
Whole-Genome Sequencing for Tracing the Genetic Diversity of Brucella abortus and Brucella melitensis Isolated from Livestock in Egypt

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Brucella Strain Isolation and Identification
2.2. Whole-Genome Sequencing
3. Results
3.1. Identification and Differentiation of Brucella Isolates
3.2. Brucella Genome
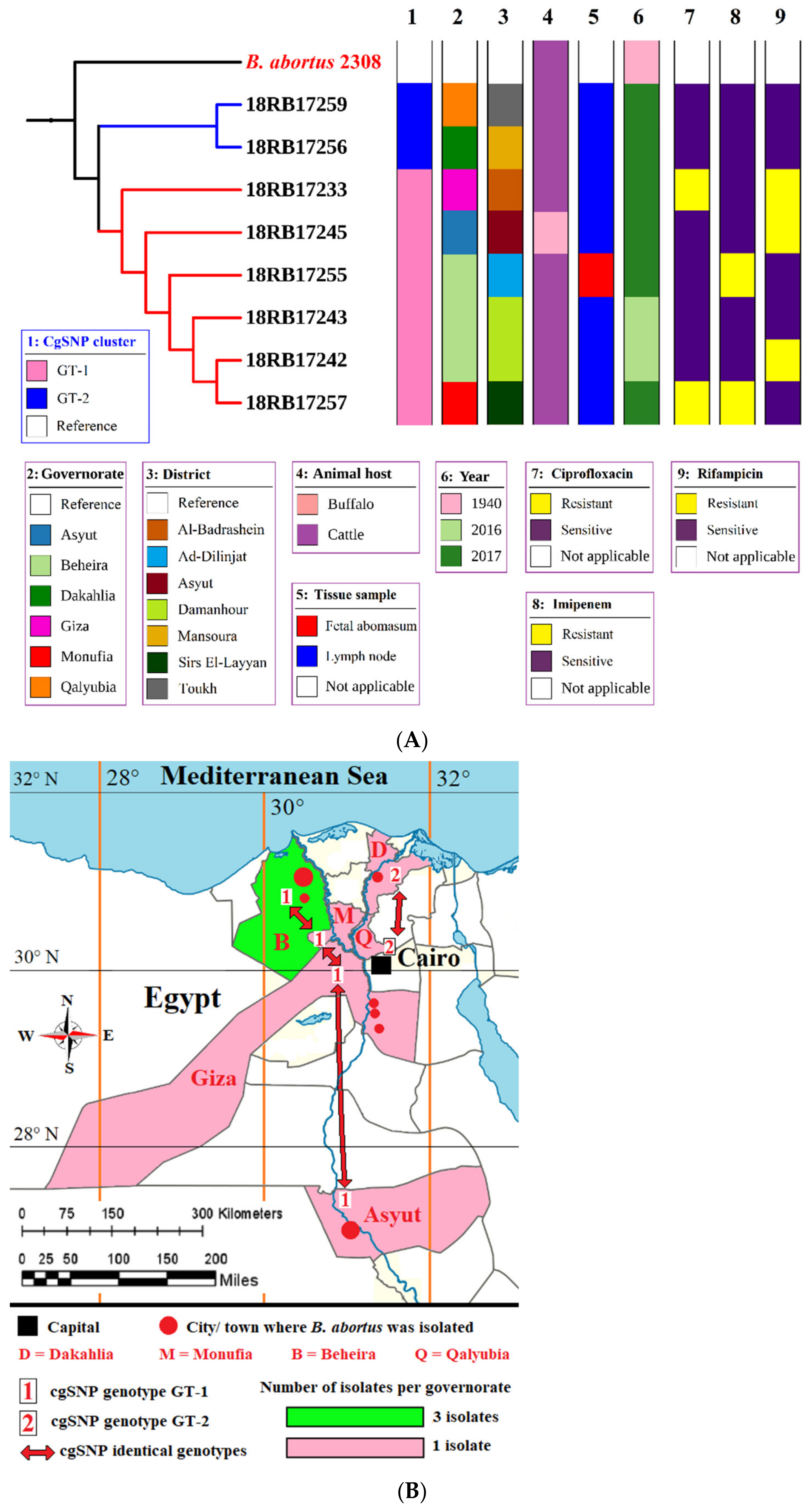
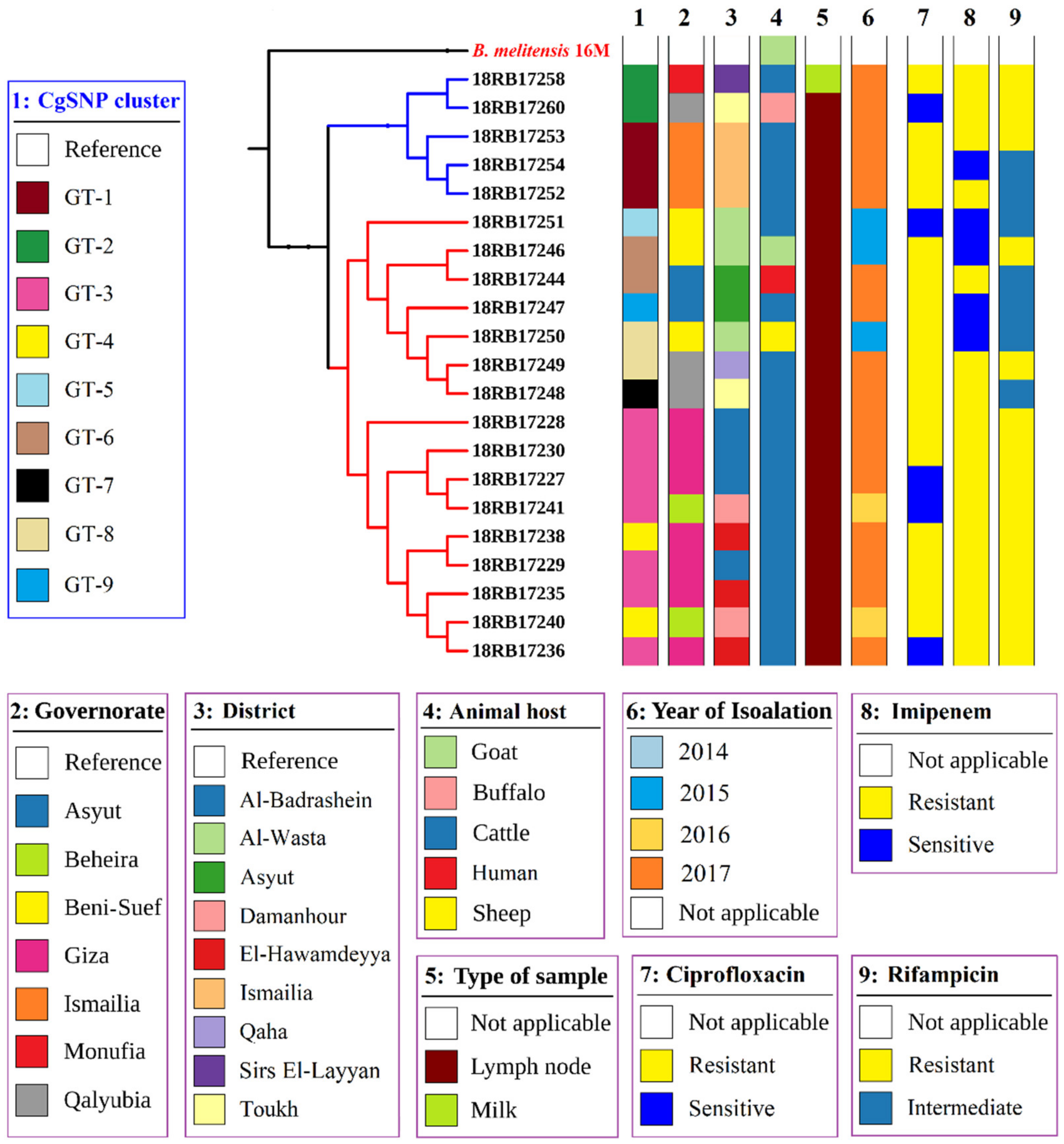
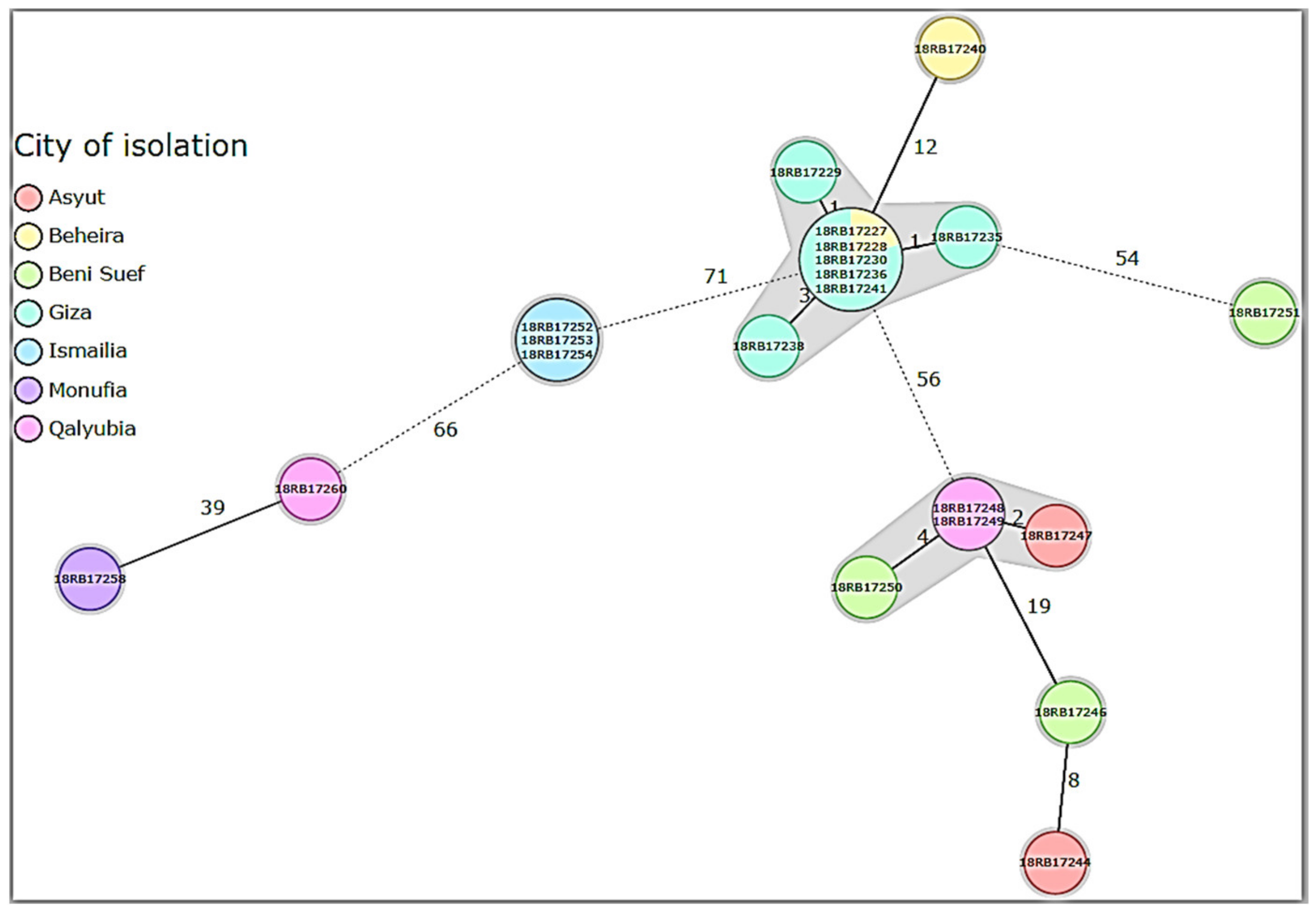
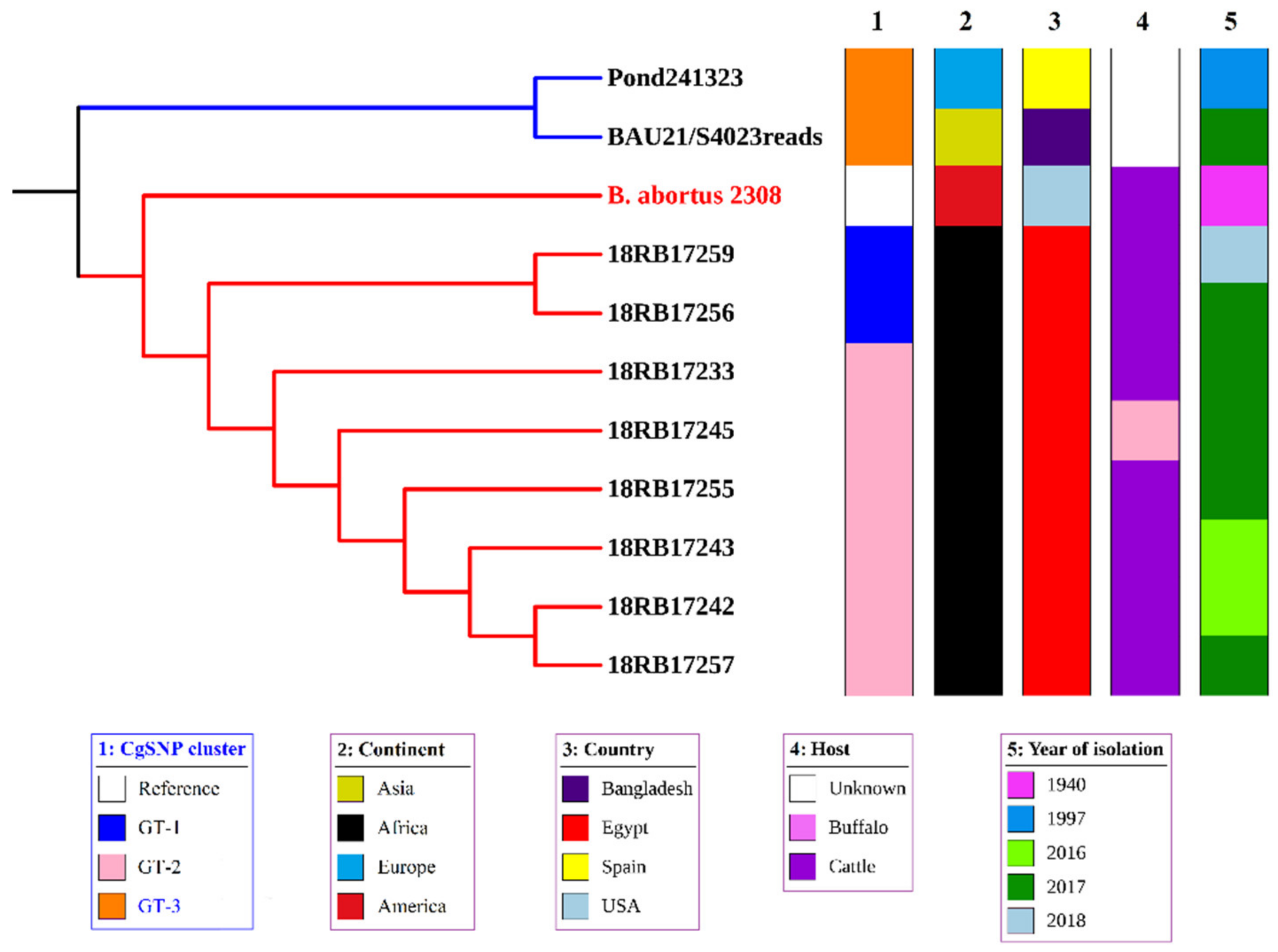
3.3. MLST, In Silico MLVA-16, and cgSNP Analysis of B. abortus and B. melitensis Isolates
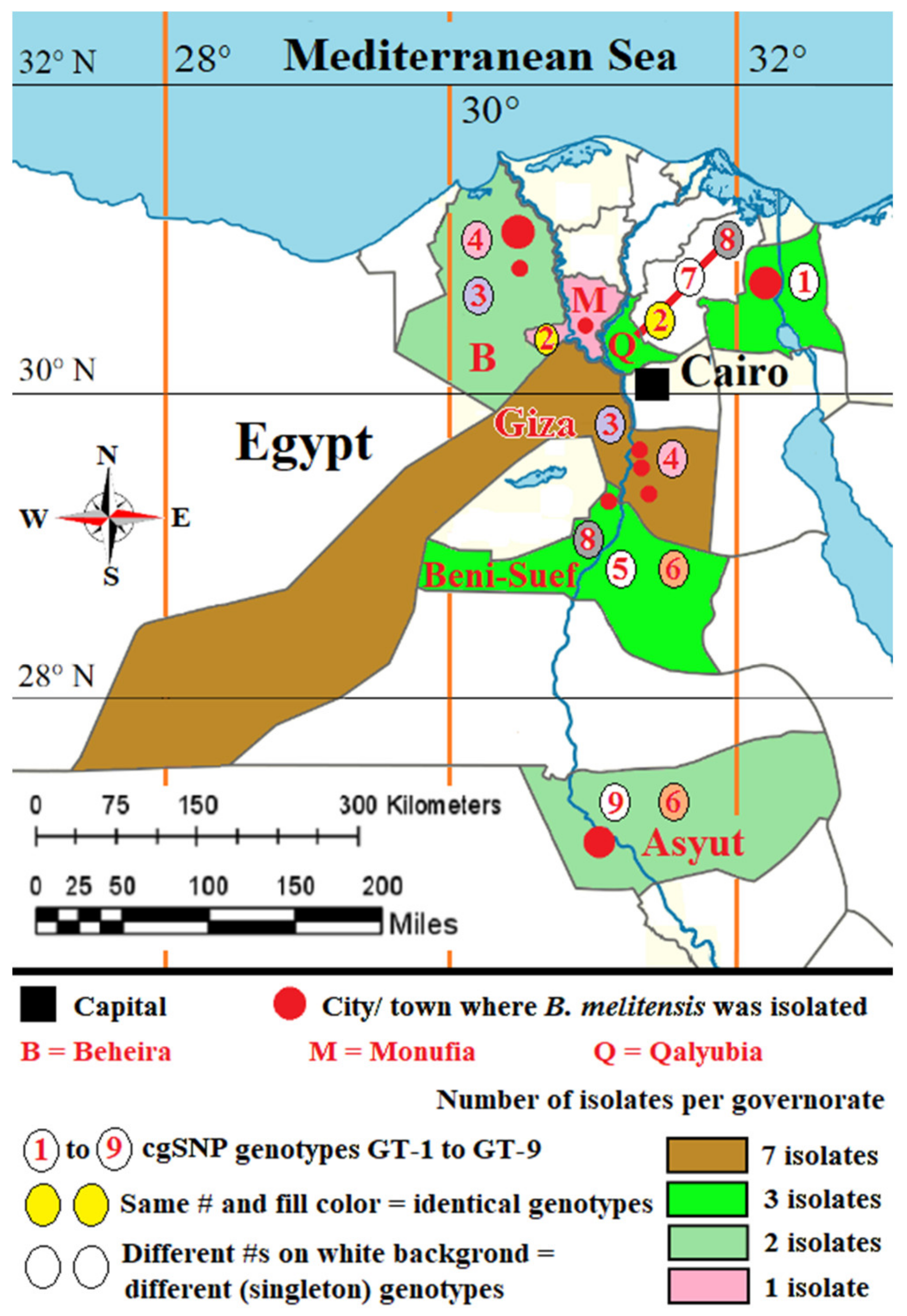
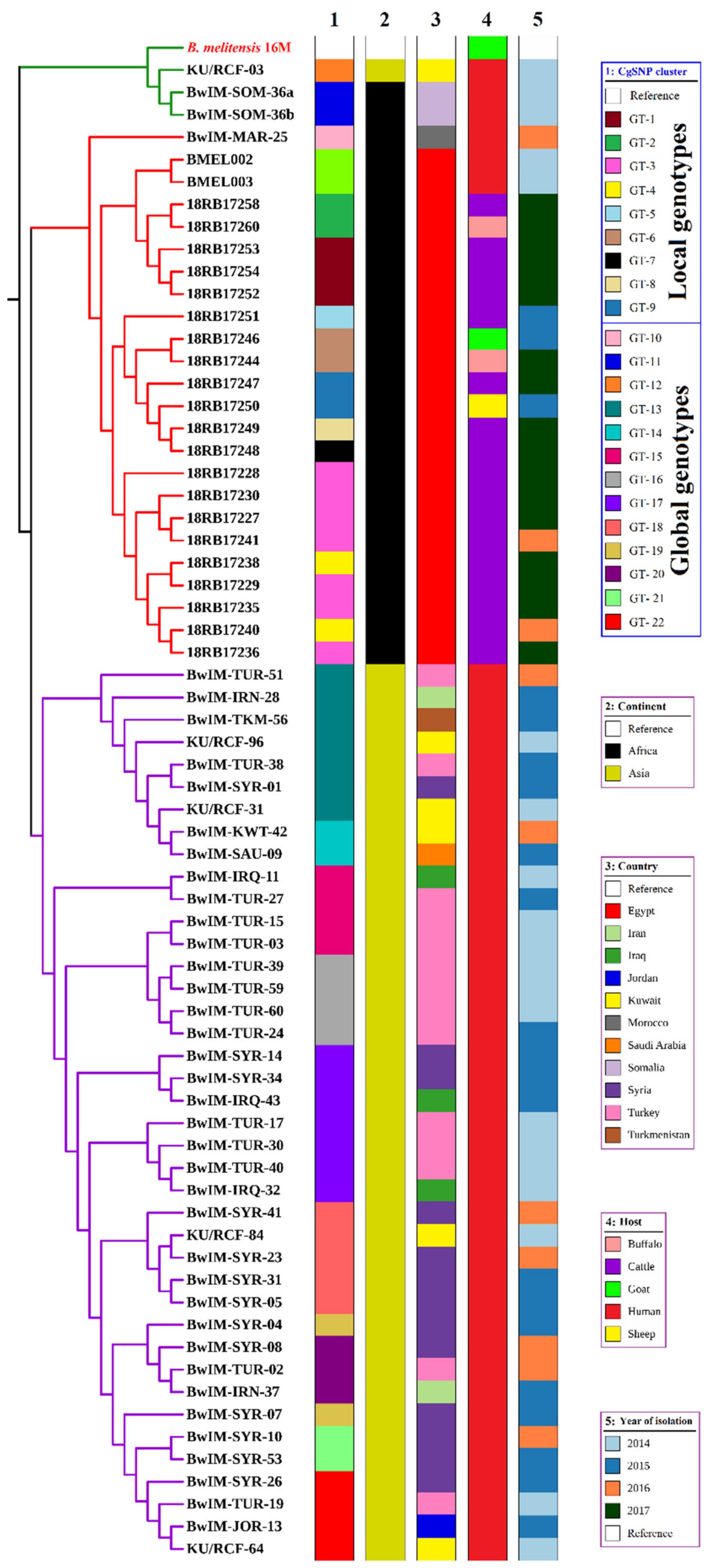
3.4. Comparison of Local Brucella Genotypes with Selected Genotypes from Neighboring Countries Using In Silico MLVA-16 and cgSNP Analyses
3.5. SNP Annotation
3.6. Factors for Antimicrobial Resistance and Virulence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janowicz, A.; De Massis, F.; Ancora, M.; Camma, C.; Patavino, C.; Battisti, A.; Prior, K.; Harmsen, D.; Scholz, H.; Zilli, K.; et al. Core genome multilocus sequence typing and single nucleotide polymorphism analysis in the epidemiology of Brucella melitensis infections. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [PubMed]
- Seleem, M.N.; Boyle, S.M.; Sriranganathan, N. Brucellosis: A re-emerging zoonosis. Vet. Microbiol. 2010, 140, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Dopfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. World Health Organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: A data synthesis. PLoS Med. 2015, 12, e1001921. [Google Scholar] [CrossRef]
- Richomme, C.; Gauthier, D.; Fromont, E. Contact rates and exposure to inter-species disease transmission in mountain ungulates. Epidemiol. Infect. 2006, 134, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Saeed, U.; Ali, S.; Khan, T.M.; El-Adawy, H.; Melzer, F.; Khan, A.U.; Iftikhar, A.; Neubauer, H. Seroepidemiology and the molecular detection of animal brucellosis in Punjab, Pakistan. Microorganisms 2019, 7, 449. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Tsolis, R.M. Brucella spp. Virulence Factors and Immunity. Annu. Rev. Anim. Biosci. 2016, 4, 111–127. [Google Scholar] [CrossRef]
- Greco, E.; El-Aguizy, O.; Ali, M.F.; Foti, S.; Cunsolo, V.; Saletti, R.; Ciliberto, E. Proteomic analyses on an ancient egyptian cheese and biomolecular evidence of brucellosis. Anal. Chem. 2018, 90, 9673–9676. [Google Scholar] [CrossRef]
- Sayour, A.E.; Elbauomy, E.; Abdel-Hamid, N.H.; Mahrous, A.; Carychao, D.; Cooley, M.B.; Elhadidy, M. MLVA fingerprinting of Brucella melitensis circulating among livestock and cases of sporadic human illness in Egypt. Transbound. Emerg. Dis. 2020, 67, 2435–2445. [Google Scholar] [CrossRef]
- Sayour, A.E.; Sayour, H.E. Binomial identification of Brucella isolates by MALDI-TOF mass spectrometry. In Proceedings of the Second Scientific Conference of Food Safety and Technology, Cairo, Egypt, 25–27 December 2018; pp. 28–49. [Google Scholar]
- Khan, A.U.; Melzer, F.; El-Soally, S.; Elschner, M.C.; Mohamed, S.A.; Sayed Ahmed, M.A.; Roesler, U.; Neubauer, H.; El-Adawy, H. Serological and Molecular Identification of Brucella spp. in Pigs from Cairo and Giza Governorates, Egypt. Pathogens 2019, 8, 248. [Google Scholar] [CrossRef]
- Khan, A.U.; Sayour, A.E.; Melzer, F.; El-Soally, S.; Elschner, M.C.; Shell, W.S.; Moawad, A.A.; Mohamed, S.A.; Hendam, A.; Roesler, U.; et al. Seroprevalence and Molecular Identification of Brucella spp. in Camels in Egypt. Microorganisms 2020, 8, 1035. [Google Scholar] [CrossRef]
- Menshawy, A.M.; Perez-Sancho, M.; Garcia-Seco, T.; Hosein, H.I.; Garcia, N.; Martinez, I.; Sayour, A.E.; Goyache, J.; Azzam, R.A.; Dominguez, L.; et al. Assessment of genetic diversity of zoonotic Brucella spp. recovered from livestock in Egypt using multiple locus VNTR analysis. Biomed Res. Int. 2014, 2014, 353876. [Google Scholar] [CrossRef]
- Samaha, H.; Al-Rowaily, M.; Khoudair, R.M.; Ashour, H.M. Multicenter study of brucellosis in Egypt. Emerg. Infect. Dis. 2008, 14, 1916–1918. [Google Scholar] [CrossRef] [PubMed]
- Eltholth, M.M.; Hegazy, Y.M.; El-Tras, W.F.; Bruce, M.; Rushton, J. Temporal analysis and costs of ruminant brucellosis control programme in Egypt between 1999 and 2011. Transbound. Emerg. Dis. 2017, 64, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- OIE. Brucellosis (Brucella abortus, B. melitensis and B. suis) (infection with B. abortus, B. melitensis and B. suis). In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 7th ed.; World Health Organization for Animal Health: Paris, France, 2018; Volume 1, pp. 355–398. [Google Scholar]
- Khan, A.U.; Shell, W.S.; Melzer, F.; Sayour, A.E.; Ramadan, E.S.; Elschner, M.C.; Moawad, A.A.; Roesler, U.; Neubauer, H.; El-Adawy, H. Identification, genotyping and antimicrobial susceptibility testing of Brucella spp. isolated from livestock in Egypt. Microorganisms 2019, 7, 603. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; El-Diasty, M.; Melzer, F.; Schmoock, G.; Moustafa, S.A.; El-Beskawy, M.; Khater, D.F.; Hamdy, M.E.R.; Zaki, H.M.; Ferreira, A.C.; et al. MLVA-16 genotyping of Brucella abortus and Brucella melitensis isolates from different animal species in Egypt: Geographical relatedness and the mediterranean lineage. Pathogens 2020, 9, 498. [Google Scholar] [CrossRef]
- Wattam, A.R.; Foster, J.T.; Mane, S.P.; Beckstrom-Sternberg, S.M.; Beckstrom-Sternberg, J.M.; Dickerman, A.W.; Keim, P.; Pearson, T.; Shukla, M.; Ward, D.V.; et al. Comparative phylogenomics and evolution of the Brucellae reveal a path to virulence. J. Bacteriol. 2014, 196, 920–930. [Google Scholar] [CrossRef]
- Pelerito, A.; Nunes, A.; Nuncio, M.S.; Gomes, J.P. Genome-scale approach to study the genetic relatedness among Brucella melitensis strains. PLoS ONE 2020, 15, e0229863. [Google Scholar] [CrossRef] [PubMed]
- Ratushna, V.G.; Sturgill, D.M.; Ramamoorthy, S.; Reichow, S.A.; He, Y.; Lathigra, R.; Sriranganathan, N.; Halling, S.M.; Boyle, S.M.; Gibas, C.J. Molecular targets for rapid identification of Brucella spp. BMC Microbiol. 2006, 6, 13. [Google Scholar] [CrossRef]
- Foster, J.T.; Beckstrom-Sternberg, S.M.; Pearson, T.; Beckstrom-Sternberg, J.S.; Chain, P.S.; Roberto, F.F.; Hnath, J.; Brettin, T.; Keim, P. Whole-genome-based phylogeny and divergence of the genus Brucella. J. Bacteriol. 2009, 191, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Whatmore, A.M. Current understanding of the genetic diversity of Brucella, an expanding genus of zoonotic pathogens. Infect. Genet. Evol. 2009, 9, 1168–1184. [Google Scholar] [CrossRef]
- Le Flèche, P.; Jacques, I.; Grayon, M.; Al Dahouk, S.; Bouchon, P.; Denoeud, F.; Nöckler, K.; Neubauer, H.; Guilloteau, L.A.; Vergnaud, G. Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. 2006, 6, 9. [Google Scholar] [CrossRef]
- Whatmore, A.M.; Perrett, L.L.; MacMillan, A.P. Characterisation of the genetic diversity of Brucella by multilocus sequencing. BMC Microbiol. 2007, 7, 34. [Google Scholar] [CrossRef]
- Vergnaud, G.; Hauck, Y.; Christiany, D.; Daoud, B.; Pourcel, C.; Jacques, I.; Cloeckaert, A.; Zygmunt, M.S. Genotypic expansion within the population structure of classical Brucella species revealed by MLVA16 Typing of 1404 Brucella isolates from different animal and geographic origins, 1974–2006. Front. Microbiol. 2018, 9, 1545. [Google Scholar] [CrossRef]
- Tan, K.K.; Tan, Y.C.; Chang, L.Y.; Lee, K.W.; Nore, S.S.; Yee, W.Y.; Mat Isa, M.N.; Jafar, F.L.; Hoh, C.C.; AbuBakar, S. Full genome SNP-based phylogenetic analysis reveals the origin and global spread of Brucella melitensis. BMC Genom. 2015, 16, 93. [Google Scholar] [CrossRef] [PubMed]
- Georgi, E.; Walter, M.C.; Pfalzgraf, M.T.; Northoff, B.H.; Holdt, L.M.; Scholz, H.C.; Zoeller, L.; Zange, S.; Antwerpen, M.H. Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PloS ONE 2017, 12, e0175425. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.; Kirkness, E.F. Whole genome sequencing. In Genetic Variation; Springer: Berlin/Heidelberg, Germany, 2010; pp. 215–226. [Google Scholar]
- Roetzer, A.; Diel, R.; Kohl, T.; Rückert, C.; Nübel, U.; Blom, J.; Wirth, T.; Jaenicke, S.; Schuback, S.; Rüsch-Gerdes, S. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLoS Med. 2013, 10, e1001387. [Google Scholar] [CrossRef]
- Sali, M.; De Maio, F.; Tarantino, M.; Garofolo, G.; Tittarelli, M.; Sacchini, L.; Zilli, K.; Pasquali, P.; Petrucci, P.; Marianelli, C.; et al. Rapid and safe one-step extraction method for the identification of Brucella strains at genus and species level by MALDI-TOF mass spectrometry. PLoS ONE 2018, 13, e0197864. [Google Scholar] [CrossRef]
- Bricker, B.J.; Halling, S.M. Differentiation of Brucella abortus bv. 1, 2, and 4, Brucella melitensis, Brucella ovis, and Brucella suis bv. 1 by PCR. J. Clin. Microbiol. 1994, 32, 2660–2666. [Google Scholar] [CrossRef]
- Bricker, B.J.; Halling, S.M. Enhancement of the Brucella AMOS PCR assay for differentiation of Brucella abortus vaccine strains S19 and RB51. J. Clin. Microbiol. 1995, 33, 1640–1642. [Google Scholar] [CrossRef]
- Garcia-Yoldi, D.; Marin, C.M.; de Miguel, M.J.; Munoz, P.M.; Vizmanos, J.L.; Lopez-Goni, I. Multiplex PCR assay for the identification and differentiation of all Brucella species and the vaccine strains Brucella abortus S19 and RB51 and Brucella melitensis Rev1. Clin. Chem. 2006, 52, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Goni, I.; Garcia-Yoldi, D.; Marin, C.M.; de Miguel, M.J.; Munoz, P.M.; Blasco, J.M.; Jacques, I.; Grayon, M.; Cloeckaert, A.; Ferreira, A.C.; et al. Evaluation of a multiplex PCR assay (Bruce-ladder) for molecular typing of all Brucella species, including the vaccine strains. J. Clin. Microbiol. 2008, 46, 3484–3487. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Marianelli, C.; Ciuchini, F.; Tarantino, M.; Pasquali, P.; Adone, R. Genetic bases of the rifampin resistance phenotype in Brucella spp. J. Clin. Microbiol. 2004, 42, 5439–5443. [Google Scholar] [CrossRef]
- Valdezate, S.; Navarro, A.; Medina-Pascual, M.J.; Carrasco, G.; Saez-Nieto, J.A. Molecular screening for rifampicin and fluoroquinolone resistance in a clinical population of Brucella melitensis. J. Antimicrob. Chemother. 2010, 65, 51–53. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 Years on. Nucleic Acids Res. 2015, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Sacchini, L.; Wahab, T.; Di Giannatale, E.; Zilli, K.; Abass, A.; Garofolo, G.; Janowicz, A. Whole genome sequencing for tracing geographical origin of imported cases of human brucellosis in Sweden. Microorganisms 2019, 7, 398. [Google Scholar] [CrossRef] [PubMed]
- Sadequl Islam, M.; El Zowalaty, M.E.; van Vliet, A.H.; Thakur, S.; Minara Khatun, M.; Saha, S.; Tanvir Rahman, M.; Noreddin, A.; Ariful Islam, M.; Dunning Hotopp, J.C. First genome sequence of Brucella abortus biovar 3 strain BAU21/S4023, isolated from a dairy cow in Bangladesh. Microbiol. Res. Announc. 2019, 8, e00446-19. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Jennings, G.J.; Hajjeh, R.A.; Girgis, F.Y.; Fadeel, M.A.; Maksoud, M.A.; Wasfy, M.O.; El-Sayed, N.; Srikantiah, P.; Luby, S.P.; Earhart, K.; et al. Brucellosis as a cause of acute febrile illness in Egypt. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; Melzer, F.; El-Diasty, M.; Schmoock, G.; Elbauomy, E.; Abdel-Hamid, N.; Sayour, A.; Neubauer, H. Isolation of Brucella abortus from a dog and a cat confirms their biological role in re-emergence and dissemination of bovine brucellosis on dairy farms. Transbound. Emerg. Dis. 2017, 64, e27–e30. [Google Scholar] [CrossRef] [PubMed]
- Wareth, G.; Melzer, F.; Tomaso, H.; Roesler, U.; Neubauer, H. Detection of Brucella abortus DNA in aborted goats and sheep in Egypt by real-time PCR. BMC Res. Notes 2015, 8, 212. [Google Scholar] [CrossRef]
- Ferreira, L.; Vega Castaño, S.; Sánchez-Juanes, F.; González-Cabrero, S.; Menegotto, F.; Orduña-Domingo, A.; González-Buitrago, J.M.; Muñoz-Bellido, J.L. Identification of Brucella by MALDI-TOF Mass Spectrometry. Fast and Reliable Identification from Agar Plates and Blood Cultures. PLoS ONE 2010, 5, e14235. [Google Scholar] [CrossRef] [PubMed]
- Lista, F.; Reubsaet, F.A.G.; De Santis, R.; Parchen, R.R.; de Jong, A.L.; Kieboom, J.; van der Laaken, A.L.; Voskamp-Visser, I.A.I.; Fillo, S.; Jansen, H.; et al. Reliable identification at the species level of Brucella isolates with MALDI-TOF-MS. BMC Microbiol. 2011, 11, 267. [Google Scholar] [CrossRef]
- Xing, R.; Wen, Y.; He, H.; Guo, Z.; Liu, Z. Recent progress in the combination of molecularly imprinted polymer-based affinity extraction and mass spectrometry for targeted proteomic analysis. TrAC Trends Anal. Chem. 2019, 110, 417–428. [Google Scholar] [CrossRef]
- Allen, A.R.; Milne, G.; Drees, K.; Presho, E.; Graham, J.; McAdam, P.; Jones, K.; Wright, L.; Skuce, R.; Whatmore, A.M.; et al. Genomic epizootiology of a Brucella abortus outbreak in Northern Ireland (1997–2012). Infect. Genet. Evol. 2020, 81, 104235. [Google Scholar] [CrossRef]
- Tiller, R.V.; De, B.K.; Boshra, M.; Huynh, L.Y.; Van Ert, M.N.; Wagner, D.M.; Klena, J.; Mohsen, T.S.; El-Shafie, S.S.; Keim, P.; et al. Comparison of two multiple-locus variable-number tandem-repeat analysis methods for molecular strain typing of human Brucella melitensis isolates from the Middle East. J. Clin. Microbiol. 2009, 47, 2226–2231. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Raoult, D.; Rolain, J.M. A bioinformatic approach to understanding antibiotic resistance in intracellular bacteria through whole genome analysis. Int. J. Antimicrob. Agents 2008, 32, 207–220. [Google Scholar] [CrossRef]
- Turkmani, A.; Psaroulaki, A.; Christidou, A.; Chochlakis, D.; Tabaa, D.; Tselentis, Y. In vitro-selected resistance to fluoroquinolones in two Brucella strains associated with mutational changes in gyrA. Int. J. Antimicrob. Agents 2008, 32, 227–232. [Google Scholar] [CrossRef]
- CLSI. Guideline M100-ED29. In Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; Clinical and Laboratory Standard Institute: Wayne, PA, USA, 2019. [Google Scholar]
- EUCAST. Breakpoint Tables for Interpretation of MICs and Zone Diameters; EUCAST: Basel, Switzerland, 2019; Volume 9. [Google Scholar]
- Lokesh, D.; Parkesh, R.; Kammara, R. Bifidobacterium adolescentis is intrinsically resistant to antitubercular drugs. Sci. Rep. 2018, 8, 11897. [Google Scholar] [CrossRef] [PubMed]
- Ernst, C.M.; Staubitz, P.; Mishra, N.N.; Yang, S.J.; Hornig, G.; Kalbacher, H.; Bayer, A.S.; Kraus, D.; Peschel, A. The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog. 2009, 5, e1000660. [Google Scholar] [CrossRef]
- Andrä, J.; Goldmann, T.; Ernst, C.M.; Peschel, A.; Gutsmann, T. Multiple peptide resistance factor (MprF)-mediated Resistance of Staphylococcus aureus against antimicrobial peptides coincides with a modulated peptide interaction with artificial membranes comprising lysyl-phosphatidylglycerol. J. Biol. Chem. 2011, 286, 18692–18700. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Seshadri, R.; Nelson, K.E.; Eisen, J.A.; Heidelberg, J.F.; Read, T.D.; Dodson, R.J.; Umayam, L.; Brinkac, L.M.; Beanan, M.J.; et al. The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc. Natl. Acad. Sci. USA 2002, 99, 13148–13153. [Google Scholar] [CrossRef]
- Mima, T.; Joshi, S.; Gomez-Escalada, M.; Schweizer, H.P. Identification and characterization of TriABC-OpmH, a triclosan efflux pump of Pseudomonas aeruginosa requiring two membrane fusion proteins. J. Bacteriol. 2007, 189, 7600–7609. [Google Scholar] [CrossRef] [PubMed]
- Ravanel, N.; Gestin, B.; Maurin, M. In vitro selection of fluoroquinolone resistance in Brucella melitensis. Int. J. Antimicrob. Agents 2009, 34, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, P.G.; Macedo, G.C.; Azevedo, V.; Oliveira, S.C. Brucella spp. noncanonical LPS: Structure, biosynthesis, and interaction with host immune system. Microb. Cell Fact. 2006, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Christopher, S.; Umapathy, B.L.; Ravikumar, K.L. Brucellosis: Review on the recent trends in pathogenicity and laboratory diagnosis. J. Lab. Physicians 2010, 2, 55–60. [Google Scholar] [CrossRef] [PubMed]
- DelVecchio, V.G.; Kapatral, V.; Redkar, R.J.; Patra, G.; Mujer, C.; Los, T.; Ivanova, N.; Anderson, I.; Bhattacharyya, A.; Lykidis, A.; et al. The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc. Natl. Acad. Sci. USA 2002, 99, 443–448. [Google Scholar] [CrossRef]
- Smith, J.A. Brucella lipopolysaccharide and pathogenicity: The core of the matter. Virulence 2018, 9, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Felix, C.; Kaplan Türköz, B.; Ranaldi, S.; Koelblen, T.; Terradot, L.; O’Callaghan, D.; Vergunst, A.C. The Brucella TIR domain containing proteins BtpA and BtpB have a structural WxxxE motif important for protection against microtubule depolymerisation. Cell Commun. Signal. 2014, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, Y.; Li, W.; Chen, Z. Type IV secretion system of Brucella spp. and its effectors. Front. Cell. Infect. Microbiol. 2015, 5, 72. [Google Scholar] [CrossRef]
- Seleem, M.N.; Boyle, S.M.; Sriranganathan, N. Brucella: A pathogen without classic virulence genes. Vet. Microbiol. 2008, 129, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Barsy, M.; Jamet, A.; Filopon, D.; Nicolas, C.; Laloux, G.; Rual, J.F.; Muller, A.; Twizere, J.C.; Nkengfac, B.; Vandenhaute, J.; et al. Identification of a Brucella spp. secreted effector specifically interacting with human small GTPase Rab2. Cell. Microbiol. 2011, 13, 1044–1058. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.U.; Melzer, F.; Sayour, A.E.; Shell, W.S.; Linde, J.; Abdel-Glil, M.; El-Soally, S.A.G.E.; Elschner, M.C.; Sayour, H.E.M.; Ramadan, E.S.; et al. Whole-Genome Sequencing for Tracing the Genetic Diversity of Brucella abortus and Brucella melitensis Isolated from Livestock in Egypt. Pathogens 2021, 10, 759. https://doi.org/10.3390/pathogens10060759
Khan AU, Melzer F, Sayour AE, Shell WS, Linde J, Abdel-Glil M, El-Soally SAGE, Elschner MC, Sayour HEM, Ramadan ES, et al. Whole-Genome Sequencing for Tracing the Genetic Diversity of Brucella abortus and Brucella melitensis Isolated from Livestock in Egypt. Pathogens. 2021; 10(6):759. https://doi.org/10.3390/pathogens10060759
Chicago/Turabian StyleKhan, Aman Ullah, Falk Melzer, Ashraf E. Sayour, Waleed S. Shell, Jörg Linde, Mostafa Abdel-Glil, Sherif A. G. E. El-Soally, Mandy C. Elschner, Hossam E. M. Sayour, Eman Shawkat Ramadan, and et al. 2021. "Whole-Genome Sequencing for Tracing the Genetic Diversity of Brucella abortus and Brucella melitensis Isolated from Livestock in Egypt" Pathogens 10, no. 6: 759. https://doi.org/10.3390/pathogens10060759
APA StyleKhan, A. U., Melzer, F., Sayour, A. E., Shell, W. S., Linde, J., Abdel-Glil, M., El-Soally, S. A. G. E., Elschner, M. C., Sayour, H. E. M., Ramadan, E. S., Mohamed, S. A., Hendam, A., Ismail, R. I., Farahat, L. F., Roesler, U., Neubauer, H., & El-Adawy, H. (2021). Whole-Genome Sequencing for Tracing the Genetic Diversity of Brucella abortus and Brucella melitensis Isolated from Livestock in Egypt. Pathogens, 10(6), 759. https://doi.org/10.3390/pathogens10060759