
The Liver and the Hepatic Immune Response in Trypanosoma cruzi Infection, a Historical and Updated View

 , and
, and 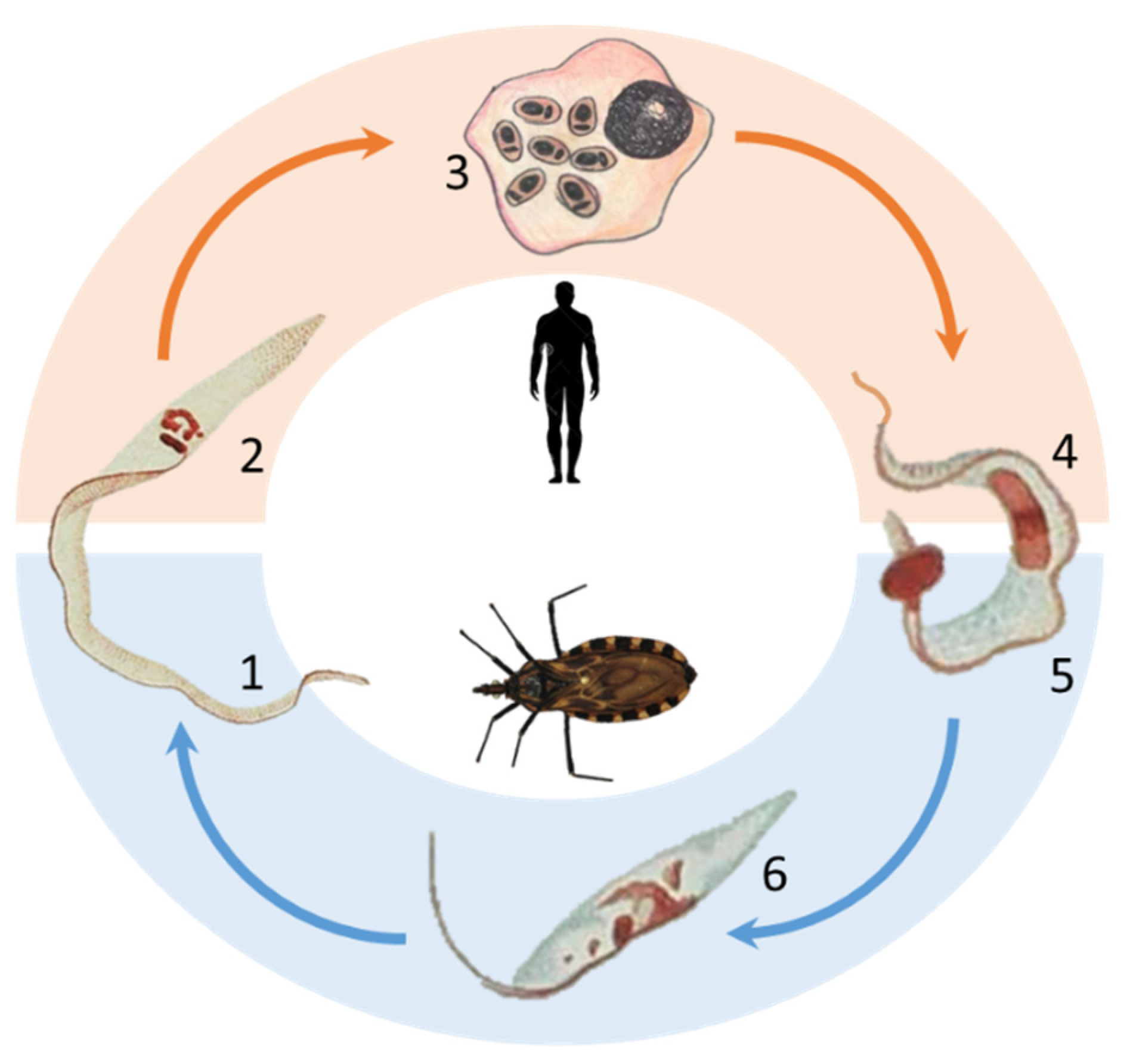{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. The History of Chagas Disease and the Description of the Trypanosoma cruzi Protozoa
1.2. Epidemiology and Clinical Aspects of Chagas Disease
1.3. The Founding Fathers of the Liver Pathology in Chagas Disease
1.4. The Participation of the Liver in T. cruzi Infection, a More Recent Perspective
1.5. The Immune Response in the Liver
1.5.1. Resident Liver Cells
Hepatocytes
Kupffer Cells
Natural Killer Cells
Natural Killer T Cells
Hepatic Dendritic Cells
Stellate Cells
Liver Sinusoidal Endothelial Cells
1.6. The Liver as a Tolerogenic Organ
1.7. Changing the Paradigm, the Trigger of Hepatic Immunogenic Responses
1.8. The Hepatic Inflammatory Response in Trypanosoma cruzi Infection
1.9. The Immunogenic Response in the Liver
1.10. The Role of Liver Cells in the Local Immunogenic Response
1.10.1. HDC, KC, HSC, and Cholangiocytes in the Hepatic Effector Response
1.10.2. Lymphoid Cells in the Liver Immunity
1.10.3. Neutrophils and Eosinophils in Liver Inflammation
2. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Steverding, D. The history of Chagas disease. Parasit Vectors 2014, 7, 1–8. [Google Scholar] [CrossRef]
- Chagas, C. Nova especie morbida do homem, produzida por um trypanozoma (Trypanozoma cruzi). Braz. Med. 1909, 16. Available online: http://www.bvschagas.coc.fiocruz.br/lildbi/docsonline/get.php?id=527 (accessed on 22 March 2021).
- Chagas, C. Nova tripanozomiaze humana: Estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Mem. Inst. Oswaldo Cruz 1909, 1, 159–228. Available online: https://archive.org/details/memriasdoinsti12inst/page/n195/mode/2up (accessed on 22 March 2021). [CrossRef]
- Thielen, E.V.; dos Santos, R.A. Belisário penna: Biographical photos. Hist. Cienc. Saude Manguinhos 2002, 9, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Chagas, C. Ueber eine neue trypanosomiasis des menschen. Arch. Fiir Schiffs Trop. 1909, 13, 120–122. [Google Scholar]
- Vianna, G. Contribuição para o estudo da anatomia patolojica da “Molestia de Carlos Chagas”: (Esquizotripanoze humana ou tireoidite parazitaria). Mem. Inst. Oswaldo Cruz 1911, 3, 276–294. [Google Scholar] [CrossRef]
- Neiva, A. Informações sobre a biolojia da Vinchuca, Triatoma infestans KLUG. Mem. Inst. Oswaldo Cruz 1913, 5, 24–31. [Google Scholar] [CrossRef][Green Version]
- Guerreiro, C.; Machado, A. Da reação de Bordet e Gengou na moléstia de Carlos Chagas como elemento diagnóstico. Bras. Med. 1913, 27, 225–226. [Google Scholar]
- Brumpt, E. O xenodignostico. Applicação ao diagnóstico de algumas infecções parasitárias e em particular a trypanosomose de Chagas. Ann. Paul. Med. Cir. 1914, 3, 97–102. [Google Scholar]
- Torres, M. Alguns fatos que interessam à epidemiolojia da molestia de Chagas. Mem. Inst. Oswaldo Cruz 1915, 7, 120–130. [Google Scholar] [CrossRef]
- Chagas, C.; Villela, E. Forma cardíaca da tripanossomíase americana. Mem. Inst. Oswaldo Cruz 1922, 14, 5–61. [Google Scholar] [CrossRef]
- Villela, E.; Bicalho, C. As pesquisas de laboratorio no diagnóstico da Molestia de Chagas. Mem. Inst. Oswaldo Cruz 1923, 16, 13–28. [Google Scholar] [CrossRef]
- Chagas, E. Forma cardiaca da Trypanosomiase americana. Mem. Inst. Oswaldo Cruz 1930, 24, 89–125. [Google Scholar] [CrossRef]
- Chagas, E. Novos estudos sobre a forma cardiaca da Trypanosomiase americana. Mem. Inst. Oswaldo Cruz 1932, 26, 329–338. [Google Scholar] [CrossRef][Green Version]
- Gascon, J.; Bern, C.; Pinazo, M.J. Chagas disease in Spain, the United States and other non-endemic countries. Acta Trop. 2010, 115, 22–27. [Google Scholar] [CrossRef] [PubMed]
- WHO. Chagas Disease (also Known as American Trypanosomiasis). Available online: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis)# (accessed on 19 April 2021).
- de Oliveira, G.M.; Diniz, R.L.; Batista, W.; Batista, M.M.; Bani Correa, C.; de Araújo-Jorge, T.C.; Henriques-Pons, A. Fas ligand-dependent inflammatory regulation in acute myocarditis induced by Trypanosoma cruzi infection. Am. J. Pathol. 2007, 171, 79–86. [Google Scholar] [CrossRef]
- Echeverria, L.E.; Morillo, C.A. American trypanosomiasis (Chagas disease). Infect. Dis. Clin. N. Am. 2019, 33, 119–134. [Google Scholar] [CrossRef]
- Ortiz, J.V.; Pereira, B.V.M.; Couceiro, K.D.N.; Silva, M.R.H.D.; Doria, S.S.; Silva, P.R.L.D.; Lira, E.D.F.; Guerra, M.D.G.V.; Guerra, J.A.O.; Ferreira, J.M.B.B. Cardiac Evaluation in the acute phase of Chagas disease with post-treatment evolution in patients attended in the state of Amazonas, Brazil. Arq. Bras. Cardiol. 2019, 112, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Chagas, C. Aspectos clinicos y anatomopatológicos de la Trypanosomiases americana. Prensa Méd. Argent. 1916, 3, 125–127, 137–138, 153–158. [Google Scholar]
- Chagas, C. Lesões hepaticas na molestia de chagas. Fac. Med. Belo. Horiz. Tese Fac. Med. Belo Horiz. 1920, 53–96. [Google Scholar]
- Crowell, B. Acute form of trypanosomiasis. Am. J. Top. Dis. 1923, 3, 425. [Google Scholar]
- Almenara, G. Las alteraciones histológicas del higado en los casos agudos de enfermedad de Chagas. Ann. VIII Congr. Bras. Med. 1925, 1, 261–266. [Google Scholar]
- Mazza, S.; Jorg, M.; Canal-Feijó, E. Primer caso cronico mortal de forma cardiaca de enfermedad de Chagas. MEPRA 1938, 38, 2–75. [Google Scholar]
- Torres, C.M.; Duarte, E. Myocarditis in the acute form of Chagas disease. Mem. Inst. Oswaldo Cruz 1948, 46, 759–793. [Google Scholar]
- Ramos, J.; Freitas, J.; Borges, S.; Villela, M.; Castro, J.; Iunes, M. Estudo clínico e epidemiológico da moléstia de Chagas; alguns aspectos da função hepatica nos portadores de miocardite chagásica. Arq. Bras. Cardiol. 1949, 2, 157–162. [Google Scholar]
- Sadek, H.M.; Vasconcelos, E. Changes in the liver in Chagas disease. Arq. Cir. Clin. Exp. 1954, 17, 69–76. [Google Scholar] [PubMed]
- Mazza, S.; Jorg, M.E. Sobre nódulos de histiocitosis en el higado de perro inoculado com Schizotrypanun cruzi Chagas, de origem humano. MEPRA 1934, 15, 3–24. [Google Scholar]
- Umekita, L.F.; Carneiro, S.M.; Sesso, A.; Mota, I. One fate of bloodstream trypomastigote forms of Trypanosoma cruzi after immune clearance: An ultrastructural study. J. Parasitol. 1999, 85, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Plata, F.; Wietzerbin, J.; Pons, F.G.; Falcoff, E.; Eisen, H. Synergistic protection by specific antibodies and interferon against infection by Trypanosoma cruzi in vitro. Eur. J. Immunol. 1984, 14, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Umekita, L.F.; Takehara, H.A.; Mota, I. Role of the antibody Fc in the immune clearance of Trypanosoma cruzi. Immunol. Lett. 1988, 17, 85–89. [Google Scholar] [CrossRef]
- Sardinha, L.R.; Mosca, T.; Elias, R.M.; do Nascimento, R.S.; Gonçalves, L.A.; Bucci, D.Z.; Marinho, C.R.; Penha-Gonçalves, C.; Lima, M.R.; Alvarez, J.M. The liver plays a major role in clearance and destruction of blood trypomastigotes in Trypanosoma cruzi chronically infected mice. PLoS Negl. Trop. Dis. 2010, 4, e578. [Google Scholar] [CrossRef]
- Mota, I.; Umekita, L.F. The effect of C3 depletion on the clearance of Trypanosoma cruzi induced by IgG antibodies. Immunol. Lett. 1989, 21, 223–225. [Google Scholar] [CrossRef]
- Cestari, I.; Ramirez, M.I. Inefficient complement system clearance of Trypanosoma cruzi metacyclic trypomastigotes enables resistant strains to invade eukaryotic cells. PLoS ONE 2010, 5, e9721. [Google Scholar] [CrossRef]
- Tambourgi, D.V.; Kipnis, T.L.; da Silva, W.D.; Joiner, K.A.; Sher, A.; Heath, S.; Hall, B.F.; Ogden, G.B. A partial cDNA clone of trypomastigote decay-accelerating factor (T-DAF), a developmentally regulated complement inhibitor of Trypanosoma cruzi, has genetic and functional similarities to the human complement inhibitor DAF. Infect. Immun. 1993, 61, 3656–3663. [Google Scholar] [CrossRef]
- Cestari, I.O.S.; Evans-Osses, I.; Freitas, J.C.; Inal, J.M.; Ramirez, M.I. Complement C2 receptor inhibitor trispanning confers an increased ability to resist complement-mediated lysis in Trypanosoma cruzi. J. Infect. Dis. 2008, 198, 1276–1283. [Google Scholar] [CrossRef] [PubMed]
- Atayde, V.D.; Neira, I.; Cortez, M.; Ferreira, D.; Freymüller, E.; Yoshida, N. Molecular basis of non-virulence of Trypanosoma cruzi clone CL-14. Int. J. Parasitol. 2004, 34, 851–860. [Google Scholar] [CrossRef]
- Miao, Q.; Ndao, M. Trypanosoma cruzi infection and host lipid metabolism. Mediat. Inflamm. 2014, 2014, 902038. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, L.M.; da Costa, K.M.; Chaves, V.S.; Freire-de-Lima, C.G.; Morrot, A.; Mendonça-Previato, L.; Previato, J.O.; Freire-de-Lima, L. Theft and reception of host cell’s sialic acid: Dynamics of Trypanosoma cruzi trans-sialidases and mucin-like molecules on Chagas’ disease immunomodulation. Front. Immunol. 2019, 10, 164. [Google Scholar] [CrossRef]
- Prioli, R.P.; Rosenberg, I.; Pereira, M.E. High- and low-density lipoproteins enhance infection of Trypanosoma cruzi in vitro. Mol. Biochem. Parasitol. 1990, 38, 191–198. [Google Scholar] [CrossRef]
- Nagajyothi, F.; Weiss, L.M.; Silver, D.L.; Desruisseaux, M.S.; Scherer, P.E.; Herz, J.; Tanowitz, H.B. Trypanosoma cruzi utilizes the host low density lipoprotein receptor in invasion. PLoS Negl. Trop. Dis. 2011, 5, e953. [Google Scholar] [CrossRef]
- Johndrow, C.; Nelson, R.; Tanowitz, H.; Weiss, L.M.; Nagajyothi, F. Trypanosoma cruzi infection results in an increase in intracellular cholesterol. Microbes Infect. 2014, 16, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Marra, C.A.; Zaidenberg, A.; de Alaniz, M.J.; Buschiazzo, H. The restoring effect of trifluralin and benznidazole on the abnormal fatty-acid pattern induced by Trypanosoma cruzi in the liver microsomes of infected mice. Ann. Trop. Med. Parasitol. 2002, 96, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Onofrio, L.I.; Arocena, A.R.; Paroli, A.F.; Cabalén, M.E.; Andrada, M.C.; Cano, R.C.; Gea, S. Trypanosoma cruzi infection is a potent risk factor for non-alcoholic steatohepatitis enhancing local and systemic inflammation associated with strong oxidative stress and metabolic disorders. PLoS Negl. Trop. Dis. 2015, 9, e0003464. [Google Scholar] [CrossRef] [PubMed]
- Sardinha, L.R.; Elias, R.M.; Mosca, T.; Bastos, K.R.; Marinho, C.R.; D’Império Lima, M.R.; Alvarez, J.M. Contribution of NK, NK T, gamma delta T, and alpha beta T cells to the gamma interferon response required for liver protection against Trypanosoma cruzi. Infect. Immun. 2006, 74, 2031–2042. [Google Scholar] [CrossRef]
- Meuser-Batista, M.; Vacani-Martins, N.; Cascabulho, C.M.; Beghini, D.G.; Henriques-Pons, A. In the presence of Trypanosoma cruzi antigens, activated peripheral T lymphocytes retained in the liver induce a proinflammatory phenotypic and functional shift in intrahepatic T lymphocyte. J. Leukoc. Biol. 2020, 107, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Ravin, H.A.; Rowley, D.; Jenkins, C.; Fine, J. On the absorption of bacterial endotoxin from the gastro-intestinal tract of the normal and shocked animal. J. Exp. Med. 1960, 112, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.B.; Henderson, J.M.; Kutner, M.H. Endotoxin levels measured by a chromogenic assay in portal, hepatic and peripheral venous blood in patients with cirrhosis. Hepatology 1988, 8, 232–236. [Google Scholar] [CrossRef]
- Crispe, I.N. The liver as a lymphoid organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Seong, S.Y.; Matzinger, P. Hydrophobicity: An ancient damage-associated molecular pattern that initiates innate immune responses. Nat. Rev. Immunol. 2004, 4, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Travers, P.; Walport, M. Imunobiologia de Janeway, 7th ed.; Artmed: Porto Alegre, Brazil, 2011. [Google Scholar]
- Wisse, E. An electron microscopic study of the fenestrated endothelial lining of rat liver sinusoids. J. Ultrastruct. Res. 1970, 31, 125–150. [Google Scholar] [CrossRef]
- Grakoui, A.; Crispe, I.N. Presentation of hepatocellular antigens. Cell. Mol. Immunol. 2016, 13, 293–300. [Google Scholar] [CrossRef]
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365. [Google Scholar] [CrossRef]
- Freudenberg, M.A.; Freudenberg, N.; Galanos, C. Time course of cellular distribution of endotoxin in liver, lungs and kidneys of rats. Br. J. Exp. Pathol. 1982, 63, 56–65. [Google Scholar] [PubMed]
- Warren, A.; Le Couteur, D.G.; Fraser, R.; Bowen, D.G.; McCaughan, G.W.; Bertolino, P. Tlymphocytes interact with hepatocytes through fenestrations in murine liver sinusoidal endothelial cells. Hepatology 2006, 44, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimkhani, M.R.; Mohar, I.; Crispe, I.N. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 2011, 54, 1379–1387. [Google Scholar] [CrossRef]
- Bertolino, P.; Trescol-Biémont, M.C.; Rabourdin-Combe, C. Hepatocytes induce functional activation of naive CD8+ T lymphocytes but fail to promote survival. Eur. J. Immunol. 1998, 28, 221–236. [Google Scholar] [CrossRef]
- Holz, L.E.; Benseler, V.; Bowen, D.G.; Bouillet, P.; Strasser, A.; O’Reilly, L.; d’Avigdor, W.M.; Bishop, A.G.; McCaughan, G.W.; Bertolino, P. Intrahepatic murine CD8 T-cell activation associates with a distinct phenotype leading to Bim-dependent death. Gastroenterology 2008, 135, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Herkel, J.; Jagemann, B.; Wiegard, C.; Lazaro, J.F.; Lueth, S.; Kanzler, S.; Blessing, M.; Schmitt, E.; Lohse, A.W. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocyutes. Hepatology 2003, 37, 1079–1085. [Google Scholar] [CrossRef]
- Santos, V.R.C.D.; Antunes, D.; Souza, D.D.S.M.; Moreira, O.C.; Lima, I.C.A.; Farias-de-Oliveira, D.A.; Lobo, J.P.; de Meis, E.; Coura, J.R.; Savino, W.; et al. Human acute Chagas disease: Changes in factor VII, activated protein C and hepatic enzymes from patients of oral outbreaks in Pará State (Brazilian Amazon). Mem. Inst. Oswaldo Cruz 2020, 115, e190364. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.Y.; Yang, M.Q.; He, Z.G.; Wei, Q.; Li, J.Y. The biological function of kupffer cells in liver disease. In Biology of Myelomonocytic Cells; IntechOpen: London, UK, 2017; Volume 2. [Google Scholar]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Mackay, I.R. Hepatoimmunology: A perspective. Immunol. Cell Biol. 2002, 80, 36–44. [Google Scholar] [CrossRef]
- Gale, R.P.; Sparkes, R.S.; Golde, D.W. Bone marrow origin of hepatic macrophages (Kupffer cells) in humans. Science 1978, 201, 937–938. [Google Scholar] [CrossRef]
- Kinoshita, M.; Uchida, T.; Sato, A.; Nakashima, M.; Nakashima, H.; Shono, S.; Habu, Y.; Miyazaki, H.; Hiroi, S.; Seki, S. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J. Hepatol. 2010, 53, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Movita, D.; Kreefft, K.; Biesta, P.; van Oudenaren, A.; Leenen, P.J.; Janssen, H.L.; Boonstra, A. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J. Leukoc. Biol. 2012, 92, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Said, E.A.; Al-Reesi, I.; Al-Riyami, M.; Al-Naamani, K.; Al-Sinawi, S.; Al-Balushi, M.S.; Koh, C.Y.; Al-Busaidi, J.Z.; Idris, M.A.; Al-Jabri, A.A. Increased CD86 but not CD80 and PD-L1 expression on liver CD68+ cells during chronic HBV infection. PLoS ONE 2016, 11, e0158265. [Google Scholar] [CrossRef]
- Blériot, C.; Dupuis, T.; Jouvion, G.; Eberl, G.; Disson, O.; Lecuit, M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 2015, 42, 145–158. [Google Scholar] [CrossRef] [PubMed]
- MacParland, S.A.; Tsoi, K.M.; Ouyang, B.; Ma, X.Z.; Manuel, J.; Fawaz, A.; Ostrowski, M.A.; Alman, B.A.; Zilman, A.; Chan, W.C.; et al. Phenotype determines nanoparticle uptake by human macrophages from liver and blood. ACS Nano. 2017, 11, 2428–2443. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gilbert, G.E.; Kokubo, Y.; Ohashi, T. Role of the liver in regulating numbers of circulating neutrophils. Blood 2001, 98, 1226–1230. [Google Scholar] [CrossRef]
- Shi, J.; Kokubo, Y.; Wake, K. Expression of P-selectin on hepatic endothelia and platelets promoting neutrophil removal by liver macrophages. Blood 1998, 92, 520–528. [Google Scholar] [CrossRef]
- Horst, A.K.; Tiegs, G.; Diehl, L. Contribution of macrophage efferocytosis to liver homeostasis and disease. Front. Immunol. 2019, 10, 2670. [Google Scholar] [CrossRef] [PubMed]
- Langers, I.; Renoux, V.M.; Thiry, M.; Delvenne, P.; Jacobs, N. Natural killer cells: Role in local tumor growth and metastasis. Biologics 2012, 6, 73–82. [Google Scholar] [PubMed]
- Doherty, D.G.; O’Farrelly, C. Innate and adaptive lymphoid cells in the human liver. Immunol. Rev. 2000, 174, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Doherty, D.G.; Norris, S.; Madrigal-Estebas, L.; McEntee, G.; Traynor, O.; Hegarty, J.E.; O’Farrelly, C. The human liver contains multiple populations of NK cells, T cells, and CD3+CD56+ natural T cells with distinct cytotoxic activities and Th1, Th2, and Th0 cytokine secretion patterns. J. Immunol. 1999, 163, 2314–2321. [Google Scholar]
- Mikulak, J.; Bruni, E.; Oriolo, F.; Di Vito, C.; Mavilio, D. Hepatic natural killer cells: Organ-Specific sentinels of liver immune homeostasis and physiopathology. Front. Immunol. 2019, 10, 946. [Google Scholar] [CrossRef] [PubMed]
- Ronco, M.T.; Francés, D.E.; Ingaramo, P.I.; Quiroga, A.D.; Alvarez, M.L.; Pisani, G.B.; Revelli, S.S.; Carnovale, C.E. Tumor necrosis factor alpha induced by Trypanosoma cruzi infection mediates inflammation and cell death in the liver of infected mice. Cytokine 2010, 49, 64–72. [Google Scholar] [CrossRef]
- Hao, J.; Dong, S.; Xia, S.; He, W.; Jia, H.; Zhang, S.; Wei, J.; O’Brien, R.L.; Born, W.K.; Wu, Z.; et al. Regulatory role of Vγ1 γδ T cells in tumor immunity through IL-4 production. J. Immunol. 2011, 187, 4979–4986. [Google Scholar] [CrossRef]
- Fauriat, C.; Long, E.O.; Ljunggren, H.G.; Bryceson, Y.T. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood 2010, 115, 2167–2176. [Google Scholar] [CrossRef]
- Viant, C.; Fenis, A.; Chicanne, G.; Payrastre, B.; Ugolini, S.; Vivier, E. SHP-1-mediated inhibitory signals promote responsiveness and anti-tumour functions of natural killer cells. Nat. Commun. 2014, 5, 5108. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef]
- Olcese, L.; Cambiaggi, A.; Semenzato, G.; Bottino, C.; Moretta, A.; Vivier, E. Human killer cell activatory receptors for MHC class I molecules are included in a multimeric complex expressed by natural killer cells. J. Immunol. 1997, 158, 5083–5086. [Google Scholar] [PubMed]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Hua, J. Immune cells in liver regeneration. Oncotarget 2017, 8, 3628–3639. [Google Scholar] [CrossRef] [PubMed]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Yamaguchi, S.; Sakamori, R.; Hiramatsu, N.; Kanto, T.; Ohkawa, K.; Hayashi, N. Natural killer cell and hepatic cell interaction via NKG2A leads to dendritic cell-mediated induction of CD4 CD25 T cells with PD-1-dependent regulatory activities. Immunology 2007, 120, 73–82. [Google Scholar] [CrossRef]
- Wu, L.; Van Kaer, L. Natural killer T cells in health and disease. Front. Biosci. 2011, 3, 236–251. [Google Scholar]
- Berzins, S.P.; Smyth, M.J.; Baxter, A.G. Presumed guilty: Natural killer T cell defects and human disease. Nat. Rev. Immunol. 2011, 11, 131–142. [Google Scholar] [CrossRef]
- Wang, H.; Yin, S. Natural killer T cells in liver injury, inflammation and cancer. Expert Rev. Gastroenterol. Hepatol. 2015, 9, 1077–1085. [Google Scholar] [CrossRef]
- Rossjohn, J.; Pellicci, D.G.; Patel, O.; Gapin, L.; Godfrey, D.I. Recognition of CD1d-restricted antigens by natural killer T cells. Nat. Rev. Immunol. 2012, 12, 845–857. [Google Scholar] [CrossRef]
- Zeissig, S.; Murata, K.; Sweet, L.; Publicover, J.; Hu, Z.; Kaser, A.; Bosse, E.; Iqbal, J.; Hussain, M.M.; Balschun, K.; et al. Hepatitis B virus-induced lipid alterations contribute to natural killer T cell-dependent protective immunity. Nat. Med. 2012, 18, 1060–1068. [Google Scholar] [CrossRef]
- Gao, B.; Radaeva, S.; Park, O. Liver natural killer and natural killer T cells: Immunobiology and emerging roles in liver diseases. J. Leukoc. Biol. 2009, 86, 513–528. [Google Scholar] [CrossRef]
- Duthie, M.S.; Kahn, M.; White, M.; Kapur, R.P.; Kahn, S.J. Critical proinflammatory and anti-inflammatory functions of different subsets of CD1d-restricted natural killer T cells during Trypanosoma cruzi infection. Infect. Immun. 2005, 73, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Kanzler, H.; Soumelis, V.; Gilliet, M. Dendritic cell lineage, plasticity and cross-regulation. Nat. Immunol. 2001, 2, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 2001, 106, 259–262. [Google Scholar] [CrossRef]
- Cabillic, F.; Rougier, N.; Basset, C.; Lecouillard, I.; Quelvennec, E.; Toujas, L.; Guguen-Guillouzo, C.; Corlu, A. Hepatic environment elicits monocyte differentiation into a dendritic cell subset directing Th2 response. J. Hepatol. 2006, 44, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Soysa, R.; Wu, X.; Crispe, I.N. Dendritic cells in hepatitis and liver transplantation. Liver Transpl. 2017, 23, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Sánchez, N.; Córdova-Gallardo, J.; Barranco-Fragoso, B.; Eslam, M. Hepatic Dendritic Cells in the Development and Progression of Metabolic Steatohepatitis. Front. Immunol. 2021, 12, 641240. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Segura, E.; Amigorena, S. Inflammatory dendritic cells in mice and humans. Trends Immunol. 2013, 34, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Liu, K.; Helft, J.; Bogunovic, M.; Greter, M.; Hashimoto, D.; Price, J.; Yin, N.; Bromberg, J.; Lira, S.A.; et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 2009, 206, 3115–3130. [Google Scholar] [CrossRef] [PubMed]
- Balan, S.; Saxena, M.; Bhardwaj, N. Dendritic cell subsets and locations. Int. Rev. Cell Mol. Biol. 2019, 348, 1–68. [Google Scholar] [PubMed]
- Kelly, A.; Fahey, R.; Fletcher, J.M.; Keogh, C.; Carroll, A.G.; Siddachari, R.; Geoghegan, J.; Hegarty, J.E.; Ryan, E.J.; O’Farrelly, C. CD141+ myeloid dendritic cells are enriched in healthy human liver. J. Hepatol. 2014, 60, 135–142. [Google Scholar] [CrossRef]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Castellaneta, A.; Sumpter, T.L.; Chen, L.; Tokita, D.; Thomson, A.W. NOD2 ligation subverts IFN-alpha production by liver plasmacytoid dendritic cells and inhibits their T cell allostimulatory activity via B7-H1 up-regulation. J. Immunol. 2009, 183, 6922–6932. [Google Scholar] [CrossRef]
- Rahman, A.H.; Aloman, C. Dendritic cells and liver fibrosis. Biochim. Biophys. Acta 2013, 1832, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Ono, Y.; Chen, Y.F.; Thomson, A.W.; Chen, X.P. Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin. Liver Dis. 2018, 38, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Lukacs-Kornek, V.; Schuppan, D. Dendritic cells in liver injury and fibrosis: Shortcomings and promises. J. Hepatol. 2013, 59, 1124–1126. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Chen, Y.; Chen, L.; Zeng, J.; Liu, J. Transforming growth factor β1 and Fas ligand synergistically enhance immune tolerance in dendritic cells in liver transplantation. J. Surg. Res. 2017, 218, 180–193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wake, K. “Sternzellen” in the liver: Perisinusoidal cells with special reference to storage of vitamin A. Am. J. Anat. 1971, 132, 429–462. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, M.P.; Jezequel, A.M.; Orlandi, F. The lipocytes in normal human liver. A quantitative study. Digestion 1981, 22, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Jezequel, A.; Novelli, G.; Venturini, C.; Orlandi, F. Quantitative analysis of the perisinusoidal cells in human liver: The lipocytes. In Liver Cirrhosis; Karger Publishers: Basel, Switzerland, 1984; Volume 8, pp. 85–90. [Google Scholar]
- Smedsrød, B.; Pertoft, H.; Gustafson, S.; Laurent, T.C. Scavenger functions of the liver endothelial cell. Biochem. J. 1990, 266, 313–327. [Google Scholar] [CrossRef]
- Senoo, H.; Mezaki, Y.; Fujiwara, M. The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 2017, 92, 387–455. [Google Scholar] [CrossRef] [PubMed]
- Ballardini, G.; Groff, P.; Badiali de Giorgi, L.; Schuppan, D.; Bianchi, F.B. Ito cell heterogeneity: Desmin-negative Ito cells in normal rat liver. Hepatology 1994, 19, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Gräff, A.; Desmoulière, A.; Gabbiani, G. Heterogeneity of myofibroblast phenotypic features: An example of fibroblastic cell plasticity. Virchows Arch. 1994, 425, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Viñas, O.; Bataller, R.; Sancho-Bru, P.; Ginès, P.; Berenguer, C.; Enrich, C.; Nicolás, J.M.; Ercilla, G.; Gallart, T.; Vives, J.; et al. Human hepatic stellate cells show features of antigen-presenting cells and stimulate lymphocyte proliferation. Hepatology 2003, 38, 919–929. [Google Scholar] [CrossRef]
- Winau, F.; Hegasy, G.; Weiskirchen, R.; Weber, S.; Cassan, C.; Sieling, P.A.; Modlin, R.L.; Liblau, R.S.; Gressner, A.M.; Kaufmann, S.H. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 2007, 26, 117–129. [Google Scholar] [CrossRef]
- Heymann, F.; Trautwein, C.; Tacke, F. Monocytes and macrophages as cellular targets in liver fibrosis. Inflamm. Allergy Drug Targets 2009, 8, 307–318. [Google Scholar] [CrossRef]
- Dunham, R.M.; Thapa, M.; Velazquez, V.M.; Elrod, E.J.; Denning, T.L.; Pulendran, B.; Grakoui, A. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J. Immunol. 2013, 190, 2009–2016. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Penas, F.N.; Cevey, Á.; Siffo, S.; Mirkin, G.A.; Goren, N.B. Hepatic injury associated with Trypanosoma cruzi infection is attenuated by treatment with 15-deoxy-Δ. Exp. Parasitol. 2016, 170, 100–108. [Google Scholar] [CrossRef]
- Magness, S.T.; Bataller, R.; Yang, L.; Brenner, D.A. A dual reporter gene transgenic mouse demonstrates heterogeneity in hepatic fibrogenic cell populations. Hepatology 2004, 40, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.N.; Walewski, J.L.; Clugston, R.D.; Berk, P.D.; Rippe, R.A.; Blaner, W.S. Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS ONE 2011, 6, e24993. [Google Scholar] [CrossRef]
- Suzuki, H.; Sugiyama, Y. Transport of drugs across the hepatic sinusoidal membrane: Sinusoidal drug influx and efflux in the liver. Semin. Liver Dis. 2000, 20, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Elvevold, K.; Smedsrød, B.; Martinez, I. The liver sinusoidal endothelial cell: A cell type of controversial and confusing identity. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G391–G400. [Google Scholar] [CrossRef]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef]
- Tang, L.; Yang, J.; Liu, W.; Tang, X.; Chen, J.; Zhao, D.; Wang, M.; Xu, F.; Lu, Y.; Liu, B.; et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T-cell immune response. Gastroenterology 2009, 137, e1491–e1495. [Google Scholar] [CrossRef]
- Berg, M.; Wingender, G.; Djandji, D.; Hegenbarth, S.; Momburg, F.; Hämmerling, G.; Limmer, A.; Knolle, P. Cross-presentation of antigens from apoptotic tumor cells by liver sinusoidal endothelial cells leads to tumor-specific CD8+ T cell tolerance. Eur. J. Immunol. 2006, 36, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrød, B. Liver Sinusoidal Endothelial Cells. Compr. Physiol. 2015, 5, 1751–1774. [Google Scholar] [PubMed]
- Qian, S.; Wang, Z.; Lee, Y.; Chiang, Y.; Bonham, C.; Fung, J.; Lu, L. Hepatocyte-induced apoptosis of activated T cells, a mechanism of liver transplant tolerance, is related to the expression of ICAM-1 and hepatic lectin. Transplant. Proc. 2001, 33, 226–227. [Google Scholar] [CrossRef]
- Sana, G.; Lombard, C.; Vosters, O.; Jazouli, N.; Andre, F.; Stephenne, X.; Smets, F.; Najimi, M.; Sokal, E.M. Adult human hepatocytes promote CD4(+) T-cell hyporesponsiveness via interleukin-10-producing allogeneic dendritic cells. Cell Transplant. 2014, 23, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Gao, B. Basic liver immunology. Cell Mol. Immunol. 2016, 13, 265–266. [Google Scholar] [CrossRef]
- Qian, S.; Demetris, A.J.; Murase, N.; Rao, A.S.; Fung, J.J.; Starzl, T.E. Murine liver allograft transplantation: Tolerance and donor cell chimerism. Hepatology 1994, 19, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Tian, Z. Liver-Mediated Adaptive Immune Tolerance. Front. Immunol. 2019, 10, 2525. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Germann, T.; Treichel, U.; Uhrig, A.; Schmitt, E.; Hegenbarth, S.; Lohse, A.W.; Gerken, G. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J. Immunol. 1999, 162, 1401–1407. [Google Scholar]
- Crispe, I.N.; Dao, T.; Klugewitz, K.; Mehal, W.Z.; Metz, D.P. The liver as a site of T-cell apoptosis: Graveyard, or killing field? Immunol. Rev. 2000, 174, 47–62. [Google Scholar] [CrossRef]
- Benseler, V.; Warren, A.; Vo, M.; Holz, L.E.; Tay, S.S.; Le Couteur, D.G.; Breen, E.; Allison, A.C.; van Rooijen, N.; McGuffog, C.; et al. Hepatocyte entry leads to degradation of autoreactive CD8 T cells. Proc.Natl. Acad Sci. USA 2011, 108, 16735–16740. [Google Scholar] [CrossRef]
- Bowen, D.G.; Zen, M.; Holz, L.; Davis, T.; McCaughan, G.W.; Bertolino, P. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J. Clin. Investig. 2004, 114, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Lang Kuhs, K.A.; Toporovski, R.; Ginsberg, A.A.; Olsen, A.L.; Shedlock, D.J.; Morrow, M.P.; Yan, J.; Wells, R.G.; Weiner, D.B. Peripheral immunization induces functional intrahepatic hepatitis C specific immunity following selective retention of vaccine-specific CD8 T cells by the liver. Hum. Vaccin. 2011, 7, 1326–1335. [Google Scholar] [CrossRef]
- Zeng, Z.; Kong, X.; Li, F.; Wei, H.; Sun, R.; Tian, Z. IL-12-based vaccination therapy reverses liver-induced systemic tolerance in a mouse model of hepatitis B virus carrier. J. Immunol. 2013, 191, 4184–4193. [Google Scholar] [CrossRef] [PubMed]
- Piché, C.; Béland, K.; Lapierre, P.; Massie, B.; Alvarez, F. Different sites of xenoantigen delivery lead to a virally induced late-onset hepatitis in mice through molecular mimicry. Liver Int. 2011, 31, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Isogawa, M.; Kakimi, K.; Kamamoto, H.; Protzer, U.; Chisari, F.V. Differential dynamics of the peripheral and intrahepatic cytotoxic T lymphocyte response to hepatitis B surface antigen. Virology 2005, 333, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef]
- Kader, M.; Alaoui-El-Azher, M.; Vorhauer, J.; Kode, B.B.; Wells, J.Z.; Stolz, D.; Michalopoulos, G.; Wells, A.; Scott, M.; Ismail, N. MyD88-dependent inflammasome activation and autophagy inhibition contributes to Ehrlichia-induced liver injury and toxic shock. PLoS Pathog. 2017, 13, e1006644. [Google Scholar] [CrossRef]
- Vacani-Martins, N.; Meuser-Batista, M.; Moreira, O.C.; Cascabulho, C.M.; Gois Beghini, D.; Horita, S.I.; Batista, M.M.; Freitas, F.C.; Guimarães, J.R.; Henriques-Pons, A. After Experimental Trypanosoma cruzi Infection, Dying Hepatic CD3+TCRαβ+B220+ T Lymphocytes Are Rescued from Death by Peripheral T Cells and Become Activated. Pathogens 2020, 9, 717. [Google Scholar] [CrossRef]
- Henriques-Pons, A.; Yu, Q.; Rayavarapu, S.; Cohen, T.V.; Ampong, B.; Cha, H.J.; Jahnke, V.; Van der Meulen, J.; Wang, D.; Jiang, W.; et al. Role of Toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum. Mol. Genet. 2014, 23, 2604–2617. [Google Scholar] [CrossRef]
- Martinon, F.; Tschopp, J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol. 2005, 26, 447–454. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Wong, J.; Johnston, B.; Lee, S.S.; Bullard, D.C.; Smith, C.W.; Beaudet, A.L.; Kubes, P. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J. Clin. Investig. 1997, 99, 2782–2790. [Google Scholar] [CrossRef]
- McDonald, B.; McAvoy, E.F.; Lam, F.; Gill, V.; de la Motte, C.; Savani, R.C.; Kubes, P. Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J. Exp. Med. 2008, 205, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Bonder, C.S.; Norman, M.U.; Swain, M.G.; Zbytnuik, L.D.; Yamanouchi, J.; Santamaria, P.; Ajuebor, M.; Salmi, M.; Jalkanen, S.; Kubes, P. Rules of recruitment for Th1 and Th2 lymphocytes in inflamed liver: A role for alpha-4 integrin and vascular adhesion protein-1. Immunity 2005, 23, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Aspinall, A.I.; Curbishley, S.M.; Lalor, P.F.; Weston, C.J.; Liaskou, E.; Adams, R.M.; Holt, A.P.; Adams, D.H.; Blahova, M. CX(3)CR1 and vascular adhesion protein-1-dependent recruitment of CD16(+) monocytes across human liver sinusoidal endothelium. Hepatology 2010, 51, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; Inverso, D.; Sironi, L.; Di Lucia, P.; Fioravanti, J.; Ganzer, L.; Fiocchi, A.; Vacca, M.; Aiolfi, R.; Sammicheli, S.; et al. Immunosurveillance of the liver by intravascular effector CD8(+) T cells. Cell 2015, 161, 486–500. [Google Scholar] [CrossRef]
- Wang, J.; Kubes, P. A Reservoir of Mature Cavity Macrophages that Can Rapidly Invade Visceral Organs to Affect Tissue Repair. Cell 2016, 165, 668–678. [Google Scholar] [CrossRef]
- Kern, M.; Popov, A.; Scholz, K.; Schumak, B.; Djandji, D.; Limmer, A.; Eggle, D.; Sacher, T.; Zawatzky, R.; Holtappels, R.; et al. Virally infected mouse liver endothelial cells trigger CD8+ T-cell immunity. Gastroenterology 2010, 138, 336–346. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Schanz, O.; Garbers, C.; Zaremba, A.; Hegenbarth, S.; Kurts, C.; Beyer, M.; Schultze, J.L.; Kastenmüller, W.; Rose-John, S.; et al. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep. 2014, 8, 1318–1327. [Google Scholar] [CrossRef]
- Wittlich, M.; Dudek, M.; Böttcher, J.P.; Schanz, O.; Hegenbarth, S.; Bopp, T.; Schmitt, E.; Kurts, C.; Garbers, C.; Rose John, S.; et al. Liver sinusoidal endothelial cell cross-priming is supported by CD4 T cell-derived IL-2. J. Hepatol. 2017, 66, 978–986. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Schanz, O.; Wohlleber, D.; Abdullah, Z.; Debey-Pascher, S.; Staratschek-Jox, A.; Höchst, B.; Hegenbarth, S.; Grell, J.; Limmer, A.; et al. Liver-primed memory T cells generated under noninflammatory conditions provide anti-infectious immunity. Cell Rep. 2013, 3, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, N.; Wu, X.; Kenerson, H.; Chen, A.; Strickland, I.; Horton, H.; Yeung, R.; Crispe, I.N. Functional responses of resident human T cells in intact liver tissue. Cell Immunol. 2021, 360, 104275. [Google Scholar] [CrossRef]
- Park, S.; Park, J.; Kim, E.; Lee, Y. The Capicua/ETS Translocation Variant 5 Axis Regulates Liver-Resident Memory CD8. Hepatology 2019, 70, 358–371. [Google Scholar] [PubMed]
- Sallusto, F.; Lanzavecchia, A. Heterogeneity of CD4+ memory T cells: Functional modules for tailored immunity. Eur. J. Immunol. 2009, 39, 2076–2082. [Google Scholar] [CrossRef]
- Rosenblum, M.D.; Way, S.S.; Abbas, A.K. Regulatory T cell memory. Nat. Rev. Immunol. 2016, 16, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Casey, K.A.; Fraser, K.A.; Schenkel, J.M.; Moran, A.; Abt, M.C.; Beura, L.K.; Lucas, P.J.; Artis, D.; Wherry, E.J.; Hogquist, K.; et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012, 188, 4866–4875. [Google Scholar] [CrossRef]
- Sathaliyawala, T.; Kubota, M.; Yudanin, N.; Turner, D.; Camp, P.; Thome, J.J.; Bickham, K.L.; Lerner, H.; Goldstein, M.; Sykes, M.; et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 2013, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.W.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, D.; Ng, W.Y.; Holz, L.E.; Ma, J.Z.; Zaid, A.; Wong, Y.C.; Lau, L.S.; Mollard, V.; Cozijnsen, A.; Collins, N.; et al. Liver-Resident Memory CD8. Immunity 2016, 45, 889–902. [Google Scholar] [CrossRef]
- Ehlting, C.; Trilling, M.; Tiedje, C.; Le-Trilling, V.T.K.; Albrecht, U.; Kluge, S.; Zimmermann, A.; Graf, D.; Gaestel, M.; Hengel, H.; et al. MAPKAP kinase 2 regulates IL-10 expression and prevents formation of intrahepatic myeloid cell aggregates during cytomegalovirus infections. J. Hepatol. 2016, 64, 380–389. [Google Scholar] [CrossRef]
- Huang, L.R.; Wohlleber, D.; Reisinger, F.; Jenne, C.N.; Cheng, R.L.; Abdullah, Z.; Schildberg, F.A.; Odenthal, M.; Dienes, H.P.; van Rooijen, N.; et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8(+) T cells and successful immunotherapy against chronic viral liver infection. Nat. Immunol. 2013, 14, 574–583. [Google Scholar] [CrossRef]
- Lin, Y.C.; Hsu, C.Y.; Huang, S.K.; Fan, Y.H.; Huang, C.H.; Yang, C.K.; Su, W.T.; Chang, P.C.; Dutta, A.; Liu, Y.J.; et al. Induction of liver-specific intrahepatic myeloid cells aggregation expands CD8 T cell and inhibits growth of murine hepatoma. Oncoimmunology 2018, 7, e1502129. [Google Scholar] [PubMed]
- Nakashima, M.; Kinoshita, M.; Nakashima, H.; Habu, Y.; Miyazaki, H.; Shono, S.; Hiroi, S.; Shinomiya, N.; Nakanishi, K.; Seki, S. Pivotal advance: Characterization of mouse liver phagocytic B cells in innate immunity. J. Leukoc. Biol. 2012, 91, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Stolz, D.B.; Chalasani, G.; Thomson, A.W. Hepatic B cells are readily activated by Toll-like receptor-4 ligation and secrete less interleukin-10 than lymphoid tissue B cells. Clin. Exp. Immunol. 2013, 173, 473–479. [Google Scholar] [CrossRef]
- Béland, K.; Marceau, G.; Labardy, A.; Bourbonnais, S.; Alvarez, F. Depletion of B cells induces remission of autoimmune hepatitis in mice through reduced antigen presentation and help to T cells. Hepatology 2015, 62, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetty, V.G.; Katz, S.C.; Bleier, J.I.; Shah, A.B.; Dematteo, R.P. Natural killer dendritic cells have both antigen presenting and lytic function and in response to CpG produce IFN-gamma via autocrine IL-12. J. Immunol. 2005, 174, 2612–2618. [Google Scholar] [CrossRef]
- Chen, L.; Calomeni, E.; Wen, J.; Ozato, K.; Shen, R.; Gao, J.X. Natural killer dendritic cells are an intermediate of developing dendritic cells. J. Leukoc. Biol. 2007, 81, 1422–1433. [Google Scholar] [CrossRef]
- Krueger, P.D.; Kim, T.S.; Sung, S.S.; Braciale, T.J.; Hahn, Y.S. Liver-resident CD103+ dendritic cells prime antiviral CD8+ T cells in situ. J. Immunol. 2015, 194, 3213–3222. [Google Scholar] [CrossRef]
- Sierro, F.; Evrard, M.; Rizzetto, S.; Melino, M.; Mitchell, A.J.; Florido, M.; Beattie, L.; Walters, S.B.; Tay, S.S.; Lu, B.; et al. A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity 2017, 47, 374–388.e376. [Google Scholar] [CrossRef]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, 6304. [Google Scholar] [CrossRef]
- Blériot, C.; Ginhoux, F. Understanding the Heterogeneity of Resident Liver Macrophages. Front. Immunol. 2019, 10, 2694. [Google Scholar] [CrossRef]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Ju, C. Hepatic Macrophages in Liver Injury. Front. Immunol. 2020, 11, 322. [Google Scholar] [CrossRef]
- Wong, C.H.; Jenne, C.N.; Petri, B.; Chrobok, N.L.; Kubes, P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat. Immunol. 2013, 14, 785–792. [Google Scholar] [CrossRef]
- Gregory, S.H.; Wing, E.J. Neutrophil-Kupffer cell interaction: A critical component of host defenses to systemic bacterial infections. J. Leukoc. Biol. 2002, 72, 239–248. [Google Scholar] [PubMed]
- Gregory, S.H.; Cousens, L.P.; van Rooijen, N.; Döpp, E.A.; Carlos, T.M.; Wing, E.J. Complementary adhesion molecules promote neutrophil-Kupffer cell interaction and the elimination of bacteria taken up by the liver. J. Immunol. 2002, 168, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Beattie, L.; Peltan, A.; Maroof, A.; Kirby, A.; Brown, N.; Coles, M.; Smith, D.F.; Kaye, P.M. Dynamic imaging of experimental Leishmania donovani-induced hepatic granulomas detects Kupffer cell-restricted antigen presentation to antigen-specific CD8 T cells. PLoS Pathog. 2010, 6, e1000805. [Google Scholar] [CrossRef] [PubMed]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363. [Google Scholar]
- Schölzel, K.; Schildberg, F.A.; Welz, M.; Börner, C.; Geiger, S.; Kurts, C.; Heikenwälder, M.; Knolle, P.A.; Wohlleber, D. Transfer of MHC-class-I molecules among liver sinusoidal cells facilitates hepatic immune surveillance. J. Hepatol. 2014, 61, 600–608. [Google Scholar] [CrossRef]
- Jeffery, H.C.; van Wilgenburg, B.; Kurioka, A.; Parekh, K.; Stirling, K.; Roberts, S.; Dutton, E.E.; Hunter, S.; Geh, D.; Braitch, M.K.; et al. Biliary epithelium and liver B cells exposed to bacteria activate intrahepatic MAIT cells through MR1. J. Hepatol. 2016, 64, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Miyake, Y.; Takaki, A.; Yasunaka, T.; Koike, K.; Ikeda, F.; Shiraha, H.; Nouso, K.; Yamamoto, K. TLR4, TLR9, and NLRP3 in biliary epithelial cells of primary sclerosing cholangitis: Relationship with clinical characteristics. J. Gastroenterol. Hepatol. 2015, 30, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Agostinelli, L.; Saccomanno, S.; Pinto, C.; Giordano, D.M.; Rychlicki, C.; De Minicis, S.; Trozzi, L.; Banales, J.M.; Melum, E.; et al. Nlrp3 Activation Induces Il-18 Synthesis and Affects the Epithelial Barrier Function in Reactive Cholangiocytes. Am. J. Pathol. 2017, 187, 366–376. [Google Scholar] [CrossRef]
- Maroni, L.; Ninfole, E.; Pinto, C.; Benedetti, A.; Marzioni, M. Gut-Liver Axis and Inflammasome Activation in Cholangiocyte Pathophysiology. Cells 2020, 9, 736. [Google Scholar] [CrossRef]
- Kimura, K.; Kakimi, K.; Wieland, S.; Guidotti, L.G.; Chisari, F.V. Interleukin-18 inhibits hepatitis B virus replication in the livers of transgenic mice. J. Virol. 2002, 76, 10702–10707. [Google Scholar] [CrossRef]
- Kimura, K.; Kakimi, K.; Wieland, S.; Guidotti, L.G.; Chisari, F.V. Activated intrahepatic antigen-presenting cells inhibit hepatitis B virus replication in the liver of transgenic mice. J. Immunol. 2002, 169, 5188–5195. [Google Scholar] [CrossRef]
- Huang, W.; He, W.; Shi, X.; He, X.; Dou, L.; Gao, Y. The Role of CD1d and MR1 Restricted T Cells in the Liver. Front. Immunol. 2018, 9, 2424. [Google Scholar] [CrossRef]
- Rha, M.S.; Han, J.W.; Kim, J.H.; Koh, J.Y.; Park, H.J.; Kim, S.I.; Kim, M.S.; Lee, J.G.; Lee, H.W.; Lee, D.H.; et al. Human liver CD8. J. Hepatol. 2020, 73, 640–650. [Google Scholar] [CrossRef]
- Toubal, A.; Lehuen, A. Lights on MAIT cells, a new immune player in liver diseases. J. Hepatol. 2016, 64, 1008–1010. [Google Scholar] [CrossRef]
- Harada, K.; Shimoda, S.; Sato, Y.; Isse, K.; Ikeda, H.; Nakanuma, Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin. Exp. Immunol. 2009, 157, 261–270. [Google Scholar] [CrossRef]
- Tang, X.Z.; Jo, J.; Tan, A.T.; Sandalova, E.; Chia, A.; Tan, K.C.; Lee, K.H.; Gehring, A.J.; De Libero, G.; Bertoletti, A. IL-7 licenses activation of human liver intrasinusoidal mucosal-associated invariant T cells. J. Immunol. 2013, 190, 3142–3152. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, Y.; Sun, R.; Wei, H.; Tian, Z. Impairment of hepatic NK cell development in IFN-γ deficient mice. Cytokine 2012, 60, 616–625. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; Long, E.O. Line of attack: NK cell specificity and integration of signals. Curr. Opin. Immunol. 2008, 20, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Minagawa, M.; Deng, Q.; Liu, Z.X.; Tsukamoto, H.; Dennert, G. Activated natural killer T cells induce liver injury by Fas and tumor necrosis factor-alpha during alcohol consumption. Gastroenterology 2004, 126, 1387–1399. [Google Scholar] [CrossRef]
- Tajiri, K.; Shimizu, Y.; Tsuneyama, K.; Sugiyama, T. Role of liver-infiltrating CD3+CD56+ natural killer T cells in the pathogenesis of nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2009, 21, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Maricic, I.; Marrero, I.; Eguchi, A.; Nakamura, R.; Johnson, C.D.; Dasgupta, S.; Hernandez, C.D.; Nguyen, P.S.; Swafford, A.D.; Knight, R.; et al. Differential Activation of Hepatic Invariant NKT Cell Subsets Plays a Key Role in Progression of Nonalcoholic Steatohepatitis. J. Immunol. 2018, 201, 3017–3035. [Google Scholar] [CrossRef]
- Jiang, W.; Sun, R.; Wei, H.; Tian, Z. Toll-like receptor 3 ligand attenuates LPS-induced liver injury by down-regulation of toll-like receptor 4 expression on macrophages. Proc. Natl. Acad. Sci. USA 2005, 102, 17077–17082. [Google Scholar] [CrossRef]
- Kakimi, K.; Guidotti, L.G.; Koezuka, Y.; Chisari, F.V. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J. Exp. Med. 2000, 192, 921–930. [Google Scholar] [CrossRef]
- Lucas, M.; Gadola, S.; Meier, U.; Young, N.T.; Harcourt, G.; Karadimitris, A.; Coumi, N.; Brown, D.; Dusheiko, G.; Cerundolo, V.; et al. Frequency and phenotype of circulating Valpha24/Vbeta11 double-positive natural killer T cells during hepatitis C virus infection. J. Virol. 2003, 77, 2251–2257. [Google Scholar] [CrossRef]
- Yamagiwa, S.; Matsuda, Y.; Ichida, T.; Honda, Y.; Takamura, M.; Sugahara, S.; Ishikawa, T.; Ohkoshi, S.; Sato, Y.; Aoyagi, Y. Sustained response to interferon-alpha plus ribavirin therapy for chronic hepatitis C is closely associated with increased dynamism of intrahepatic natural killer and natural killer T cells. Hepatol. Res. 2008, 38, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Moriarty, T.J.; Wong, C.H.; Zhou, H.; Strieter, R.M.; van Rooijen, N.; Chaconas, G.; Kubes, P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat. Immunol. 2010, 11, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Bangen, J.M.; Govaere, O.; Zimmermann, H.W.; Gassler, N.; Huss, S.; Liedtke, C.; Prinz, I.; Lira, S.A.; Luedde, T.; et al. Chemokine receptor CCR6-dependent accumulation of γδ T cells in injured liver restricts hepatic inflammation and fibrosis. Hepatology 2014, 59, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Tacke, F. Role of gamma-delta T cells in liver inflammation and fibrosis. World J. Gastrointest. Pathophysiol. 2014, 5, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Kenna, T.; Golden-Mason, L.; Norris, S.; Hegarty, J.E.; O’Farrelly, C.; Doherty, D.G. Distinct subpopulations of gamma delta T cells are present in normal and tumor-bearing human liver. Clin. Immunol. 2004, 113, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Agrati, C.; D’Offizi, G.; Narciso, P.; Abrignani, S.; Ippolito, G.; Colizzi, V.; Poccia, F. Vdelta1 T lymphocytes expressing a Th1 phenotype are the major gammadelta T cell subset infiltrating the liver of HCV-infected persons. Mol. Med. 2001, 7, 11–19. [Google Scholar] [CrossRef]
- Wu, D.; Yan, W.M.; Wang, H.W.; Huang, D.; Luo, X.P.; Ning, Q. γδ T Cells Contribute to the Outcome of Murine Fulminant Viral Hepatitis via Effector Cytokines TNF-α and IFN-γ. Curr. Med. Sci. 2018, 38, 648–655. [Google Scholar] [CrossRef]
- Hou, W.; Wu, X. Diverse Functions of γδ T Cells in the Progression of Hepatitis B Virus and Hepatitis C Virus Infection. Front. Immunol. 2020, 11, 619872. [Google Scholar] [CrossRef]
- Tsuji, M.; Mombaerts, P.; Lefrancois, L.; Nussenzweig, R.S.; Zavala, F.; Tonegawa, S. Gamma delta T cells contribute to immunity against the liver stages of malaria in alpha beta T-cell-deficient mice. Proc. Natl. Acad. Sci. USA 1994, 91, 345–349. [Google Scholar] [CrossRef]
- Ribot, J.C.; Neres, R.; Zuzarte-Luís, V.; Gomes, A.Q.; Mancio-Silva, L.; Mensurado, S.; Pinto-Neves, D.; Santos, M.M.; Carvalho, T.; Landry, J.J.M.; et al. γδ-T cells promote IFN-γ-dependent. Proc. Natl. Acad. Sci. USA 2019, 116, 9979–9988. [Google Scholar] [CrossRef]
- McDonald, B.; Kubes, P. Neutrophils and intravascular immunity in the liver during infection and sterile inflammation. Toxicol. Pathol. 2012, 40, 157–165. [Google Scholar] [CrossRef]
- Weber, C. Liver: Neutrophil extracellular traps mediate bacterial liver damage. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 251. [Google Scholar] [CrossRef]
- Hilscher, M.B.; Shah, V.H. Neutrophil Extracellular Traps and Liver Disease. Semin. Liver Dis. 2020, 40, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Marchese, P.; Castro, M.G.; Lowenstein, P.R.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat. Med. 2005, 11, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, B.; Massberg, S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013, 13, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Louis, H.; Le Moine, A.; Flamand, V.; Nagy, N.; Quertinmont, E.; Paulart, F.; Abramowicz, D.; Le Moine, O.; Goldman, M.; Devière, J. Critical role of interleukin 5 and eosinophils in concanavalin A-induced hepatitis in mice. Gastroenterology 2002, 122, 2001–2010. [Google Scholar] [CrossRef]
- Pham, B.N.; Bemuau, J.; Durand, F.; Sauvanet, A.; Degott, C.; Prin, L.; Janin, A. Eotaxin expression and eosinophil infiltrate in the liver of patients with drug-induced liver disease. J. Hepatol. 2001, 34, 537–547. [Google Scholar] [CrossRef]
- Tsuda, K.; Maeda, T.; Tominaga, A.; Watanabe, Y.; Miyazaki, E.; Enzan, H.; Akisawa, N.; Iwasaki, S.; Saibara, T.; Onishi, S. Eosinophil-induced liver injury: An experimental model using IL-5 transgenic mice. J. Hepatol. 2001, 34, 270–277. [Google Scholar] [CrossRef]
- dos Santos, D.C.; da Silva Gomes Martinho, J.M.; Pacheco-Moreira, L.F.; Carvalho Viana de Araújo, C.; Caroli-Bottino, A.; Pannain, V.L.; Soares Trinta, K.; Gandini, M.; da Costa Neves, P.C.; de Souza Matos, D.C.; et al. Eosinophils involved in fulminant hepatic failure are associated with high interleukin-6 expression and absence of interleukin-5 in liver and peripheral blood. Liver Int. 2009, 29, 544–551. [Google Scholar] [CrossRef]
- Palacios-Macapagal, D.; Connor, J.; Mustelin, T.; Ramalingam, T.R.; Wynn, T.A.; Davidson, T.S. Cutting Edge: Eosinophils Undergo Caspase-1-Mediated Pyroptosis in Response to Necrotic Liver Cells. J. Immunol. 2017, 199, 847–853. [Google Scholar] [CrossRef]
- Proctor, W.R.; Chakraborty, M.; Chea, L.S.; Morrison, J.C.; Berkson, J.D.; Semple, K.; Bourdi, M.; Pohl, L.R. Eosinophils mediate the pathogenesis of halothane-induced liver injury in mice. Hepatology 2013, 57, 2026–2036. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Hogan, S.P. The eosinophil. Annu. Rev. Immunol. 2006, 24, 147–174. [Google Scholar] [CrossRef]
- Fisicaro, P.; Valdatta, C.; Massari, M.; Loggi, E.; Biasini, E.; Sacchelli, L.; Cavallo, M.C.; Silini, E.M.; Andreone, P.; Missale, G.; et al. Antiviral intrahepatic T-cell responses can be restored by blocking programmed death-1 pathway in chronic hepatitis B. Gastroenterology 2010, 138, 682–693. [Google Scholar] [CrossRef]
- Tzeng, H.T.; Tsai, H.F.; Liao, H.J.; Lin, Y.J.; Chen, L.; Chen, P.J.; Hsu, P.N. PD-1 blockage reverses immune dysfunction and hepatitis B viral persistence in a mouse animal model. PLoS ONE 2012, 7, e39179. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhang, H.; Sun, B.; Karin, M. The immunobiology of hepatocellular carcinoma in humans and mice: Basic concepts and therapeutic implications. J. Hepatol. 2020, 72, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- El-Khoueiry, A. The Promise of Immunotherapy in the Treatment of Hepatocellular Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Carambia, A.; Freund, B.; Schwinge, D.; Bruns, O.T.; Salmen, S.C.; Ittrich, H.; Reimer, R.; Heine, M.; Huber, S.; Waurisch, C.; et al. Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice. J. Hepatol. 2015, 62, 1349–1356. [Google Scholar] [CrossRef]
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J. Hepatol. 2014, 61, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Lüth, S.; Huber, S.; Schramm, C.; Buch, T.; Zander, S.; Stadelmann, C.; Brück, W.; Wraith, D.C.; Herkel, J.; Lohse, A.W. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J. Clin. Investig. 2008, 118, 3403–3410. [Google Scholar] [CrossRef]
- Alissafi, T.; Hatzioannou, A.; Ioannou, M.; Sparwasser, T.; Grün, J.R.; Grützkau, A.; Verginis, P. De novo-induced self-antigen-specific Foxp3+ regulatory T cells impair the accumulation of inflammatory dendritic cells in draining lymph nodes. J. Immunol. 2015, 194, 5812–5824. [Google Scholar] [CrossRef] [PubMed]
- Moorman, C.D.; Sohn, S.J.; Phee, H. Emerging Therapeutics for Immune Tolerance: Tolerogenic Vaccines, T cell Therapy, and IL-2 Therapy. Front. Immunol. 2021, 12, 657768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tu, E.; Kasagi, S.; Zanvit, P.; Chen, Q.; Chen, W. Manipulating regulatory T cells: A promising strategy to treat autoimmunity. Immunotherapy 2015, 7, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Richardson, N.; Ng, S.T.H.; Wraith, D.C. Antigen-Specific Immunotherapy for Treatment of Autoimmune Liver Diseases. Front. Immunol. 2020, 11, 1586. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, S87–S97. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vacani-Martins, N.; Meuser-Batista, M.; dos Santos, C.d.L.P.; Hasslocher-Moreno, A.M.; Henriques-Pons, A. The Liver and the Hepatic Immune Response in Trypanosoma cruzi Infection, a Historical and Updated View. Pathogens 2021, 10, 1074. https://doi.org/10.3390/pathogens10091074
Vacani-Martins N, Meuser-Batista M, dos Santos CdLP, Hasslocher-Moreno AM, Henriques-Pons A. The Liver and the Hepatic Immune Response in Trypanosoma cruzi Infection, a Historical and Updated View. Pathogens. 2021; 10(9):1074. https://doi.org/10.3390/pathogens10091074
Chicago/Turabian StyleVacani-Martins, Natalia, Marcelo Meuser-Batista, Carina de Lima Pereira dos Santos, Alejandro Marcel Hasslocher-Moreno, and Andrea Henriques-Pons. 2021. "The Liver and the Hepatic Immune Response in Trypanosoma cruzi Infection, a Historical and Updated View" Pathogens 10, no. 9: 1074. https://doi.org/10.3390/pathogens10091074
APA StyleVacani-Martins, N., Meuser-Batista, M., dos Santos, C. d. L. P., Hasslocher-Moreno, A. M., & Henriques-Pons, A. (2021). The Liver and the Hepatic Immune Response in Trypanosoma cruzi Infection, a Historical and Updated View. Pathogens, 10(9), 1074. https://doi.org/10.3390/pathogens10091074