
Coevolved Multidrug-Resistant HIV-1 Protease and Reverse Transcriptase Influences Integrase Drug Susceptibility and Replication Fitness


Abstract
:1. Introduction
2. Results
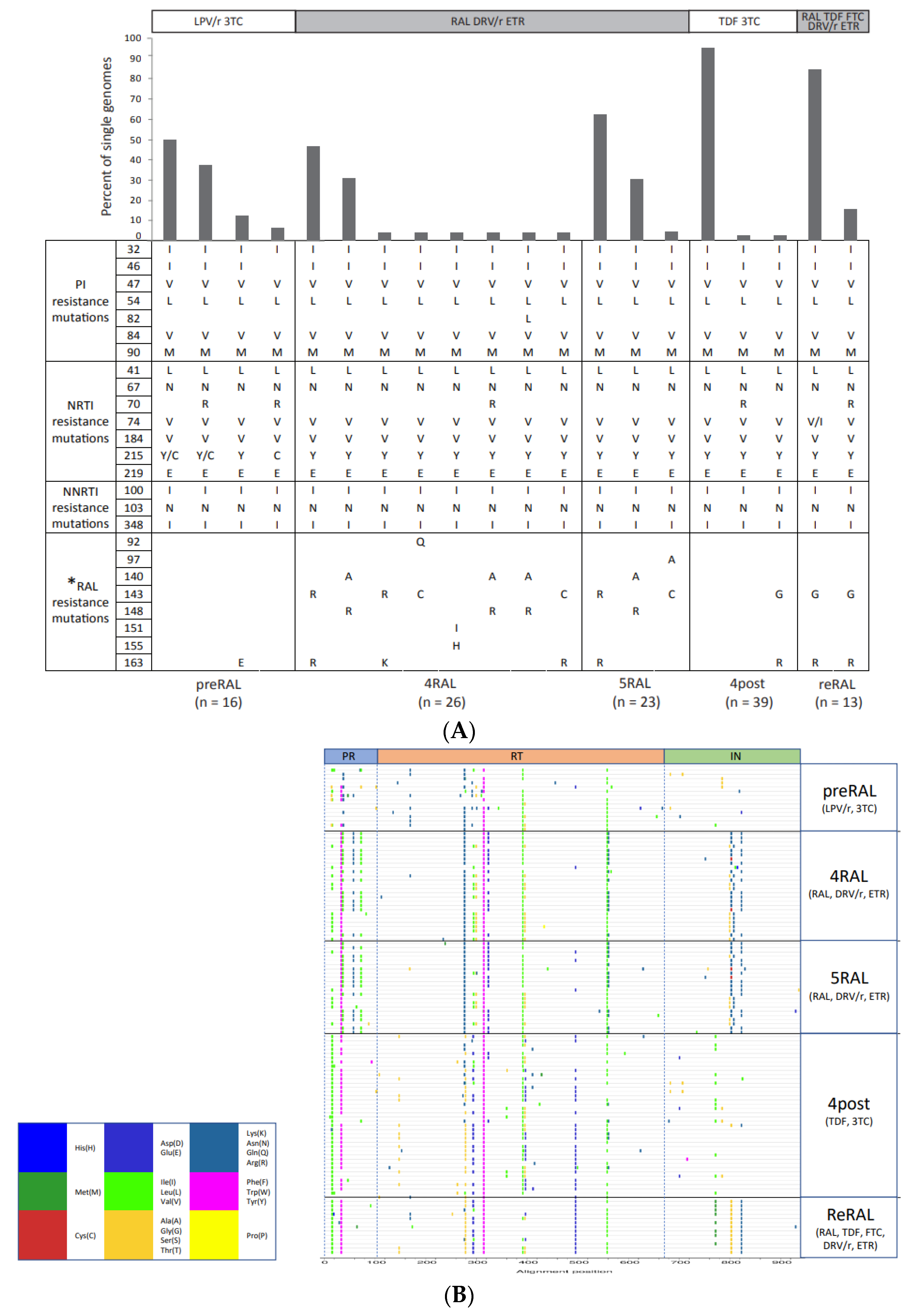
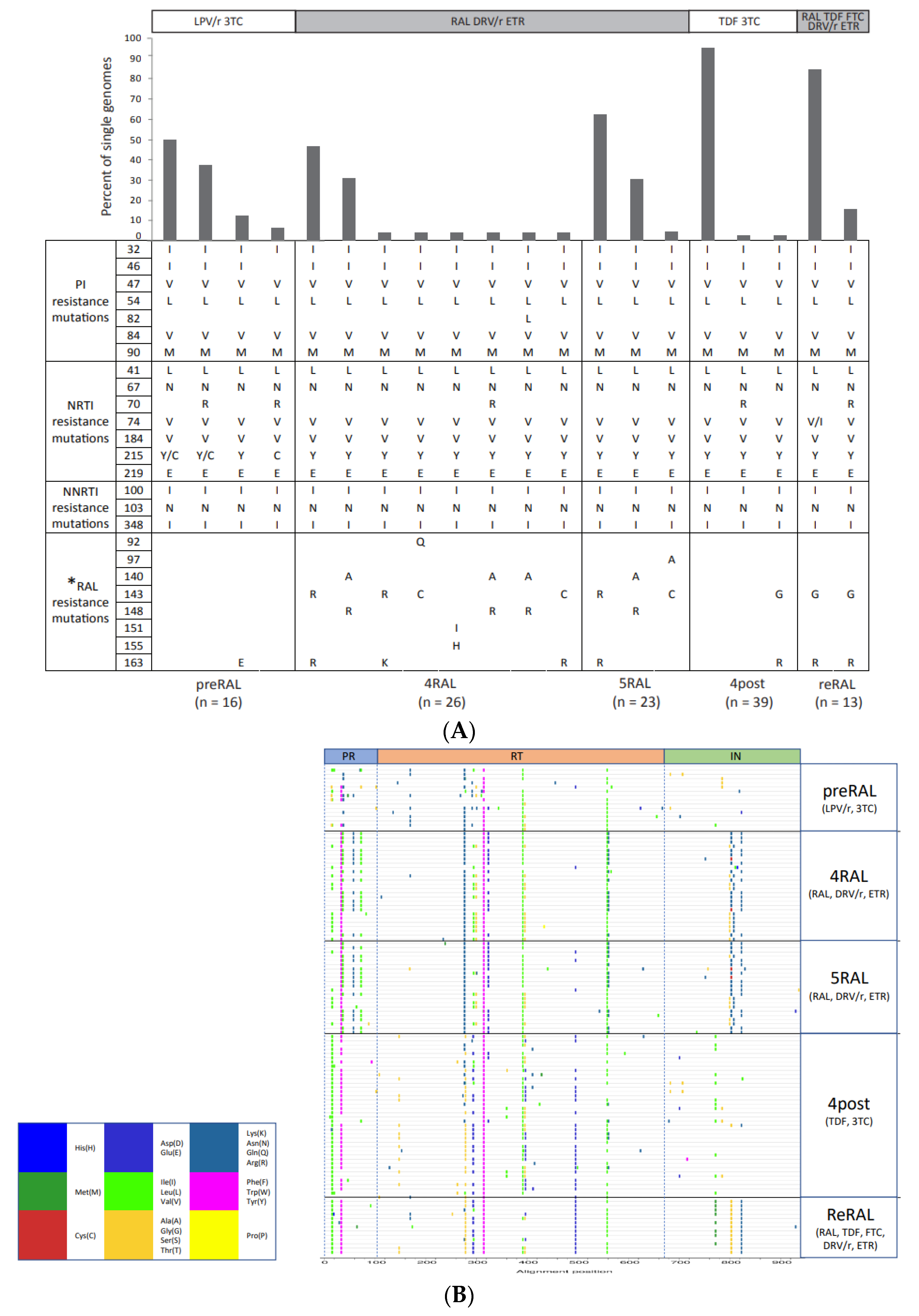
2.1. Single-Genome Sequencing of Full-Length HIV-1 pol Gene in Longitudinal Samples from a Patient Failing RAL-Containing Therapy
2.2. The Development and Linkage of Drug Resistance Mutations in Full-Length HIV-1 pol Gene
2.3. Intrapatient Evolution of InSTi Susceptibility
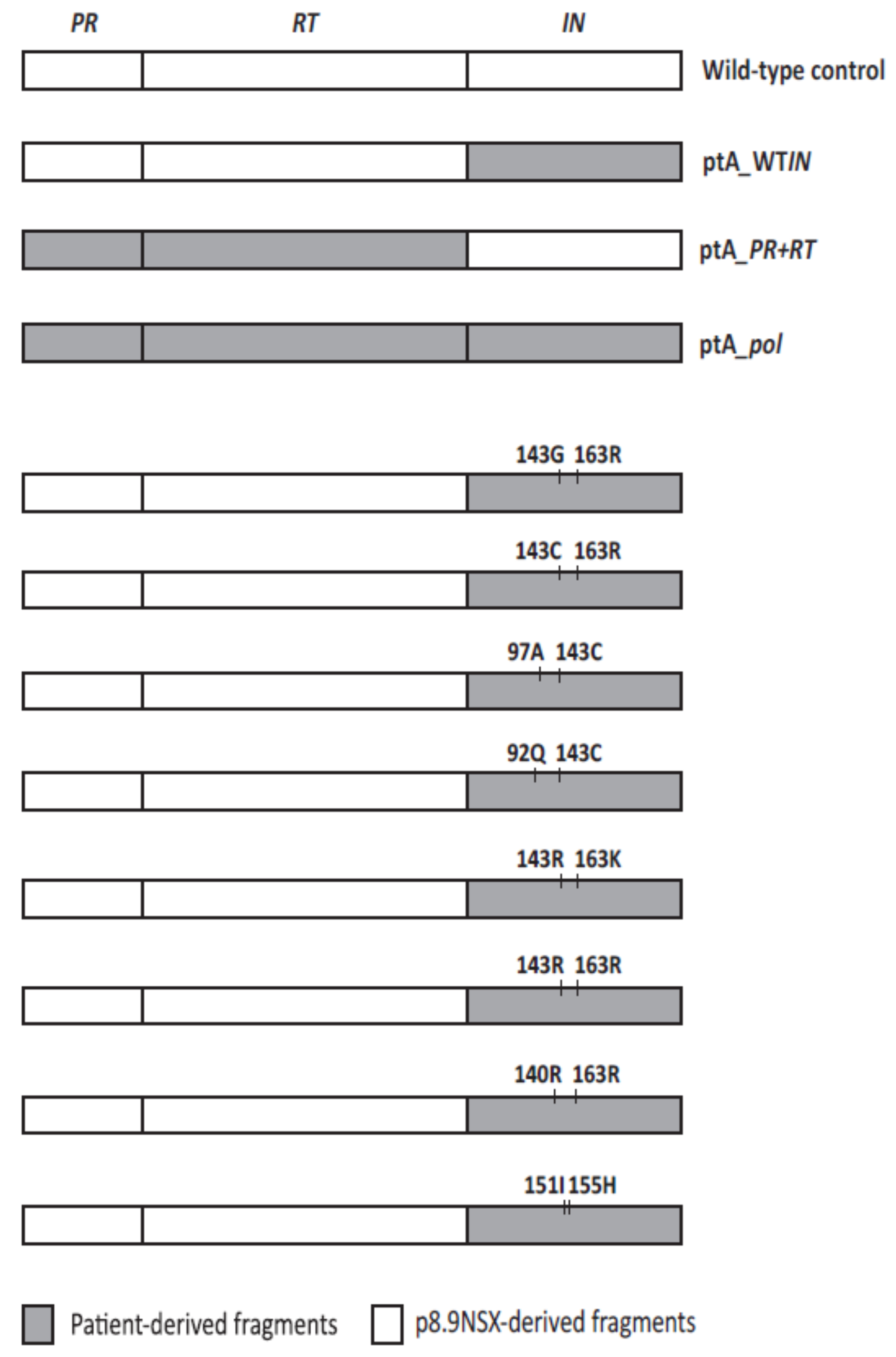
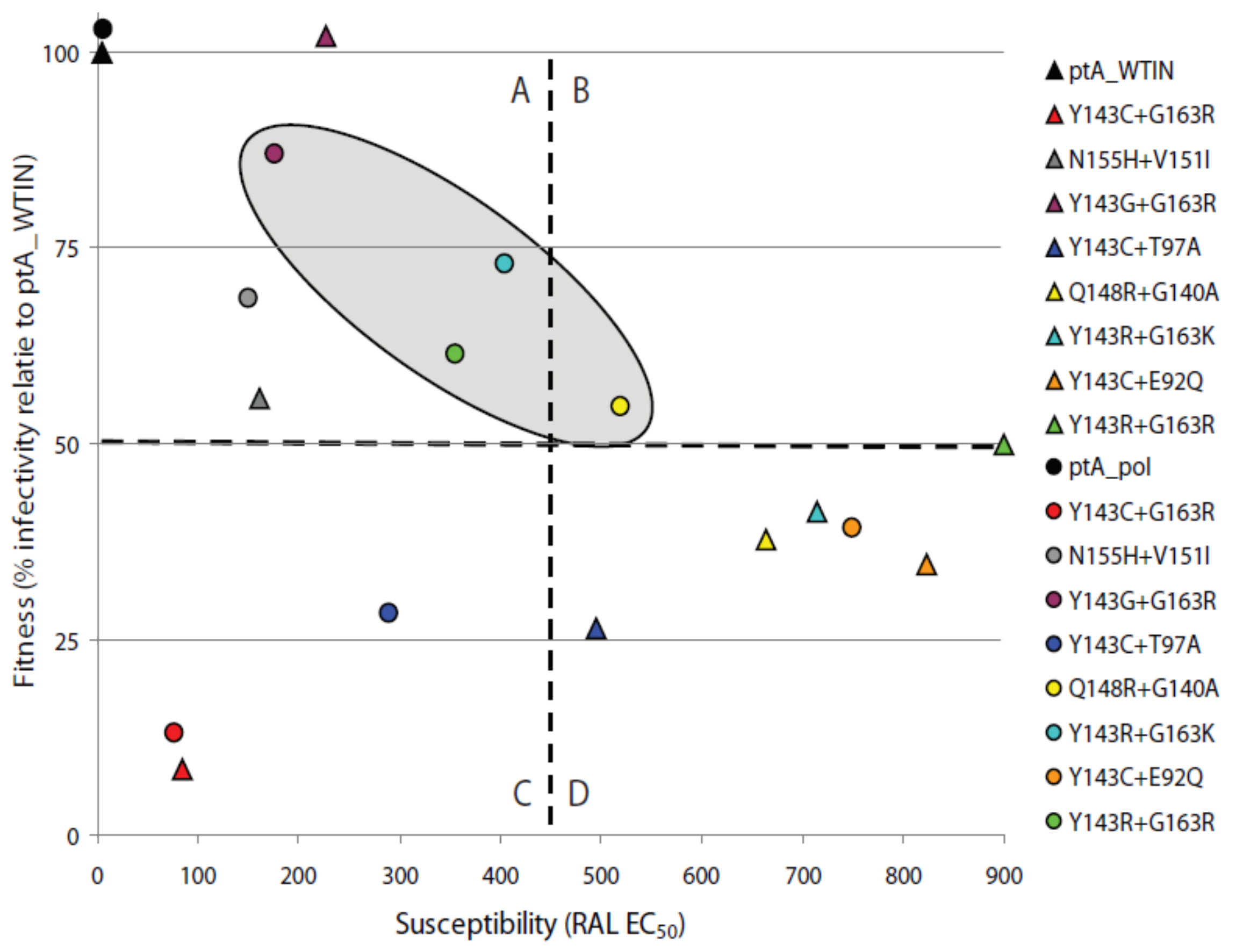
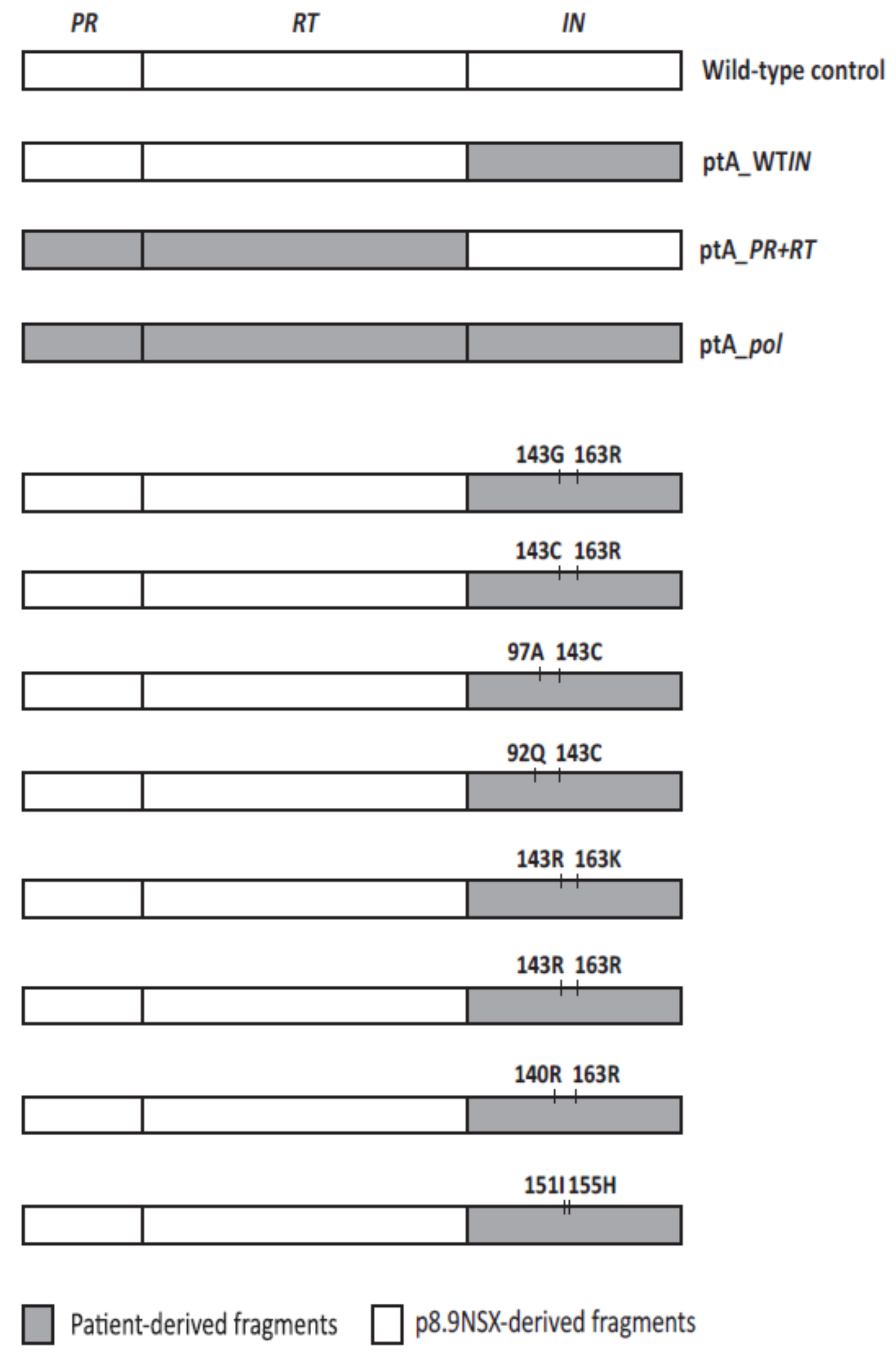
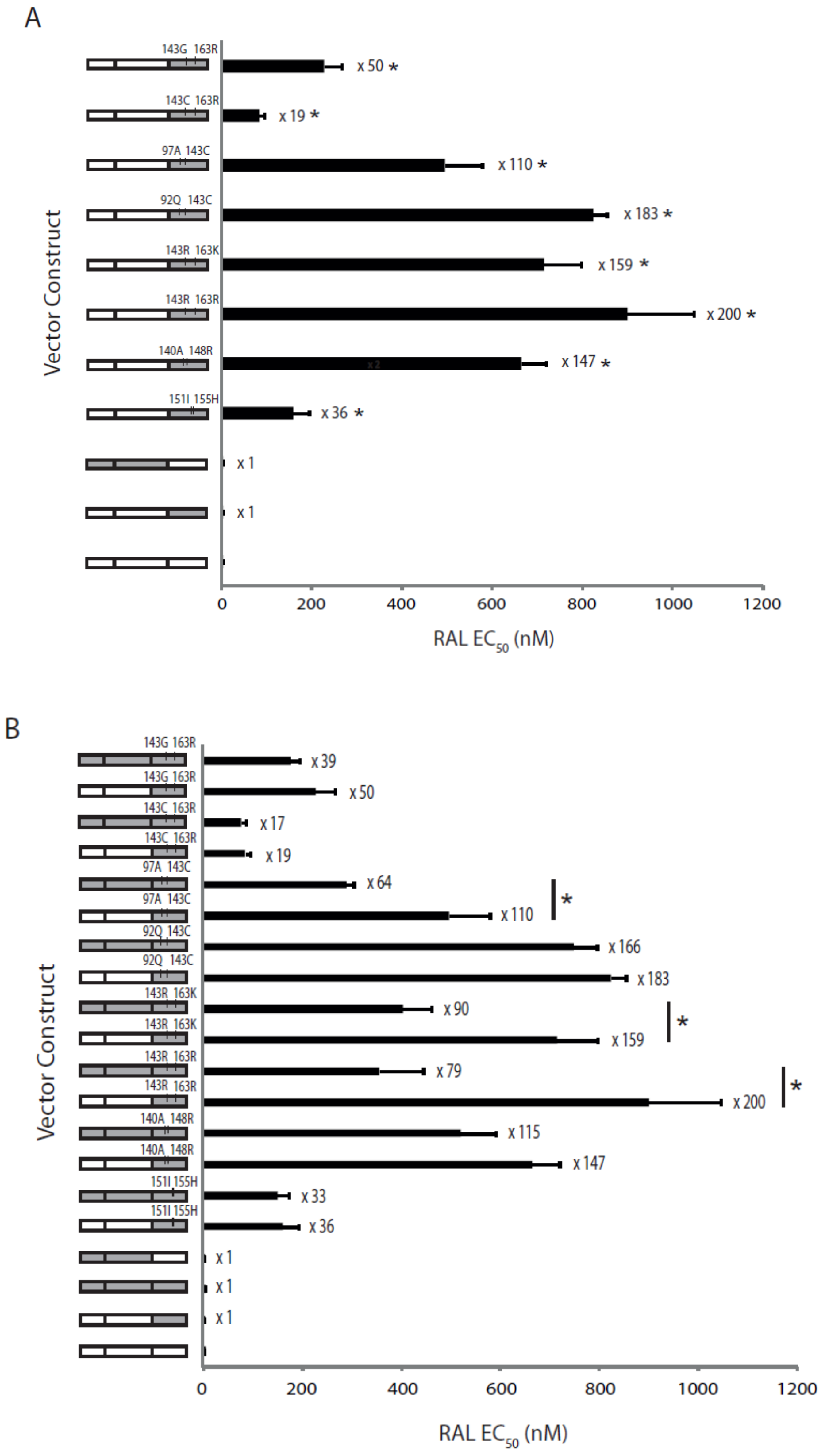
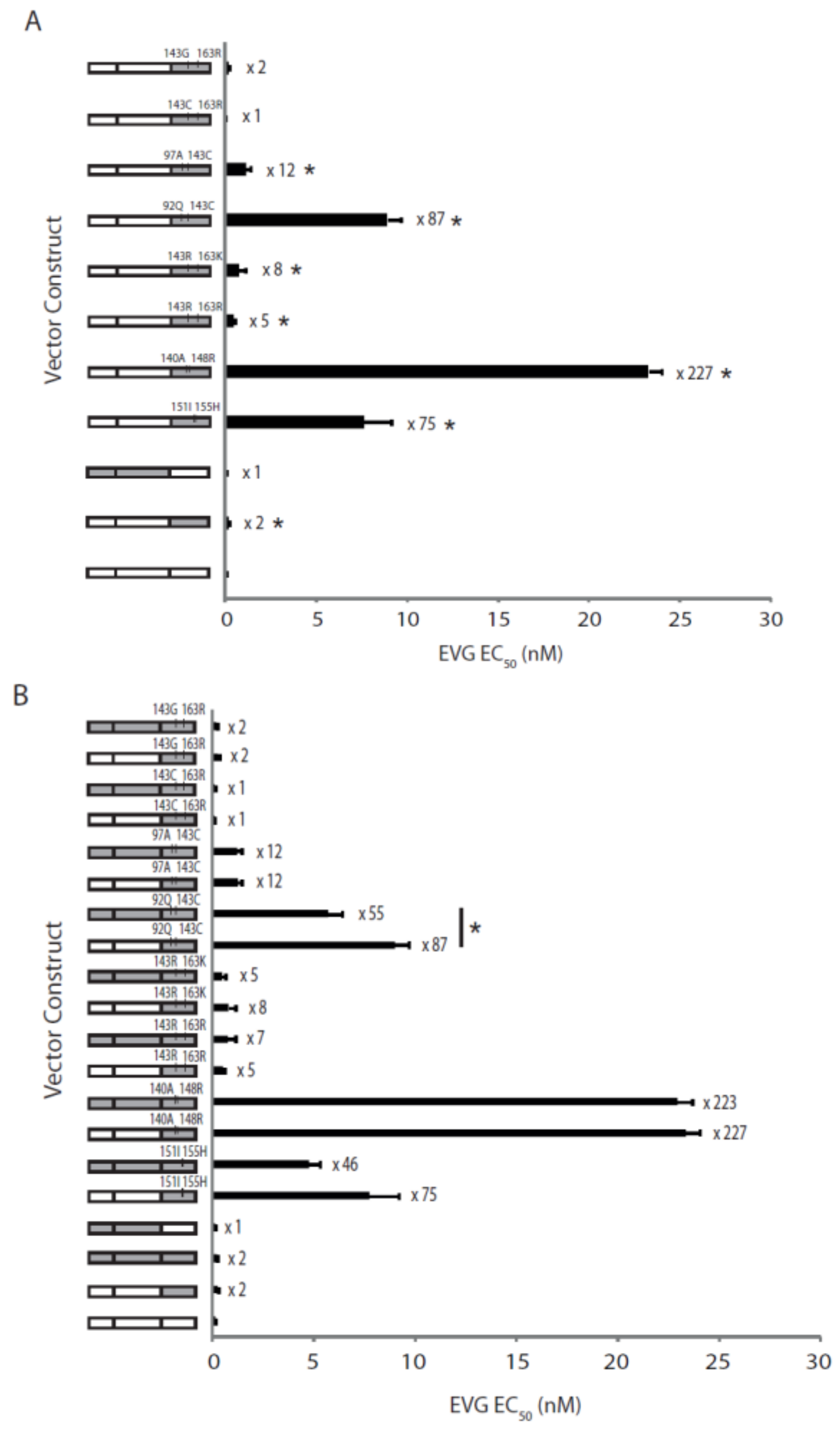
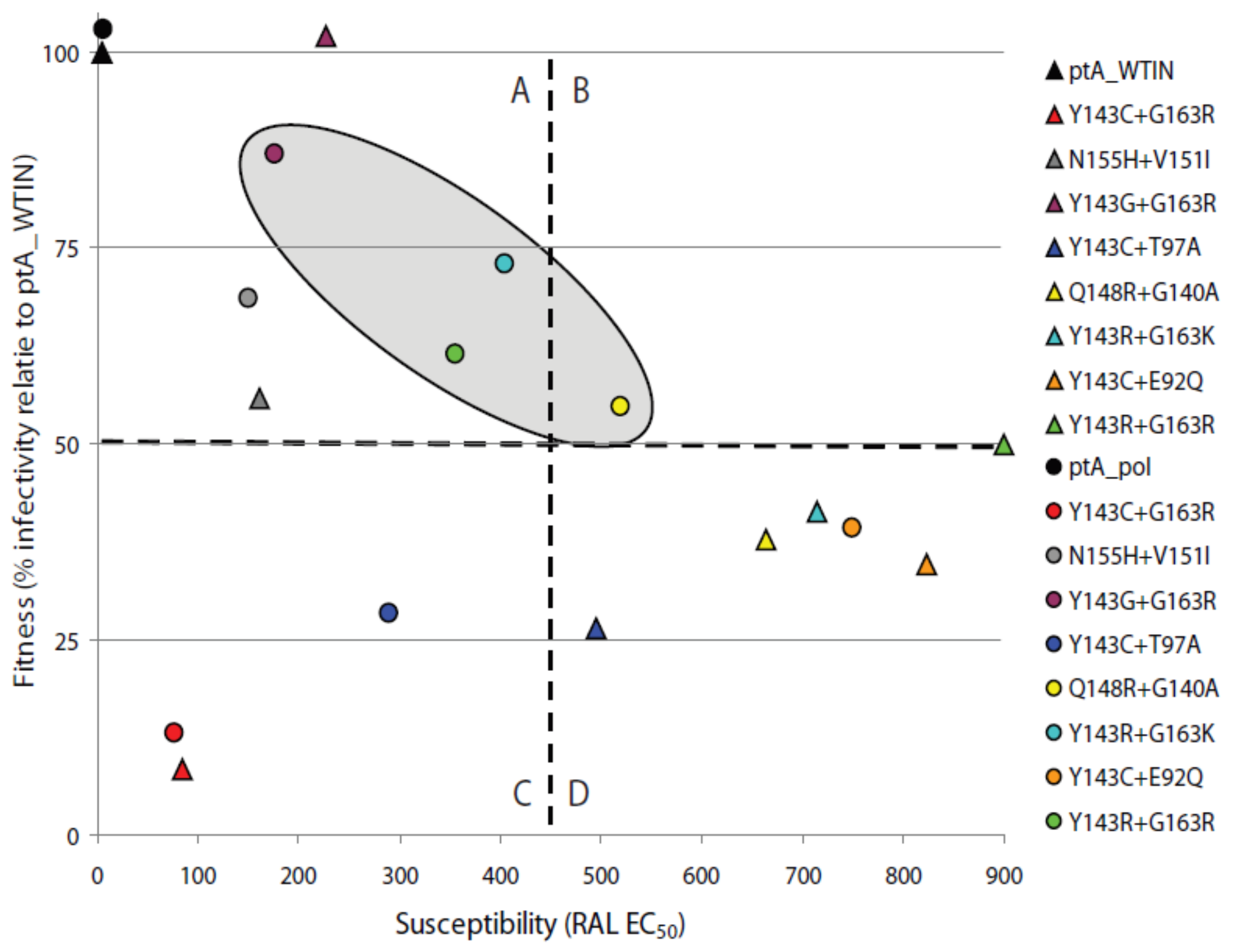
2.4. The Effects of Coevolved PR and RT Genes on Susceptibility to InSTIs
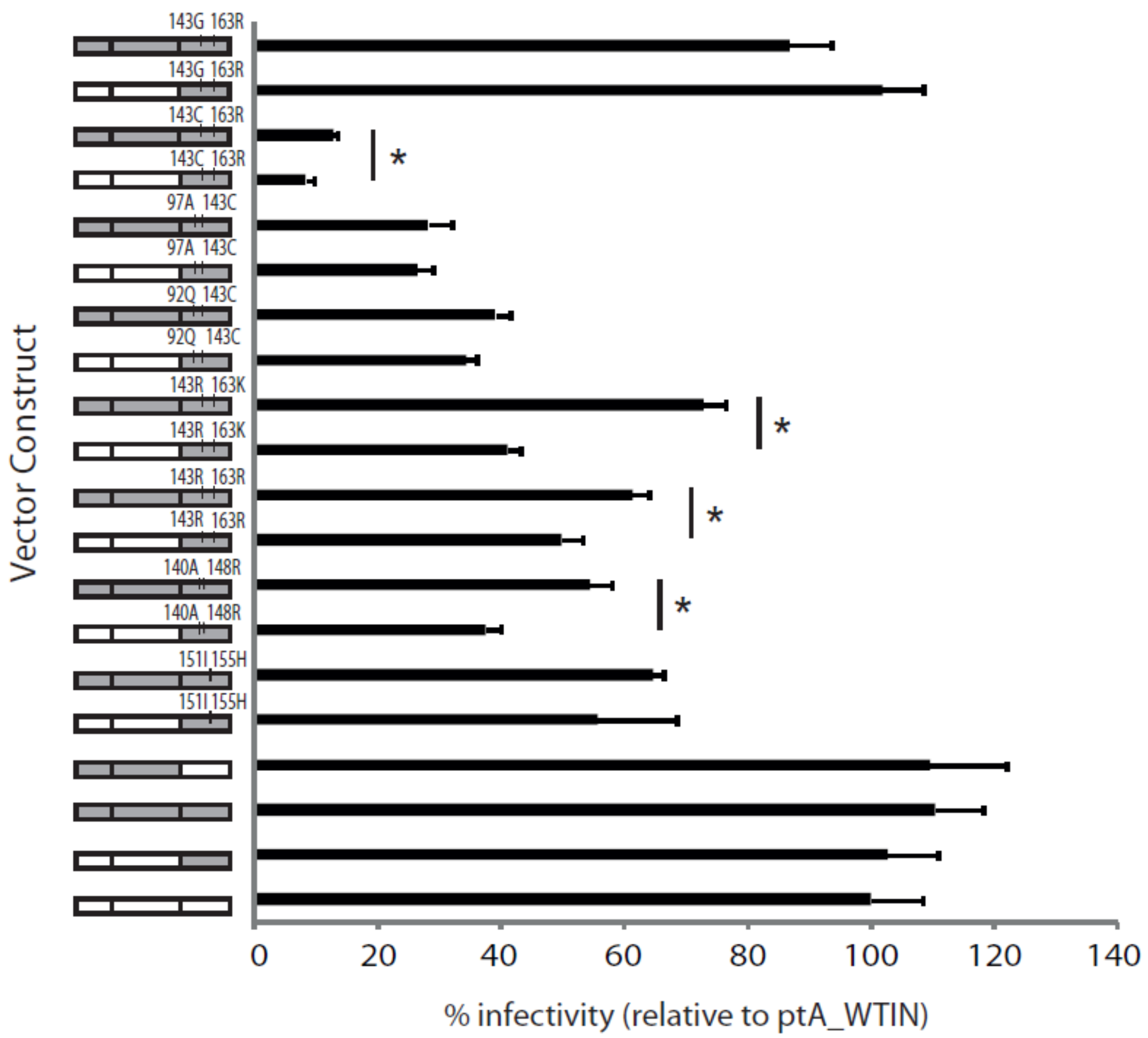
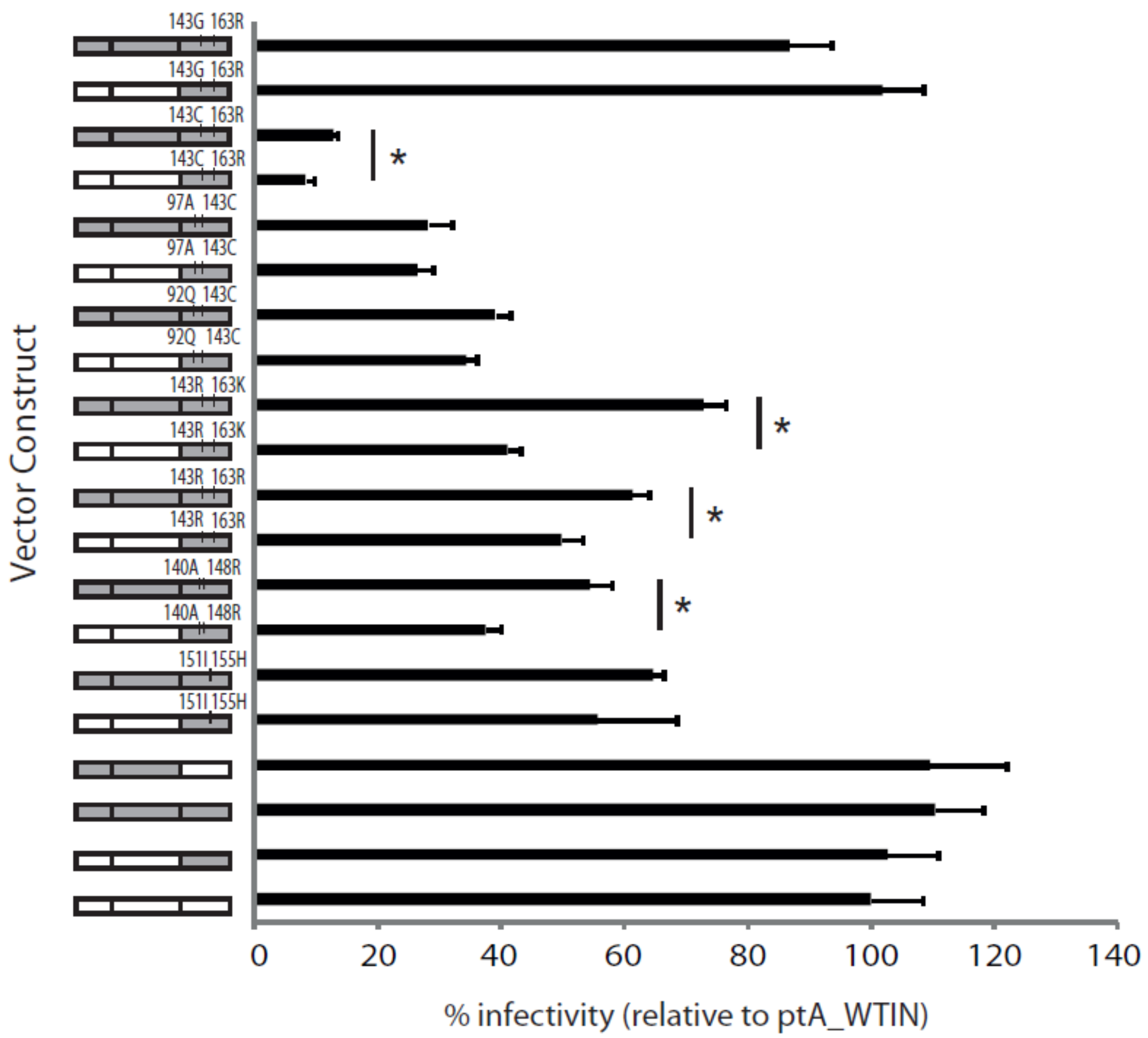
2.5. The Effects of Patient-Derived pol Gene Fragments on Viral Replicative Fitness
3. Discussion
4. Materials and Methods
4.1. Clinical Samples
4.2. Single-Genome Sequencing
4.3. Single-Replication Cycle Drug Susceptibility Assay
4.4. Antiretroviral Drugs
4.5. Statistical Analyses
4.6. Nucleotide Sequence Accession Numbers
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS Joint United Nations Programme on HIV/AIDS. UNAIDS Data 2020; UNAIDS: Geneva, Switzerland, 2020. [Google Scholar]
- Grobler, J.A.; Stillmock, K.; Hu, B.; Witmer, M.; Felock, P.; Espeseth, A.S.; Wolfe, A.; Egbertson, M.; Bourgeois, M.; Melamed, J.; et al. Diketo acid inhibitor mechanism and HIV-1 integrase: Implications for metal binding in the active site of phosphotransferase enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 6661–6666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchand, C.; Johnson, A.A.; Karki, R.G.; Pais, G.C.; Zhang, X.; Cowansage, K.; Patel, T.A.; Nicklaus, M.C.; Burke, T.R., Jr.; Pommier, Y. Metal-dependent inhibition of HIV-1 integrase by beta-diketo acids and resistance of the soluble double-mutant (F185K/C280S). Mol. Pharmacol. 2003, 64, 600–609. [Google Scholar] [CrossRef] [Green Version]
- Arribas, J.R.; Eron, J. Advances in antiretroviral therapy. Curr. Opin. HIV AIDS 2013, 8, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Steigbigel, R.T.; Cooper, D.A.; Teppler, H.; Eron, J.J.; Gatell, J.M.; Kumar, P.N.; Rockstroh, J.K.; Schechter, M.; Katlama, C.; Markowitz, M.; et al. Long-term efficacy and safety of Raltegravir combined with optimized background therapy in treatment-experienced patients with drug-resistant HIV infection: Week 96 results of the BENCHMRK 1 and 2 Phase III trials. Clin. Infect. Dis. 2010, 50, 605–612. [Google Scholar] [CrossRef]
- Churchill, D.; Waters, L.; Ahmed, N.; Angus, B.; Boffito, M.; Bower, M.; Dunn, D.; Edwards, S.; Emerson, C.; Fidler, S.; et al. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2015. HIV Med. 2016, 17 (Suppl. 4), s2–s104. [Google Scholar] [CrossRef]
- European AIDS Clinical Society. Guidelines 2018. Available online: https://www.eacsociety.org/files/2018_guidelines-9.1-english.pdf (accessed on 4 November 2019).
- Mbisa, J.L. Antiviral Resistance Testing. eLS. 2013. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/9780470015902.a0024795 (accessed on 10 June 2021).
- Avila-Rios, S.; Parkin, N.; Swanstrom, R.; Paredes, R.; Shafer, R.; Ji, H.; Kantor, R. Next-Generation Sequencing for HIV Drug Resistance Testing: Laboratory, Clinical, and Implementation Considerations. Viruses 2020, 12, 617. [Google Scholar] [CrossRef]
- Hertogs, K.; de Bethune, M.P.; Miller, V.; Ivens, T.; Schel, P.; Van Cauwenberge, A.; Van Den Eynde, C.; Van Gerwen, V.; Azijn, H.; Van Houtte, M.; et al. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicrob. Agents Chemother. 1998, 42, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Kellam, P.; Larder, B.A. Recombinant virus assay: A rapid, phenotypic assay for assessment of drug susceptibility of human immunodeficiency virus type 1 isolates. Antimicrob. Agents Chemother. 1994, 38, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Petropoulos, C.J.; Parkin, N.T.; Limoli, K.L.; Lie, Y.S.; Wrin, T.; Huang, W.; Tian, H.; Smith, D.; Winslow, G.A.; Capon, D.J.; et al. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 2000, 44, 920–928. [Google Scholar] [CrossRef] [Green Version]
- Parry, C.M.; Kohli, A.; Boinett, C.J.; Towers, G.J.; McCormick, A.L.; Pillay, D. Gag determinants of fitness and drug susceptibility in protease inhibitor-resistant human immunodeficiency virus type 1. J. Virol. 2009, 83, 9094–9101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.; Vazquez, A.C.; Winner, D.; Rose, J.D.; Wylie, D.; Rhea, A.M.; Henry, K.; Pappas, J.; Wright, A.; Mohamed, N.; et al. Novel method for simultaneous quantification of phenotypic resistance to maturation, protease, reverse transcriptase, and integrase HIV inhibitors based on 3'Gag(p2/p7/p1/p6)/PR/RT/INT-recombinant viruses: A useful tool in the multitarget era of antiretroviral therapy. Antimicrob. Agents Chemother. 2011, 55, 3729–3742. [Google Scholar]
- Winters, M.A.; Lloyd, R.M., Jr.; Shafer, R.W.; Kozal, M.J.; Miller, M.D.; Holodniy, M. Development of elvitegravir resistance and linkage of integrase inhibitor mutations with protease and reverse transcriptase resistance mutations. PLoS ONE 2012, 7, e40514. [Google Scholar] [CrossRef] [Green Version]
- Van Baelen, K.; Rondelez, E.; Van Eygen, V.; Arien, K.; Clynhens, M.; Van den Zegel, P.; Winters, B.; Stuyver, L.J. A combined genotypic and phenotypic human immunodeficiency virus type 1 recombinant virus assay for the reverse transcriptase and integrase genes. J. Virol. Methods 2009, 161, 231–239. [Google Scholar] [CrossRef]
- Low, A.; Prada, N.; Topper, M.; Vaida, F.; Castor, D.; Mohri, H.; Hazuda, D.; Muesing, M.; Markowitz, M. Natural polymorphisms of human immunodeficiency virus type 1 integrase and inherent susceptibilities to a panel of integrase inhibitors. Antimicrob. Agents Chemother. 2009, 53, 4275–4282. [Google Scholar] [CrossRef] [Green Version]
- Garrido, C.; Villacian, J.; Zahonero, N.; Pattery, T.; Garcia, F.; Gutierrez, F.; Caballero, E.; Van Houtte, M.; Soriano, V.; de Mendoza, C.; et al. Broad phenotypic cross-resistance to elvitegravir in HIV-infected patients failing on raltegravir-containing regimens. Antimicrob. Agents Chemother. 2012, 56, 2873–2878. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, C.; Karmochkine, M.; Laureillard, D.; Tisserand, P.; Belec, L.; Weiss, L.; Si-Mohamed, A.; Piketty, C. Drug resistance profiles for the HIV integrase gene in patients failing raltegravir salvage therapy. HIV Med. 2008, 9, 765–770. [Google Scholar] [CrossRef]
- Malet, I.; Delelis, O.; Soulie, C.; Wirden, M.; Tchertanov, L.; Mottaz, P.; Peytavin, G.; Katlama, C.; Mouscadet, J.F.; Calvez, V.; et al. Quasispecies variant dynamics during emergence of resistance to raltegravir in HIV-1-infected patients. J. Antimicrob. Chemother. 2009, 63, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Reigadas, S.; Anies, G.; Masquelier, B.; Calmels, C.; Stuyver, L.J.; Parissi, V.; Fleury, H.; Andreola, M.L. The HIV-1 integrase mutations Y143C/R are an alternative pathway for resistance to Raltegravir and impact the enzyme functions. PLoS ONE 2010, 5, e10311. [Google Scholar] [CrossRef] [PubMed]
- Fransen, S.; Gupta, S.; Danovich, R.; Hazuda, D.; Miller, M.; Witmer, M.; Petropoulos, C.J.; Huang, W. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J. Virol. 2009, 83, 11440–11446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferns, R.B.; Kirk, S.; Bennett, J.; Cook, P.M.; Williams, I.; Edwards, S.; Pillay, D. The dynamics of appearance and disappearance of HIV-1 integrase mutations during and after withdrawal of raltegravir therapy. AIDS 2009, 23, 2159–2164. [Google Scholar] [CrossRef]
- Canducci, F.; Marinozzi, M.C.; Sampaolo, M.; Boeri, E.; Spagnuolo, V.; Gianotti, N.; Castagna, A.; Paolucci, S.; Baldanti, F.; Lazzarin, A.; et al. Genotypic/phenotypic patterns of HIV-1 integrase resistance to raltegravir. J. Antimicrob. Chemother. 2010, 65, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Baldanti, F.; Paolucci, S.; Gulminetti, R.; Brandolini, M.; Barbarini, G.; Maserati, R. Early emergence of raltegravir resistance mutations in patients receiving HAART salvage regimens. J. Med. Virol. 2010, 82, 116–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fun, A.; Van Baelen, K.; van Lelyveld, S.F.; Schipper, P.J.; Stuyver, L.J.; Wensing, A.M.; Nijhuis, M. Mutation Q95K enhances N155H-mediated integrase inhibitor resistance and improves viral replication capacity. J. Antimicrob. Chemother. 2010, 65, 2300–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delelis, O.; Malet, I.; Na, L.; Tchertanov, L.; Calvez, V.; Marcelin, A.G.; Subra, F.; Deprez, E.; Mouscadet, J.F. The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation. Nucleic Acids Res. 2009, 37, 1193–1201. [Google Scholar] [CrossRef]
- Metifiot, M.; Maddali, K.; Naumova, A.; Zhang, X.; Marchand, C.; Pommier, Y. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry 2010, 49, 3715–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, K.; Wakasa-Morimoto, C.; Kobayashi, M.; Miki, S.; Noshi, T.; Seki, T.; Kanamori-Koyama, M.; Kawauchi, S.; Suyama, A.; Fujishita, T.; et al. Secondary mutations in viruses resistant to HIV-1 integrase inhibitors that restore viral infectivity and replication kinetics. Antivir. Res. 2009, 81, 141–146. [Google Scholar] [CrossRef]
- Delelis, O.; Thierry, S.; Subra, F.; Simon, F.; Malet, I.; Alloui, C.; Sayon, S.; Calvez, V.; Deprez, E.; Marcelin, A.G.; et al. Impact of Y143 HIV-1 integrase mutations on resistance to raltegravir in vitro and in vivo. Antimicrob. Agents Chemother. 2010, 54, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Mbisa, J.L.; Martin, S.A.; Cane, P.A. Patterns of resistance development with integrase inhibitors in HIV. Infect. Drug Resist. 2011, 4, 65–76. [Google Scholar]
- Mbisa, J.L.; Gupta, R.K.; Kabamba, D.; Mulenga, V.; Kalumbi, M.; Chintu, C.; Parry, C.M.; Gibb, D.M.; Walker, S.A.; Cane, P.A.; et al. The evolution of HIV-1 reverse transcriptase in route to acquisition of Q151M multi-drug resistance is complex and involves mutations in multiple domains. Retrovirology 2011, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Malet, I.; Delelis, O.; Nguyen, T.; Leducq, V.; Abdi, B.; Morand-Joubert, L.; Calvez, V.; Marcelin, A.G. Variability of the HIV-1 3′ polypurine tract (3′PPT) region and implication in integrase inhibitor resistance. J. Antimicrob. Chemother. 2019, 74, 3440–3444. [Google Scholar] [CrossRef]
- Hu, Z.; Kuritzkes, D.R. Altered viral fitness and drug susceptibility in HIV-1 carrying mutations that confer resistance to nonnucleoside reverse transcriptase and integrase strand transfer inhibitors. J. Virol. 2014, 88, 9268–9276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.F.; Frantzell, A.; Chappey, C.; Petropoulos, C.; Huang, W. Combinations of primary NNRTI- and integrase inhibitor-resistance mutations do not alter HIV-1 drug susceptibility but impair replication capacity. In Proceedings of the 16th Conference on Retroviruses and Opportunistic Infections, Montreal, QC, Canada, 8–11 February 2009. [Google Scholar]
- Van Duyne, R.; Kuo, L.S.; Pham, P.; Fujii, K.; Freed, E.O. Mutations in the HIV-1 envelope glycoprotein can broadly rescue blocks at multiple steps in the virus replication cycle. Proc. Natl. Acad. Sci. USA 2019, 116, 9040–9049. [Google Scholar] [CrossRef] [Green Version]
- Malet, I.; Subra, F.; Charpentier, C.; Collin, G.; Descamps, D.; Calvez, V.; Marcelin, A.G.; Delelis, O. Mutations Located outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. MBio 2017, 8, e00922-17. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T., Jr.; Dewar, R.L.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laethem, K.; Schrooten, Y.; Covens, K.; Dekeersmaeker, N.; De Munter, P.; Van Wijngaerden, E.; Van Ranst, M.; Vandamme, A.M. A genotypic assay for the amplification and sequencing of integrase from diverse HIV-1 group M subtypes. J. Virol. Methods 2008, 153, 176–181. [Google Scholar] [CrossRef]
- Gupta, R.K.; Kohli, A.; McCormick, A.L.; Towers, G.J.; Pillay, D.; Parry, C.M. Full-length HIV-1 Gag determines protease inhibitor susceptibility within in vitro assays. AIDS 2010, 24, 1651–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainbridge, J.W.; Stephens, C.; Parsley, K.; Demaison, C.; Halfyard, A.; Thrasher, A.J.; Ali, R.R. In vivo gene transfer to the mouse eye using an HIV-based lentiviral vector; efficient long-term transduction of corneal endothelium and retinal pigment epithelium. Gene Ther. 2001, 8, 1665–1668. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.; Temperton, N.J.; Marston, D.A.; McElhinney, L.M.; Fooks, A.R.; Weiss, R.A. Investigating antibody neutralization of lyssaviruses using lentiviral pseudotypes: A cross-species comparison. J. Gen. Virol. 2008, 89 Pt 9, 2204–2213. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Months Before or After Initiation of RAL Therapy | Viral Load (Copies/mL) | CD4 Count (Cells/mm3) | Antiretroviral Treatment | Number of Single Genomes |
|---|---|---|---|---|---|
| A | −3 (preRAL) | 59,000 | 150 | LPVr, 3TC | 16 |
| 2 (2RAL) | 140 | 230 | DRVr, ETR, RAL | Na a | |
| 4 (4RAL) | 39,000 | 280 | DRVr, ETR, RAL | 26 | |
| 5 (5RAL) | 63,000 | 200 | DRVr, ETR, RAL | 23 | |
| 9 (4post) | 77,300 | 140 | TDF, 3TC | 39 | |
| 14 (ReRAL) | 1900 | 140 | DRVr, TDF, FTC, ETR, RAL | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, S.A.; Cane, P.A.; Pillay, D.; Mbisa, J.L. Coevolved Multidrug-Resistant HIV-1 Protease and Reverse Transcriptase Influences Integrase Drug Susceptibility and Replication Fitness. Pathogens 2021, 10, 1070. https://doi.org/10.3390/pathogens10091070
Martin SA, Cane PA, Pillay D, Mbisa JL. Coevolved Multidrug-Resistant HIV-1 Protease and Reverse Transcriptase Influences Integrase Drug Susceptibility and Replication Fitness. Pathogens. 2021; 10(9):1070. https://doi.org/10.3390/pathogens10091070
Chicago/Turabian StyleMartin, Supang A., Patricia A. Cane, Deenan Pillay, and Jean L. Mbisa. 2021. "Coevolved Multidrug-Resistant HIV-1 Protease and Reverse Transcriptase Influences Integrase Drug Susceptibility and Replication Fitness" Pathogens 10, no. 9: 1070. https://doi.org/10.3390/pathogens10091070
APA StyleMartin, S. A., Cane, P. A., Pillay, D., & Mbisa, J. L. (2021). Coevolved Multidrug-Resistant HIV-1 Protease and Reverse Transcriptase Influences Integrase Drug Susceptibility and Replication Fitness. Pathogens, 10(9), 1070. https://doi.org/10.3390/pathogens10091070