
Modulation of Neuroinflammation by the Gut Microbiota in Prion and Prion-Like Diseases


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Gut Microbiota and Neuroinflammation
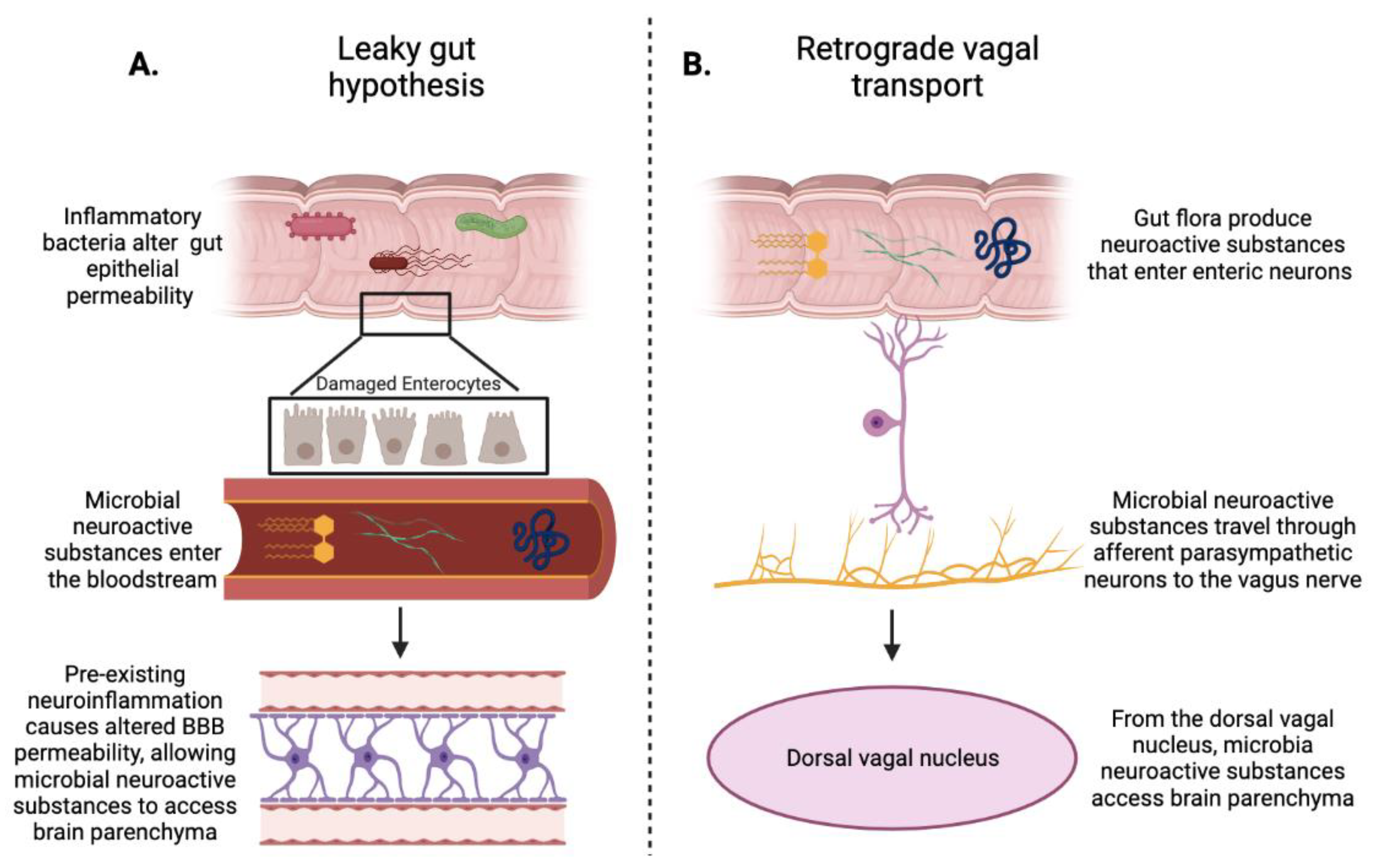
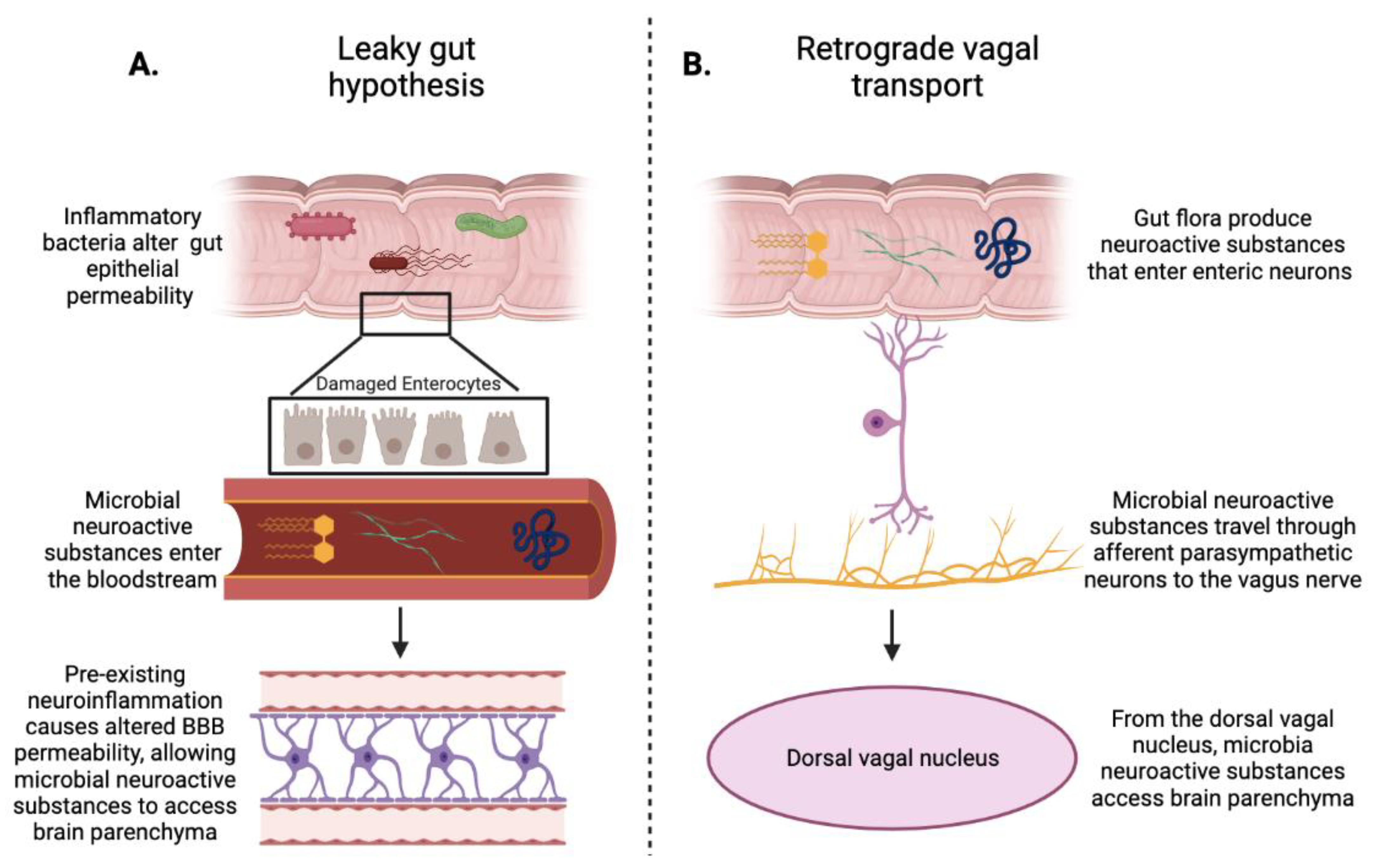
3. Gut Microbiota and Blood–Brain Barrier
4. Gut–Brain Axis
5. Parkinson’s Disease
6. Alzheimer’s Disease
7. Prion Diseases
8. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Singh, V.; Sadler, R.; Heindl, S.; Llovera, G.; Roth, S.; Benakis, C.; Liesz, A. The gut microbiome primes a cerebroprotective immune response after stroke. Br. J. Pharmacol. 2018, 38, 1293–1298. [Google Scholar] [CrossRef]
- Hoffman, J.D.; Yanckello, L.M.; Chlipala, G.; Hammond, T.C.; McCulloch, S.D.; Parikh, I.; Sun, S.; Morganti, J.M.; Green, S.J.; Lin, A.-L. Dietary inulin alters the gut microbiome, enhances systemic metabolism and reduces neuroinflammation in an APOE4 mouse model. PLoS ONE 2019, 14, e0221828. [Google Scholar] [CrossRef] [PubMed]
- Hocquart, M.; Lagier, J.-C.; Cassir, N.; Saidani, N.; Eldin, C.; Kerbaj, J.; Delord, M.; Valles, C.; Brouqui, P.; Raoult, D.; et al. Early Fecal Microbiota Transplantation Improves Survival in Severe Clostridium difficile Infections. Clin. Infect. Dis. 2018, 66, 645–650. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients with Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schain, M.; Kreisl, W.C. Neuroinflammation in Neurodegenerative Disorders—A Review. Curr. Neurol. Neurosci. Rep. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Ashford, B.A.; Boche, D.; Cooper-Knock, J.; Heath, P.R.; Simpson, J.E.; Highley, J. Review: Microglia in motor neuron disease. Neuropathol. Appl. Neurobiol. 2021, 47, 179–197. [Google Scholar] [CrossRef]
- Colombo, E.; Farina, C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016, 37, 608–620. [Google Scholar] [CrossRef]
- Erny, D.; De Angelis, A.L.H.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Chen, W.-W.; Zhang, X.; Huang, W.-J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Zhou, J. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Luissint, A.-C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.-O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Arellano, L.; Maldonado-Bernal, C. Helicobacter pyloriand neurological diseases: Married by the laws of inflammation. World J. Gastrointest. Pathophysiol. 2014, 5, 400–404. [Google Scholar] [CrossRef]
- Roubaud-Baudron, C.; Krolak-Salmon, P.; Quadrio, I.; Mégraud, F.; Salles, N. Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: Preliminary results. Neurobiol. Aging 2012, 33, 1009.e11–1009.e19. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.H.; Mahadeva, S.; Marras, C.; Thalha, A.M.; Kiew, C.K.; Yeat, C.M.; Ng, S.W.; Ang, S.P.; Chow, S.K.; Loke, M.F.; et al. Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Park. Relat. Disord. 2015, 21, 221–225. [Google Scholar] [CrossRef]
- Pascal, M.; Perez-Gordo, M.; Caballero, T.; Escribese, M.M.; Longo, M.N.L.; Luengo, O.; Manso, L.; Matheu, V.; Seoane, E.; Zamorano, M.; et al. Microbiome and Allergic Diseases. Front. Immunol. 2018, 9, 1584. [Google Scholar] [CrossRef]
- Wilkins, L.J.; Monga, M.; Miller, A.W. Defining Dysbiosis for a Cluster of Chronic Diseases. Sci. Rep. 2019, 9, 12918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.; Abel, T.; et al. Histone Deacetylase Inhibitors Enhance Memory and Synaptic Plasticity via CREB: CBP-Dependent Transcriptional Activation. J. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [CrossRef] [PubMed]
- Sperringer, J.E.; Addington, A.; Hutson, S.M. Branched-Chain Amino Acids and Brain Metabolism. Neurochem. Res. 2017, 42, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Fleck, A.-K.; Schuppan, D.; Wiendl, H.; Klotz, L. Gut–CNS-Axis as Possibility to Modulate Inflammatory Disease Activity—Implications for Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan Metabolism by Gut Microbiome and Gut-Brain-Axis: An in silico Analysis. Front. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [Green Version]
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution ofα-synuclein pathology. Neuropathol. Appl. Neurobiol. 2015, 42, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, D.B.; Parsons, R.B.; Ho, S.L.; Waring, R.H. The aetiology of idiopathic Parkinson’s disease. Mol. Pathol. 2001, 54, 369–380. [Google Scholar]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef]
- Wallen, Z.D.; Stone, W.J.; Factor, S.A.; Molho, E.; Zabetian, C.P.; Standaert, D.G.; Payami, H. Human-genome gut-microbiome interaction in Parkinson’s disease. Genet. Prepr. 2021. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nehru, B. Characterization of the lipopolysaccharide induced model of Parkinson’s disease: Role of oxidative stress and neuroinflammation. Neurochem. Int. 2015, 87, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.P.; Carvey, P.M.; Keshavarzian, A.; Shannon, K.M.; Shaikh, M.; Bakay, R.A.E.; Kordower, J.H. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov. Disord. 2014, 29, 999–1009. [Google Scholar] [CrossRef]
- Choi, J.G.; Kim, N.; Ju, I.G.; Eo, H.; Lim, S.-M.; Jang, S.-E.; Kim, D.-H.; Oh, M.S. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Sci. Rep. 2018, 8, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorecki, A.M.; Preskey, L.; Bakeberg, M.C.; Kenna, J.E.; Gildenhuys, C.; MacDougall, G.; Dunlop, S.A.; Mastaglia, F.L.; Akkari, P.A.; Koengten, F.; et al. Altered gut microbiome in Parkinson’s disease and the influence of lipopolysaccharide in a human α-synuclein over-expressing mouse model. Front. Neurosci. 2019, 13, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paiva, I.; Pinho, R.; Pavlou, M.A.; Hennion, M.; Wales, P.; Schütz, A.-L.; Rajput, A.; Szegő, É.M.; Kerimoglu, C.; Gerhardt, E.; et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum. Mol. Genet. 2017, 26, 2231–2246. [Google Scholar] [CrossRef]
- Laurent, R.S.; O’Brien, L.; Ahmad, S. Sodium butyrate improves locomotor impairment and early mortality in a rotenone-induced Drosophila model of Parkinson’s disease. Neuroscience 2013, 246, 382–390. [Google Scholar] [CrossRef] [Green Version]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids from Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Robinson, S.M. Minimal penetration of lipopolysaccharide across the murine blood–brain barrier. Brain Behav. Immun. 2010, 24, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, S.M.; Empadinhas, N. The Microbiome-Mitochondria Dance in Prodromal Parkinson’s Disease. Front. Physiol. 2018, 9, 471. [Google Scholar] [CrossRef] [Green Version]
- Cersosimo, M.G.; Benarroch, E.E. Pathological correlates of gastrointestinal dysfunction in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 559–564. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef]
- Sampson, T.R.; Challis, C.; Jain, N.; Moiseyenko, A.; Ladinsky, M.S.; Shastri, G.G.; Thron, T.; Needham, B.; Horvath, I.; Debelius, J.W.; et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. eLife 2020, 9, e53111. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef]
- Horsager, J.; Andersen, K.B.; Knudsen, K.; Skjærbæk, C.; Fedorova, T.D.; Okkels, N.; Schaeffer, E.; Bonkat, S.K.; Geday, J.; Otto, M.; et al. Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 2020, 143, 3077–3088. [Google Scholar] [CrossRef]
- Nair, A.T.; Ramachandran, V.; Joghee, N.M.; Antony, S.; Ramalingam, G. Gut Microbiota Dysfunction as Reliable Non-invasive Early Diagnostic Biomarkers in the Pathophysiology of Parkinson’s Disease: A Critical Review. J. Neurogastroenterol. Motil. 2018, 24, 30–42. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register–based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef] [Green Version]
- Tamtaji, O.R.; Taghizadeh, M.; Kakhaki, R.D.; Kouchaki, E.; Bahmani, F.; Borzabadi, S.; Oryan, S.; Mafi, A.; Asemi, Z. Clinical and metabolic response to probiotic administration in people with Parkinson’s disease: A randomized, double-blind, placebo-controlled trial. Clin. Nutr. 2019, 38, 1031–1035. [Google Scholar] [CrossRef]
- Sun, M.-F.; Zhu, Y.-L.; Zhou, Z.-L.; Jia, X.-B.; Xu, Y.-D.; Yang, Q.; Cui, C.; Shen, Y.-Q. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav. Immun. 2018, 70, 48–60. [Google Scholar] [CrossRef]
- Alcalay, R.N.; Gu, Y.; Mejia-Santana, H.; Cote, L.; Marder, K.S.; Scarmeas, N. The association between Mediterranean diet adherence and Parkinson’s disease. Mov. Disord. 2012, 27, 771–774. [Google Scholar] [CrossRef] [Green Version]
- Boulos, C.; Yaghi, N.; El Hayeck, R.; Heraoui, G.N.; Fakhoury-Sayegh, N. Nutritional Risk Factors, Microbiota and Parkinson’s Disease: What Is the Current Evidence? Nutrients 2019, 11, 1896. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.; Jacova, C.; Cummings, J.L.; DeKosky, S.; Barberger-Gateau, P.; Delacourte, A.; Frisoni, G.B.; Fox, N.; Galasko, D.; et al. Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol. 2010, 9, 1118–1127. [Google Scholar] [CrossRef]
- Rubio-Perez, J.M.; Morillas-Ruiz, J.M. A Review: Inflammatory Process in Alzheimer’s Disease, Role of Cytokines. Sci. World J. 2012, 2012, 756357. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.-Q.; Shen, L.-L.; Li, W.-W.; Fu, X.; Zeng, F.; Gui, L.; Lü, Y.; Cai, M.; Zhu, C.; Tan, Y.-L.; et al. Gut Microbiota is Altered in Patients with Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 63, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Kim, Y.; Choi, H.; Kim, W.; Park, S.; Lee, D.; Kim, D.K.; Kim, H.J.; Choi, H.; Hyun, D.-W.; et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 2020, 69, 283–294. [Google Scholar] [CrossRef]
- Brandscheid, C.; Schuck, F.; Reinhardt, S.; Schäfer, K.-H.; Pietrzik, C.U.; Grimm, M.; Hartmann, T.; Schwiertz, A.; Endres, K. Altered Gut Microbiome Composition and Tryptic Activity of the 5xFAD Alzheimer’s Mouse Model. J. Alzheimer’s Dis. 2017, 56, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, L.; Ji, H.-F. Alzheimer’s Disease Histological and Behavioral Manifestations in Transgenic Mice Correlate with Specific Gut Microbiome State. J. Alzheimer’s Dis. 2017, 56, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef] [PubMed]
- Syed, A.; Boles, B.R. Fold modulating function: Bacterial toxins to functional amyloids. Front. Microbiol. 2014, 5, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Dua, P.; Lukiw, W.J. Microbial Sources of Amyloid and Relevance to Amyloidogenesis and Alzheimer’s Disease (AD). J. Alzheimer’s Dis. Park. 2015, 5, 177. [Google Scholar] [CrossRef] [Green Version]
- Chapman, M.; Robinson, L.S.; Pinkner, J.S.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S.J. Role of Escherichia coli Curli Operons in Directing Amyloid Fiber Formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Asti, A.; Gioglio, L. Can a Bacterial Endotoxin be a Key Factor in the Kinetics of Amyloid Fibril Formation? J. Alzheimer’s Dis. 2014, 39, 169–179. [Google Scholar] [CrossRef]
- Deng, X.; Li, M.; Ai, W.; He, L.; Lu, D.; Patrylo, P.R.; Cai, H.; Luo, X.; Li, Z.; Yan, X.-X. Lipolysaccharide-Induced Neuroinflammation Is Associated with Alzheimer-Like Amyloidogenic Axonal Pathology and Dendritic Degeneration in Rats. Adv. Alzheimer’s Dis. 2014, 3, 78–93. [Google Scholar] [CrossRef] [Green Version]
- Tükel, Ç.; Wilson, R.P.; Nishimori, J.H.; Pezeshki, M.; Chromy, B.A.; Bäumler, A.J. Responses to Amyloids of Microbial and Host Origin Are Mediated through Toll-like Receptor 2. Cell Host Microbe 2009, 6, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Fassbender, K.; Walter, S.; Kühl, S.; Landmann, R.; Ishii, K.; Bertsch, T.; Stalder, A.K.; Muehlhauser, F.; Liu, Y.; Ulmer, A.J.; et al. The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J. 2003, 18, 203–205. [Google Scholar] [CrossRef]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Xu, J.; Ling, Y.; Wang, F.; Gong, T.; Yang, C.; Ye, S.; Ye, K.; Wei, D.; Song, Z.; et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 2019, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Hazan, S. Rapid improvement in Alzheimer’s disease symptoms following fecal microbiota transplantation: A case report. J. Int. Med. Res. 2020, 48, 030006052092593. [Google Scholar] [CrossRef]
- Yuan, T.; Ma, H.; Liu, W.; Niesen, D.B.; Shah, N.; Crews, R.; Rose, K.N.; Vattem, D.A.; Seeram, N.P. Pomegranate’s Neuroprotective Effects against Alzheimer’s Disease Are Mediated by Urolithins, Its Ellagitannin-Gut Microbial Derived Metabolites. ACS Chem. Neurosci. 2016, 7, 26–33. [Google Scholar] [CrossRef]
- Berti, V.; Murray, J.; Davies, M.; Spector, N.; Tsui, W.H.; Li, Y.; Williams, S.; Pirraglia, E.; Vallabhajosula, S.; McHugh, P.; et al. Nutrient patterns and brain biomarkers of Alzheimer’s disease in cognitively normal individuals. J. Nutr. Health Aging 2015, 19, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Murray, J.; Davies, M.; Williams, S.; Pirraglia, E.; Spector, N.; Tsui, W.H.; Li, Y.; Butler, T.; Osorio, R.; et al. Nutrient intake and brain biomarkers of Alzheimer’s disease in at-risk cognitively normal individuals: A cross-sectional neuroimaging pilot study. BMJ Open 2014, 4, e004850. [Google Scholar] [CrossRef] [Green Version]
- Stefani, M.; Rigacci, S. Beneficial properties of natural phenols: Highlight on protection against pathological conditions associated with amyloid aggregation: Phenols Protection against Amyloid Diseases. BioFactors 2014, 40, 482–493. [Google Scholar] [CrossRef]
- Rigacci, S.; Stefani, M. Nutraceuticals and amyloid neurodegenerative diseases: A focus on natural phenols. Expert Rev. Neurother. 2015, 15, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Rigacci, S. Olive Oil Phenols as Promising Multi-targeting Agents against Alzheimer’s Disease. In Natural Compounds as Therapeutic Agents for Amyloidogenic Diseases; Vassallo, N., Ed.; Springer International Publishing: Cham, Switzerland, 2015; Volume 863, pp. 1–20. [Google Scholar] [CrossRef]
- Ladiwala, A.R.A.; Dordick, J.S.; Tessier, P.M. Aromatic Small Molecules Remodel Toxic Soluble Oligomers of Amyloid β through Three Independent Pathways. J. Biol. Chem. 2011, 286, 3209–3218. [Google Scholar] [CrossRef] [Green Version]
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Nichino, D.; Relini, A.; Berti, A.; Stefani, M. Aβ(1-42) Aggregates into Non-Toxic Amyloid Assemblies in the Presence of the Natural Polyphenol Oleuropein Aglycon. Curr. Alzheimer Res. 2011, 8, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S.B. Nobel Lecture: Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [Green Version]
- Das, A.S.; Zou, W.-Q. Prions: Beyond a Single Protein. Clin. Microbiol. Rev. 2016, 29, 633–658. [Google Scholar] [CrossRef] [Green Version]
- Will, R.; Ironside, J.; Zeidler, M.; Estibeiro, K.; Cousens, S.; Smith, P.; Alperovitch, A.; Poser, S.; Pocchiari, M.; Hofman, A. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 1996, 347, 921–925. [Google Scholar] [CrossRef]
- Hill, A.; Desbruslais, M.; Joiner, S.; Sidle, K.C.L.; Gowland, I.; Collinge, J.; Doey, L.J.; Lantos, P. The same prion strain causes vCJD and BSE. Nature 1997, 389, 448–450. [Google Scholar] [CrossRef]
- Donaldson, D.S.; Mabbott, N.A. The influence of the commensal and pathogenic gut microbiota on prion disease pathogenesis. J. Gen. Virol. 2016, 97, 1725–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev, M.; Raine, C.S.; Levenson, S.M. Enhanced survival of germfree mice after infection with irradiated scrapie brain. Cell Mol. Life Sci. 1971, 27, 1358–1359. [Google Scholar] [CrossRef] [PubMed]
- Wade, W.; Dees, C.; German, T.; Marsh, R. Effect of Bacterial Flora and Mouse Genotype (Euthymic or Athymic) on Scrapie Pathogenesis. J. Leukoc. Biol. 1986, 40, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Bradford, B.M.; Tetlow, L.; Mabbott, N.A. Prion disease pathogenesis in the absence of the commensal microbiota. J. Gen. Virol. 2017, 98, 1943–1952. [Google Scholar] [CrossRef]
- Yang, D.; Zhao, D.; Shah, S.Z.A.; Wu, W.; Lai, M.; Zhang, X.; Li, J.; Guan, Z.; Zhao, H.; Li, W.; et al. Implications of gut microbiota dysbiosis and metabolic changes in prion disease. Neurobiol. Dis. 2020, 135, 104704. [Google Scholar] [CrossRef]
- Obrenovich, M.E.M. Leaky Gut, Leaky Brain? Microorganisms 2018, 6, 107. [Google Scholar] [CrossRef] [Green Version]
- Marizzoni, M.; Cattaneo, A.; Mirabelli, P.; Festari, C.; Lopizzo, N.; Nicolosi, V.; Mombelli, E.; Mazzelli, M.; Luongo, D.; Naviglio, D.; et al. Short-Chain Fatty Acids and Lipopolysaccharide as Mediators Between Gut Dysbiosis and Amyloid Pathology in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 78, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Van Beurden, Y.H.; De Groot, P.F.; Van Nood, E.; Nieuwdorp, M.; Keller, J.J.; Goorhuis, A. Complications, effectiveness, and long term follow-up of fecal microbiota transfer by nasoduodenal tube for treatment of recurrent Clostridium difficile infection. United Eur. Gastroenterol. J. 2017, 5, 868–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcella, C.; Cui, B.; Kelly, C.R.; Ianiro, G.; Cammarota, G.; Zhang, F. Systematic review: The global incidence of faecal microbiota transplantation-related adverse events from 2000 to 2020. Aliment. Pharmacol. Ther. 2020, 53, 33–42. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trichka, J.; Zou, W.-Q. Modulation of Neuroinflammation by the Gut Microbiota in Prion and Prion-Like Diseases. Pathogens 2021, 10, 887. https://doi.org/10.3390/pathogens10070887
Trichka J, Zou W-Q. Modulation of Neuroinflammation by the Gut Microbiota in Prion and Prion-Like Diseases. Pathogens. 2021; 10(7):887. https://doi.org/10.3390/pathogens10070887
Chicago/Turabian StyleTrichka, Josephine, and Wen-Quan Zou. 2021. "Modulation of Neuroinflammation by the Gut Microbiota in Prion and Prion-Like Diseases" Pathogens 10, no. 7: 887. https://doi.org/10.3390/pathogens10070887
APA StyleTrichka, J., & Zou, W. -Q. (2021). Modulation of Neuroinflammation by the Gut Microbiota in Prion and Prion-Like Diseases. Pathogens, 10(7), 887. https://doi.org/10.3390/pathogens10070887