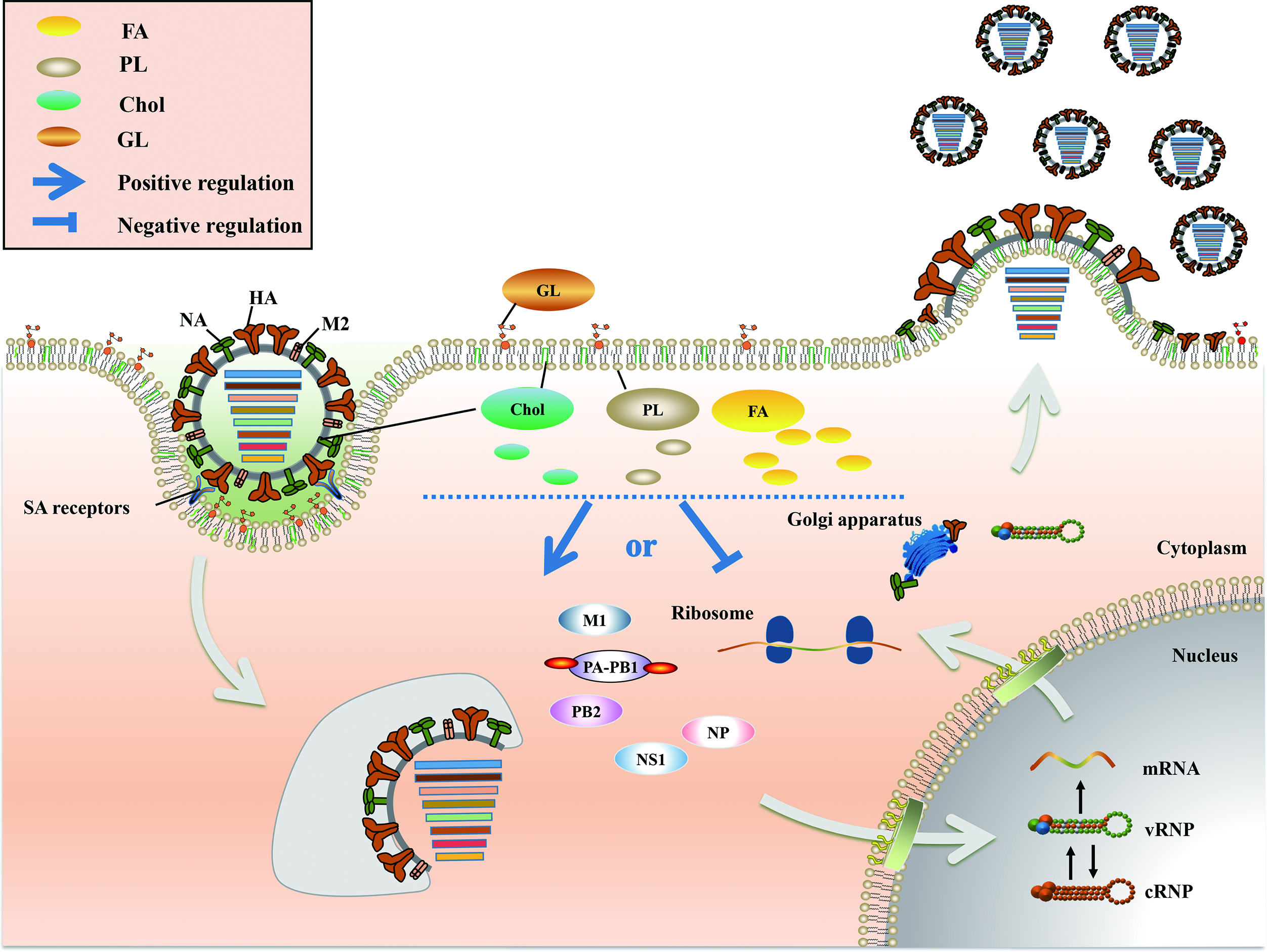
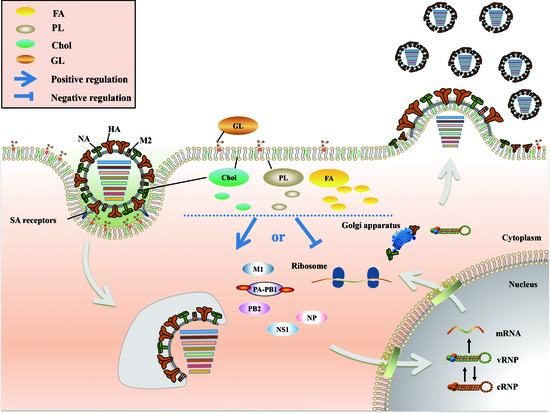
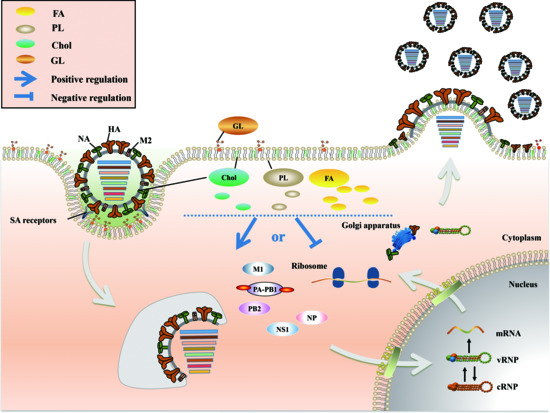
2. Positive and Negative Regulation of FA Metabolism in IAV Infection
3. Chol Plays a Major Role in Fusion and Release during the IAV Life Cycle
4. PL Metabolism Is Involved in Multiple Stages of IAV Infection
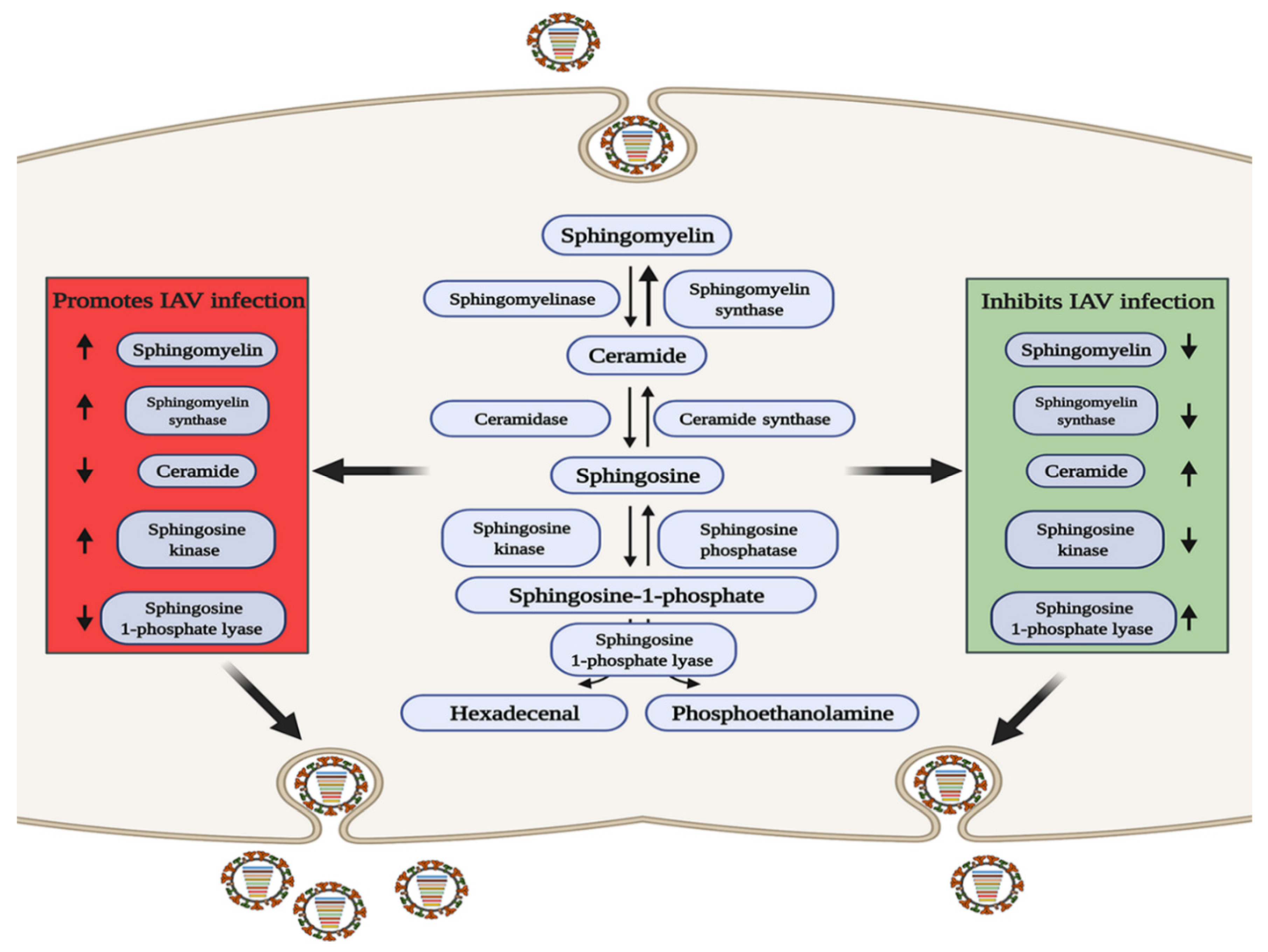
5. An Appropriate GL Concentration Is Important for IAV Infection
6. Conclusions and Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A Complicated Message: Identification of a Novel PB1-Related Protein Translated from Influenza A Virus Segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.S.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza a virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.; Wills, N.M.; Xiao, Y.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Wise, H.M.; Hutchinson, E.C.; Jagger, B.W.; Stuart, A.D.; Kang, Z.H.; Robb, N.; Schwartzman, L.M.; Kash, J.C.; Fodor, E.; Firth, A.E.; et al. Identification of a Novel Splice Variant Form of the Influenza A Virus M2 Ion Channel with an Antigenically Distinct Ectodomain. PLoS Pathog. 2012, 8, e1002998. [Google Scholar] [CrossRef] [Green Version]
- Muramoto, Y.; Noda, T.; Kawakami, E.; Akkina, R.; Kawaoka, Y. Identification of Novel Influenza a Virus Proteins Translated from PA mRNA. J. Virol. 2013, 87, 2455–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Alvarez Castillo, D.A.; Chen, L.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza a virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouchier, R.; Munster, V.; Wallensten, A.; Bestebroer, T.M.; Herfst, S.; Smith, D.; Rimmelzwaan, G.F.; Olsen, B.; Osterhaus, A. Characterization of a novel influenza a virus hemagglutinin subtype (H16) obtained from black-headed gulls. J. Virol. 2005, 79, 2814–2822. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [Green Version]
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Vijaykrishna, D.; Poon, L.L.M.; Zhu, H.C.; Ma, S.K.; Li, O.T.W.; Cheung, C.L.; Smith, G.J.D.; Peiris, J.S.M.; Guan, Y. Reassortment of Pandemic H1N1/2009 Influenza A Virus in Swine. Science 2010, 328, 1529. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Xiao, Y.; Liu, J.; Wang, D.; Li, F.; Wang, C.; Li, C.; Zhu, J.; Song, J.; Sun, H.; et al. Prevalent Eurasian avian-like H1N1 swine influenza virus with 2009 pandemic viral genes facilitating human infection. Proc. Natl. Acad. Sci. USA 2020, 117, 17204–17210. [Google Scholar] [CrossRef]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Whittaker, G.R. Role for influenza virus envelope cholesterol in virus entry and infection. J. Virol. 2003, 77, 12543–12551. [Google Scholar] [CrossRef] [Green Version]
- Chazal, N.; Gerlier, D. Virus entry, assembly, budding, and membrane rafts. Microbiol. Mol. Biol. Rev. 2003, 67, 226–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, D.P.; Balogun, R.A.; Yamada, H.; Zhou, Z.H.; Barman, S. Influenza virus morphogenesis and budding. Virus Res. 2009, 143, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.; Lee, Y.; Lee, Y.; Kim, K.; Kang, S. Distinct Effects of Monophosphoryl Lipid A, Oligodeoxynucleotide CpG, and Combination Adjuvants on Modulating Innate and Adaptive Immune Responses to Influenza Vaccination. Immune Net. 2017, 17, 326–342. [Google Scholar] [CrossRef] [Green Version]
- Oguin, T.; Lindsley, C.; Thomas, P.; Brown, H. Effects of lipid signaling on innate immune networks during influenza infection. J. Immunol. 2015, 194 (Suppl. 1), 127.1. [Google Scholar]
- Schoggins, J.W.; Randall, G. Lipids in Innate Antiviral Defense. Cell Host Microbe 2013, 14, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Zheng, D.; Lee, Y.H.; Chan, T.K.; Kumar, Y.; Ho, W.E.; Chen, J.Z.; Tannenbaum, S.R.; Ong, C.N. Metabolomics Investigation Reveals Metabolite Mediators Associated with Acute Lung Injury and Repair in a Murine Model of Influenza Pneumonia. Sci. Rep. 2016, 6, 26076. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.; Methling, K.; Rothe, M.; Lalk, M. Eicosanoid Profile of Influenza A Virus Infected Pigs. Metabolites 2019, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, P.T.; Myers, D.S.; Milne, S.B.; McClaren, J.L.; Thomas, P.G.; Brown, H.A. Lipid Composition of the Viral Envelope of Three Strains of Influenza Virus—Not All Viruses Are Created Equal. ACS Infect. Dis. 2015, 1, 435–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milner, J.J.; Rebeles, J.; Dhungana, S.; Stewart, D.A.; Sumner, S.C.J.; Meyers, M.H.; Mancuso, P.; Beck, M.A. Obesity Increases Mortality and Modulates the Lung Metabolome during Pandemic H1N1 Influenza Virus Infection in Mice. J. Immunol. 2015, 194, 4846–4859. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Kuba, K.; Ichikawa, A.; Nakayama, M.; Katahira, J.; Iwamoto, R.; Watanebe, T.; Sakabe, S.; Daidoji, T.; Nakamura, S.; et al. The Lipid Mediator Protectin D1 Inhibits Influenza Virus Replication and Improves Severe Influenza. Cell 2013, 153, 112–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Lannan, K.L.; Thatcher, T.H.; Pollock, S.J.; Woeller, C.F.; Phipps, R.P. Lipoxin B-4 Enhances Human Memory B Cell Antibody Production via Upregulating Cyclooxygenase-2 Expression. J. Immunol. 2018, 201, 3343–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Song, L.; Feng, S.; Li, L.; Yu, H.; Wang, Q.; Wang, X.; Hou, Z.; Li, X.; Li, Y.; et al. Fatty Acid Metabolism is Associated with Disease Severity After H7N9 Infection. EBioMedicine 2018, 33, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Byleveld, P.M.; Pang, G.T.; Clancy, R.L.; Roberts, D. Fish oil feeding delays influenza virus clearance and impairs production of interferon-gamma and virus-specific immunoglobulin A in the lungs of mice. J. Nutr. 1999, 129, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Schwerbrock, N.M.J.; Karlsson, E.A.; Shi, Q.; Sheridan, P.A.; Beck, M.A. Fish oil-fed mice have impaired resistance to influenza infection. J. Nutr. 2009, 139, 1588–1594. [Google Scholar] [CrossRef] [Green Version]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Smith, W.L. The eicosanoids and their biochemical-mechanisms of action. Biochem. J. 1989, 259, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Full, F.; Gack, M.U. Prostaglandin E-2: The Villain in the Host Response to Influenza Virus. Immunity 2014, 40, 453–454. [Google Scholar] [CrossRef] [Green Version]
- Coulombe, F.; Jaworska, J.; Verway, M.; Tzelepis, F.; Massoud, A.; Gillard, J.; Wong, G.; Kobinger, G.; Xing, Z.; Couture, C.; et al. Targeted Prostaglandin E-2 Inhibition Enhances Antiviral Immunity through Induction of Type I Interferon and Apoptosis in Macrophages. Immunity 2014, 40, 554–568. [Google Scholar] [CrossRef] [Green Version]
- Oguin, T.H.I.; Sharma, S.; Stuart, A.D.; Duan, S.; Scott, S.A.; Jones, C.K.; Daniels, J.S.; Lindsley, C.W.; Thomas, P.G.; Brown, H.A. Phospholipase D Facilitates Efficient Entry of Influenza Virus, Allowing Escape from Innate Immune Inhibition. J. Biol. Chem. 2014, 289, 25405–25417. [Google Scholar] [CrossRef] [Green Version]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 132–818. [Google Scholar] [CrossRef]
- Behera, D.K.; Behera, P.M.; Acharya, L.; Dixit, A. Pharmacophore modelling, virtual screening and molecular docking studies on PLD1 inhibitors. SAR QSAR Environ. Res. 2017, 28, 991–1009. [Google Scholar] [CrossRef] [PubMed]
- Lenard, J.; Compans, R.W. Mmbrabe structure of lipid-containing viruses. Biochim. Biophys. Acta 1974, 344, 51–94. [Google Scholar] [CrossRef]
- Domanska, M.K.; Wrona, D.; Kasson, P.M. Multiphasic Effects of Cholesterol on Influenza Fusion Kinetics Reflect Multiple Mechanistic Roles. Biophys. J. 2013, 105, 1383–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, P.M.; Lawrence, M.C. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003, 4, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Dunning, R.A.; Domanska, M.K.; Dryden, K.; Yeager, M.; Kasson, P.M. Effect of Cholesterol Depletion on HA Distribution in the Viral Membrane of Influenza. Biophys. J. 2015, 1081, 406A–407A. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.N.; Boxer, S.G. Target Membrane Cholesterol Modulates Single Influenza Virus Membrane Fusion Efficiency but Not Rate. Biophys. J. 2020, 118, 2426–2433. [Google Scholar] [CrossRef]
- Bajimaya, S.; Frankl, T.; Hayashi, T.; Takimoto, T. Cholesterol is required for stability and infectivity of influenza A and respiratory syncytial viruses. Virology 2017, 510, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, I.N.; Rawle, R.J.; Boxer, S.G.; Kasson, P.M. Cholesterol enhances influenza binding avidity by controlling nanoscale receptor clustering. Chem. Sci. 2018, 9, 2340–2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef] [Green Version]
- Cubells, L.; de Muga, S.V.; Tebar, F.; Wood, P.; Evans, R.; Ingelmo-Torres, M.; Calvo, M.; Gaus, K.; Pol, A.; Grewal, T.; et al. Annexin A6-induced alterations in cholesterol transport and caveolin export from the golgi complex. Traffic 2007, 8, 1568–1589. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Creutz, C.E.; Moss, S.E. Annexins: Linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 2005, 6, 449–461. [Google Scholar] [CrossRef] [PubMed]
- De Diego, I.; Schwartz, F.; Siegfried, H.; Dauterstedt, P.; Heeren, J.; Beisiegel, U.; Enrich, C.; Grewal, T. Cholesterol modulates the membrane binding and intracellular distribution of annexin 6. J. Biol. Chem. 2002, 277, 32187–32194. [Google Scholar] [CrossRef] [Green Version]
- Musiol, A.; Gran, S.; Ehrhardt, C.; Ludwig, S.; Grewal, T.; Gerke, V.; Rescher, U. Annexin A6-balanced late endosomal cholesterol controls influenza A replication and propagation. MBio 2013, 4, e00608-13. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Kien, F.; Maniere, M.; Zhang, Y.; Lagarde, N.; Tse, K.S.; Poon, L.L.M.; Nal, B. Human Annexin A6 Interacts with Influenza A Virus Protein M2 and Negatively Modulates Infection. J. Virol. 2012, 86, 1789–1801. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, C. Cholesterol-binding viral proteins in virus entry and morphogenesis. Subcell Biochem. 2010, 51, 77–108. [Google Scholar]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The Epidermal Growth Factor Receptor (EGFR) Promotes Uptake of Influenza A Viruses (IAV) into Host Cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.K.; Gupta, D.; Lal, S.K. Host Lipid Rafts Play a Major Role in Binding and Endocytosis of Influenza A Virus. Viruses 2018, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Pekosz, A.; Lamb, R.A. Influenza virus assembly and lipid raft microdomains: A role for the cytoplasmic tails of the spike glycoproteins. J. Virol. 2000, 74, 4634–4644. [Google Scholar] [CrossRef]
- Lewis, J.F.; Jobe, A.H. Surfactant and the adult respiratory-distress syndrome. Am. Rev. Respir. Dis. 1993, 147, 218–233. [Google Scholar] [CrossRef]
- Woods, P.S.; Doolittle, L.M.; Rosas, L.E.; Joseph, L.M.; Calomeni, E.P.; Davis, I.C. Lethal H1N1 influenza A virus infection alters the murine alveolar type II cell surfactant lipidome. Am. J. Physiol.-Lung C 2016, 311, L1160–L1169. [Google Scholar] [CrossRef] [PubMed]
- Coil, D.A.; Miller, A.D. Phosphatidylserine treatment relieves the block to retrovirus infection of cells expressing glycosylated virus receptors. Retrovirology 2005, 2, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelker, D.R.; Numata, M. Phospholipid regulation of innate immunity and respiratory viral infection. J. Biol. Chem. 2019, 294, 4282–4289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, M.; Mitchell, J.R.; Tipper, J.L.; Brand, J.D.; Trombley, J.E.; Nagashima, Y.; Kandasamy, P.; Chu, H.W.; Harrod, K.S.; Voelker, D.R. Pulmonary surfactant lipids inhibit infections with the pandemic H1N1 influenza virus in several animal models. J. Biol. Chem. 2020, 295, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Numata, M.; Kandasamy, P.; Nagashima, Y.; Posey, J.; Hartshorn, K.; Woodland, D.; Voelker, D.R. Phosphatidylglycerol Suppresses Influenza A Virus Infection. Am. J. Respir. Cell Mol. Boil. 2012, 46, 479–487. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201. [Google Scholar] [CrossRef]
- Soudani, N.; Hage-Sleiman, R.; Karam, W.; Dbaibo, G.; Zaraket, H. Ceramide Suppresses Influenza A Virus Replication In Vitro. J. Virol. 2019, 93, e00053-19. [Google Scholar] [CrossRef] [Green Version]
- Santinha, D.R.; Marques, D.R.; Maciel, E.A.; Simoes, C.S.O.; Rosa, S.; Neves, B.M.; Macedo, B.; Domingues, P.; Cruz, M.T.; Domingues, M.R.M. Profiling changes triggered during maturation of dendritic cells: A lipidomic approach. Anal. Bioanal. Chem. 2012, 403, 457–471. [Google Scholar] [CrossRef]
- Chiba, N.; Masuda, A.; Yoshikai, Y.; Matsuguchi, T. Ceramide inhibits LPS-Induced production of IL-5, IL-10, and IL-13 from mast cells. J. Cell. Physiol. 2007, 213, 126–136. [Google Scholar] [CrossRef]
- Audi, A.; Soudani, N.; Dbaibo, G.; Zaraket, H. Depletion of Host and Viral Sphingomyelin Impairs Influenza Virus Infection. Front. Microbiol. 2020, 11, 612. [Google Scholar] [CrossRef]
- Tafesse, F.G.; Sanyal, S.; Ashour, J.; Guimaraes, C.P.; Hermansson, M.; Somerharju, P.; Ploegh, H.L. Intact sphingomyelin biosynthetic pathway is essential for intracellular transport of influenza virus glycoproteins. Proc. Natl. Acad. Sci. USA 2013, 110, 6406–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritzl, C.J.; Seo, Y.; Xia, C.; Vijayan, M.; Stokes, Z.D.; Hahm, B. A Ceramide Analogue Stimulates Dendritic Cells to Promote T Cell Responses upon Virus Infections. J. Immunol. 2015, 194, 4339–4349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhu, M.; Jiang, H.; Shen, S.; Su, X.; Shi, Y. Combination of sphingosine-1-phosphate receptor 1 (S1PR1) agonist and antiviral drug: A potential therapy against pathogenic influenza virus. Sci. Rep. 2019, 9, 5272. [Google Scholar] [CrossRef]
- Oldstone, M.B.; Rosen, H. Cytokine storm plays a direct role in the morbidity and mortality from influenza virus infection and is chemically treatable with a single sphingosine-1-phosphate agonist molecule. Curr. Top. Microbiol. Immunol. 2014, 378, 129–147. [Google Scholar] [PubMed]
- Jiang, H.; Shen, S.M.; Yin, J.; Zhang, P.P.; Shi, Y. Sphingosine 1-phosphate receptor 1 (S1PR1) agonist CYM5442 inhibits expression of intracellular adhesion molecule 1 (ICAM1) in endothelial cells infected with influenza A viruses. PLoS ONE 2017, 12, e175188. [Google Scholar] [CrossRef]
- Gandy, K.A.O.; Obeid, L.M. Regulation of the sphingosine kinase/sphingosine 1-phosphate pathway. Handb. Exp. Pharmacol. 2013, 216, 275–303. [Google Scholar]
- Pitson, S.M. Regulation of sphingosine kinase and sphingolipid signaling. Trends Biochem. Sci. 2011, 36, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Seo, Y.; Studstill, C.J.; Vijayan, M.; Wolf, J.J.; Hahm, B. Transient inhibition of sphingosine kinases confers protection to influenza A virus infected mice. Antivir. Res. 2018, 158, 171–177. [Google Scholar] [CrossRef]
- Hait, N.C.; Oskeritzian, C.A.; Paugh, S.W.; Milstien, S.; Spiegel, S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim. Biophys. Acta 2006, 1758, 2016–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.; Pritzl, C.J.; Vijayan, M.; Bomb, K.; McClain, M.E.; Alexander, S.; Hahm, B. Sphingosine Kinase 1 Serves as a Pro-Viral Factor by Regulating Viral RNA Synthesis and Nuclear Export of Viral Ribonucleoprotein Complex upon Influenza Virus Infection. PLoS ONE 2013, 8, e75005. [Google Scholar] [CrossRef]
- Carr, J.M.; Mahalingam, S.; Bonder, C.S.; Pitson, S.M. Sphingosine kinase 1 in viral infections. Rev. Med. Virol. 2013, 23, 73–84. [Google Scholar] [CrossRef]
- Kumar, N.; Xin, Z.; Liang, Y.; Ly, H.; Liang, Y. NF-kappa B Signaling Differentially Regulates Influenza Virus RNA Synthesis. J. Virol. 2008, 82, 9880–9889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.; Blake, C.; Alexander, S.; Hahm, B. Sphingosine 1-Phosphate-Metabolizing Enzymes Control Influenza Virus Propagation and Viral Cytopathogenicity. J. Virol. 2010, 84, 8124–8131. [Google Scholar] [CrossRef] [Green Version]
- Gambaryan, A.S.; Tuzikov, A.B.; Pazynina, G.V.; Webster, R.G.; Matrosovich, M.N.; Bovin, N.V. H5N1 chicken influenza viruses display a high binding affinity for Neu5Ac alpha 2-3Gal beta 1-4(6-HSO3) GlcNAc-containing receptors. Virology 2004, 326, 310–316. [Google Scholar] [CrossRef] [Green Version]
- Hidari, K.I.P.J.; Shimada, S.; Suzuki, Y.; Suzuki, T. Binding kinetics of influenza viruses to sialic acid-containing carbohydrates. Glycoconj. J. 2007, 24, 583–590. [Google Scholar] [CrossRef]
- Ablan, S.; Rawat, S.S.; Blumenthal, R.; Puri, A. Entry of influenza virus into a glycosphingolipid-deficient mouse skin fibroblast cell line. Arch. Virol. 2001, 146, 2227–2238. [Google Scholar] [CrossRef]
- Chu, V.C.; Whittaker, G.R. Influenza virus entry and infection require host cell N-linked glycoprotein. Proc. Natl. Acad. Sci. USA 2004, 101, 18153–18158. [Google Scholar] [CrossRef] [Green Version]
- Kasson, P.M.; Pande, V.S. Structural basis for influence of viral glycans on ligand binding by influenza hemagglutinin. Biophys. J. 2008, 95, L48–L50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishibashi, Y.; Kohyama-Koganeya, A.; Hirabayashi, Y. New insights on glucosylated lipids: Metabolism and functions. Biochim. Biophys. Acta 2013, 1831, 1475–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Nelson, E.A.; Han, J.D.; Fox, T.; et al. Glucosylceramidase Maintains Influenza Virus Infection by Regulating Endocytosis. J. Virol. 2019, 93, e00017-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drews, K.; Calgi, M.P.; Harrison, W.C.; Drews, C.M.; Costa-Pinheiro, P.; Shaw, J.J.P.; Jobe, K.A.; Han, J.D.; Fox, T.E.; White, J.M.; et al. Glucosylceramide synthase maintains influenza virus entry and infection. PLoS ONE 2020, 15, e0228735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, L.H.; Lamb, R.A. Understanding the mechanism of action of the antiinfluenza virus drug amantadine. Trends Microbiol. 1995, 3, 271. [Google Scholar] [CrossRef]
- Gubareva, L.V.; Kaiser, L.; Hayden, F.G. Influenza virus neuraminidase inhibitors. Lancet 2000, 355, 827–835. [Google Scholar] [CrossRef]
- Shankaran, S.; Bearman, G.M.L. Influenza Virus Resistance to Neuraminidase Inhibitors: Implications for Treatment. Curr. Infect. Dis. Rep. 2012, 14, 155–160. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Nayak, D.P. Lipid raft disruption by cholesterol depletion enhances influenza a virus budding from MDCK cells. J. Virol. 2007, 81, 12169–12178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Schroeder, C.; Schlechtingen, G.; Braxmeier, T.; Jennings, G.; Knoelker, H. Evaluation of steroidal amines as lipid raft modulators and potential anti-influenza agents. Bioorg. Med. Chem. Lett. 2013, 23, 5165–5169. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Metabolism | Name of Component | Role in IAV Infection | Reference |
|---|---|---|---|
| FA | 12-HETE, 15-HETE, 17-HDoHE | Inhibits IAV infection in vitro | [21] |
| PD1 | Inhibits nuclear export of IAV mRNA | [21,24] | |
| LXB4 | Promotes the production of effector B cells and IgG | [25] | |
| EPA, DHA | Inhibits innate and adaptive immunity | [28] | |
| PGE2 | Inhibits IFN-mediated innate immunity and impairs adaptive immunity | [32] | |
| PLD | Promotes host innate immune evasion by IAV | [33] | |
| Chol | Viral envelope Chol | Promotes the fusion of IAV particles | [39] |
| Cell membrane Chol | Improves the infectivity and stability of progeny virions | [41] | |
| PL | PS | Promotes the susceptibility of cells to IAV infection | [57] |
| PI | Binds to IAV with high affinity and inhibits inflammation induced by IAV infection | [60] | |
| SM | Promotes the fusion and infectivity of progeny virions | [61,66] | |
| Cer | Promotes the adaptive immune response after viral infection | [63] | |
| SGMS1 | Promotes the release of progeny virions | [67] | |
| SPHK1 | Promotes vRNA synthesis and vRNP nuclear export | [74,77,78] | |
| SPL | Enhances the immune response to IAV | [79] | |
| GL | GlcCer | Maintains the optimal state of IAV infection | [86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Pu, J.; Wu, Y. The Role of Lipid Metabolism in Influenza A Virus Infection. Pathogens 2021, 10, 303. https://doi.org/10.3390/pathogens10030303
Zhou Y, Pu J, Wu Y. The Role of Lipid Metabolism in Influenza A Virus Infection. Pathogens. 2021; 10(3):303. https://doi.org/10.3390/pathogens10030303
Chicago/Turabian StyleZhou, Yong, Juan Pu, and Yuping Wu. 2021. "The Role of Lipid Metabolism in Influenza A Virus Infection" Pathogens 10, no. 3: 303. https://doi.org/10.3390/pathogens10030303
APA StyleZhou, Y., Pu, J., & Wu, Y. (2021). The Role of Lipid Metabolism in Influenza A Virus Infection. Pathogens, 10(3), 303. https://doi.org/10.3390/pathogens10030303