
Biliary Migration, Colonization, and Pathogenesis of O. viverrini Co-Infected with CagA+ Helicobacter pylori

 ,
, 
Abstract
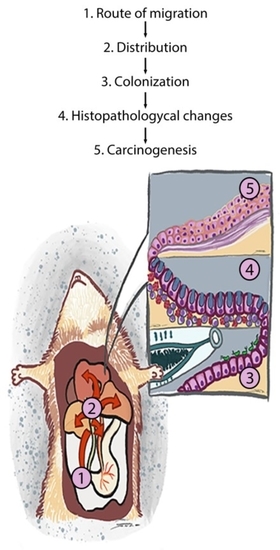
1. Introduction
2. Materials and Methods
2.1. Metacercaria Preparation
2.2. Bacterial Preparation
2.3. Experimental Design
2.4. DNA Extraction
2.5. Real-Time Polymerase Chain Reaction (qPCR) for gfp Gene Detection
2.6. Immunofluorescence for Localization of H. pylori-GFP
2.7. O. viverrini Excretory-Secretory Products Preparation
2.8. H. pylori Adhesion Assay
2.9. Epithelial Transmigration Assay
2.10. Histopathology and E-cadherin Immunohistochemistry
2.11. Data Analysis
3. Results
3.1. Organ Distribution of H. pylori in Infected Hamsters
3.2. Route of Migration of H. pylori in O. viverrini Co-Infection
3.3. O. viverrini Enhances H. pylori Colonization In Vivo
3.4. O. viverrini Excretory-Secretory Products Enhance H. pylori Binding to Bile Duct Epithelial Cells In Vitro
3.5. Effect of O. viverrini Excretory-Secretory Products on H. pylori Trans-Epithelial Migration and E-Cadherin Expression In Vitro
3.6. Co-Infection of cagA-Positive H. pylori and O. viverrini Enhances Biliary Epithelial Pathological Changes
3.7. Co-Infection of cagA Positive H. pylori and O. viverrini Reduces E-Cadherin Expression In Vivo
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sithithaworn, P.; Andrews, R.H.; Van De, N.; Wongsaroj, T.; Sinuon, M.; Odermatt, P.; Nawa, Y.; Liang, S.; Brindley, P.J.; Sripa, B. The current status of opisthorchiasis and clonorchiasis in the Mekong Basin. Parasitol. Int. 2012, 61, 10–16. [Google Scholar] [CrossRef]
- Sithithaworn, P.; Yongvanit, P.; Duenngai, K.; Kiatsopit, N.; Pairojkul, C. Roles of liver fluke infection as risk factor for cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2014, 21, 301–308. [Google Scholar] [CrossRef]
- Sripa, B.; Brindley, P.J.; Mulvenna, J.; Laha, T.; Smout, M.J.; Mairiang, E.; Bethony, J.M.; Loukas, A. The tumorigenic liver fluke Opisthorchis viverrini—multiple pathways to cancer. Trends. Parasitol. 2013, 28, 395–407. [Google Scholar] [CrossRef] [PubMed]
- IARC. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241. [Google Scholar]
- Sripa, B.; Deenonpoe, R.; Brindley, P.J. Co-infections with liver fluke and Helicobacter species: A paradigm change in pathogenesis of opisthorchiasis and cholangiocarcinoma? Parasitol. Int. 2017, 66, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Plieskatt, J.L.; Deenonpoe, R.; Mulvenna, J.P.; Krause, L.; Sripa, B.; Bethony, J.M.; Brindley, P.J. Infection with the carcinogenic liver fluke Opisthorchis viverrini modifies intestinal and biliary microbiome. FASEB J. 2013, 27, 4572–4584. [Google Scholar] [CrossRef]
- Deenonpoe, R.; Chomvarin, C.; Pairojkul, C.; Loukas, A.; Brindley, P.J.; Sripa, B. The carcinogenic liver fluke Opisthorchis viverrini is a reservoir for species of Helicobacter. Asian. Pac. J. Cancer. Prev. 2015, 16, 1751–1758. [Google Scholar] [CrossRef] [PubMed]
- Dangtakot, R.; Pinlaor, S.; Itthitaetrakool, U.; Chaidee, A.; Chomvarin, C.; Sangka, A.; Wilailuckana, C.; Pinlaor, P. Coinfection with Helicobacter pylori and Opisthorchis viverrini enhances the severity of hepatobiliary abnormalities in hamsters. Infect. Immun. 2017, 85, e00009–e00017. [Google Scholar] [CrossRef]
- Deenonpoe, R.; Mairiang, E.; Mairiang, P.; Pairojkul, C.; Chamgramol, Y.; Rinaldi, G.; Loukas, A.; Brindley, P.J.; Sripa, B. Elevated prevalence of Helicobacter species and virulence factors in opisthorchiasis and associated hepatobiliary disease. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Boonyanugomol, W.; Chomvarin, C.; Sripa, B.; Bhudhisawasdi, V.; Khuntikeo, N.; Hahnvajanawong, C.; Chumsuwan, A. Helicobacter pylori in Thai patients with cholangiocarcinoma and its association with biliary inflammation and proliferation. HPB 2012, 14, 177–184. [Google Scholar] [CrossRef]
- Bhamarapravati, N.; Thammavit, W.; Vajrasthira, S. Liver changes in hamsters infected with a liver fluke of man, Opisthorchis viverrini. Am. J. Trop. Med. Hyg. 1978, 27, 787–794. [Google Scholar] [CrossRef]
- Kaewkes, S.; Sripa, B. Migratory pattern of Opisthorchis viverrini in hamsters. Southeast Asian. J. Trop. Med. 2004, 35, 306–308. [Google Scholar]
- Heuermann, D.; Haas, R. A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Mol. Gen. Genet. 1998, 257, 519–528. [Google Scholar] [CrossRef]
- Schmitt, W.; Odenbreit, S.; Heuermann, D.; Haas, R. Cloning of the Helicobacter pylori recA gene and functional characterization of its product. Mol. Gen. Genet. 1995, 248, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Tatematsu, M.; Yamamoto, M.; Shimizu, N.; Yoshikawa, A.; Fukami, H.; Kaminishi, M.; Oohara, T.; Sugiyama, A.; Ikeno, T. Induction of Glandular Stomach Cancers in Helicobacter pylori-sensitive Mongolian Gerbils Treated with N-Methyl-N-nitrosourea and N-Methyl-N ′′ ′ -nitro-N-nitrosoguanidine in Drinking Water. Jpn. J. Cancer. Res. 1998, 89, 97–104. [Google Scholar] [CrossRef]
- Hussain, I.; Tasneem, F.; Umer, M.; Pervaiz, A.; Raza, M.; Arshad, M.I.; Shahzad, N. Specific and Quantitative Detection of Human Polyomaviruses BKPyV and JCPyV in the Healthy Pakistani Population. Virol. J. 2017, 14, 86. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Purification of Nucleic Acids by Extraction With Phenol:chloroform. CSH Protoc. 2006, 2006, 4455. [Google Scholar] [CrossRef]
- Whelan, J.A.; Russell, N.B.; Whelan, M.A. A method for the absolute quantification of cDNA using real-time PCR. J. Immunol. Methods 2003, 278, 261–269. [Google Scholar] [CrossRef]
- Aida, Y.; Pabst, M.J. Removal of endotoxin from protein solutions by phase separation using Triton X-114. J. Immunol. Methods 1990, 132, 191–195. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Sripa, B.; Tangkawattana, S.; Brindley, P.J. Update on Pathogenesis of Opisthorchiasis and Cholangiocarcinoma. Adv. Parasitol. 2018, 102, 97–113. [Google Scholar] [PubMed]
- Peek, R.M.; Crabtree, J.E. Helicobacter infection and gastric neoplasia. J. Pathol. 2006, 208, 233–248. [Google Scholar] [CrossRef]
- Ramirez, A.; Sánchez, R. Helicobacter pylori persistence: Biology and disease. Rev. Gastroenterol. Perú. 2008, 18, 258–266. [Google Scholar]
- Franceschi, F.; Gasbarrini, A.; Polyzos, S.A.; Kountouras, J. Extragastric diseases and Helicobacter pylori. Helicobacter 2015, 20, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, F.; Zuccalà, G.; Roccarina, D.; Gasbarrini, A. Clinical effects of Helicobacter pylori outside the stomach. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.A.N.; Fan, X.; Tang, Z.; Liu, L.I.; Tian, X.; Li, N. Detection of Helicobacter pylori DNA in peripheral blood from patients with peptic ulcer or gastritis. APMIS 2006, 114, 851–856. [Google Scholar] [CrossRef]
- Kalia, N.; Bardhan, K.D. Of blood and guts: Association between Helicobacter pylori and the gastric microcirculation. J. Gastroenterol. Hepatol. 2003, 18, 1010–1017. [Google Scholar] [CrossRef]
- Nath, G.; Gulati, A.K.; Shukla, V.K. Role of bacteria in carcinogenesis, with special reference to carcinoma of the gallbladder. World J. Gastroenterol. 2010, 16, 5395–5404. [Google Scholar] [CrossRef]
- Pellicano, R.; Ménard, A.; Rizzetto, M.; Mégraud, F. Helicobacter Species and Liver Diseases: Association or Causation? Lancet Infect. Dis. 2008, 8, 254–260. [Google Scholar] [CrossRef]
- Mannion, A.; Shen, Z.; Fox, J.G. Comparative genomics analysis to differentiate metabolic and virulence gene potential in gastric versus enterohepatic Helicobacter species. BMC Genom. 2018, 19, 830. [Google Scholar] [CrossRef]
- Camorlinga-Ponce, M.; Romo, C.; González-Valencia, G.; Muñoz, O.; Torres, J. Topographical localisation of cagA positive and cagA negative Helicobacter pylori strains in the gastric mucosa; an in situ hybridisation study. J. Clin. Pathol. 2004, 57, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Salama, N.R.; Hartung, M.L.; Müller, A. Life in the Human Stomach: Persistence Strategies of the Bacterial Pathogen Helicobacter pylori. Nat. Rev. Microbiol. 2013, 11, 385–399. [Google Scholar] [CrossRef]
- Ilver, D.; Arnqvist, A.; Ögren, J.; Frick, I.M.; Kersulyte, D.; Incecik, E.T.; Berg, E.D.; Covacci, A.; Engstrand, L.; Borén, T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 1998, 279, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, J.; Sondén, B.; Hurtig, M.; Olfat, F.O.; Forsberg, L.; Roche, N.; Ångström, J.; Larsson, T.; Teneberg, S.; Karlsson, K.A.; et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 2002, 297, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Sheu, B.S.; Wu, J.J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 4–23. [Google Scholar] [CrossRef] [PubMed]
- Pachathundikandi, S.K.; Tegtmeyer, N.; Backert, S. Signal transduction of Helicobacter pylori during interaction with host cell protein receptors of epithelial and immune cells. Gut. Microbes 2013, 4, 454–474. [Google Scholar] [CrossRef]
- Binh, T.T.T.; Nguyen, D.L.; Jala, I.; Dontumprai, R.; Plumworasawat, S.; Aighewi, O.; Ong, E.; Shawley, A.; Potriquet, J.; Saichua, P.; et al. Identification, recombinant protein production, and functional analysis of a M60-like metallopeptidase, secreted by the liver fluke Opisthorchis viverrini. Parasitol. Int. 2020, 75, 102050. [Google Scholar]
- Chen, X.M.; O’Hara, S.P.; LaRusso, N.F. The Immunobiology of Cholangiocytes. Immunol. Cell Biol. 2008, 86, 497–505. [Google Scholar] [CrossRef]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Chmiela, M.; Kupcinskas, J. Review: Pathogenesis of Helicobacter pylori Infection. Helicobacter 2019, 24, e12638. [Google Scholar] [CrossRef]
- Li, H.; Liao, T.; Debowski, A.W.; Tang, H.; Nilsson, H.O.; Stubbs, K.A.; Marshall, B.J.; Benghezal, M. Lipopolysaccharide Structure and Biosynthesis in Helicobacter pylori. Helicobacter 2016, 21, 445–461. [Google Scholar] [CrossRef]
- Ninlawan, K.; O’Hara, S.P.; Splinter, P.L.; Yongvanit, P.; Kaewkes, S.; Surapaitoon, A.; LaRusso, N.F.; Sripa, B. Opisthorchis viverrini excretory/secretory products induce toll-like receptor 4 upregulation and production of interleukin 6 and 8 in cholangiocyte. Parasitol. Int. 2010, 59, 616–621. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sawanyawisuth, K.; Silsirivanit, A.; Kunlabut, K.; Tantapotinan, N.; Vaeteewoottacharn, K.; Wongkham, S. Novel Carbohydrate Antigen Expression During Development of Opisthorchis Viverrini-Associated Cholangiocarcinoma in Golden Hamster: A Potential Marker for Early Diagnosis. Parasitol. Int. 2012, 61, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Waskito, L.A.; Salama, N.R.; Yamaoka, Y. Pathogenesis of Helicobacter pylori Infection. Helicobacter 2018, 23, e12516. [Google Scholar] [CrossRef]
- Jittimanee, J.; Sermswan, R.W.; Puapairoj, A.; Maleewong, W.; Wongratanacheewin, S. Cytokine Expression in Hamsters Experimentally Infected with Opisthorchis viverrini. Parasite Immunol. 2007, 29, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Cho, Y.; Lee, D.; Jeon, B.Y.; Ju, J.W.; Chung, S.; Pak, J.H. Clonorchis sinensis Excretory-Secretory Products Increase Malignant Characteristics of Cholangiocarcinoma Cells in Three-Dimensional Co-Culture with Biliary Ductal Plates. PLoS Pathog. 2019, 15, e1007818. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Tegtmeyer, N. Type IV Secretion and signal transduction of Helicobacter pylori CagA through interactions with host cell receptors. Toxins 2017, 9, 115. [Google Scholar] [CrossRef]
- Kaur, J.; Sawhney, M.; DattaGupta, S.; Shukla, N.K.; Srivastava, A.; Walfish, P.G.; Ralhan, R. Clinical significance of altered expression of β-catenin and E-cadherin in oral dysplasia and cancer: Potential link with ALCAM expression. PLoS ONE 2013, 8, e67361. [Google Scholar] [CrossRef] [PubMed]
- von Zeidler, S.V.; Souza Botelho, T.D.; Mendonça, E.F.; Batista, A.C. E-cadherin as a potential biomarker of malignant transformation in oral leukoplakia: A retrospective cohort study. BMC Cancer 2014, 17, 972. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Guan, W.B.; Wang, J.D.; Zhang, Y.; Gong, W.; Quan, Z.W. A Comparative Study of clinicopathological features between chronic cholecystitis patients with and without Helicobacter pylori Infection in gallbladder mucosa. PLoS ONE 2013, 8, e70265. [Google Scholar] [CrossRef]
- Choi, Y.L.; Xuan, Y.H.; Shin, Y.K.; Chae, S.W.; Kook, M.C.; Sung, P.H.; Youn, S.J.; Choi, J.W.; Kim, S.H. An immunohistochemical study of the expression of adhesion molecules in gallbladder lesions. J. Histochem. Cytochem. 2004, 52, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Techasen, A.; Loilome, W.; Namwat, N.; Khuntikeo, N.; Puapairoj, A.; Jearanaikoon, P.; Saya, H.; Yongvanit, P. Loss of E-cadherin Promotes Migration and Invasion of Cholangiocarcinoma Cells and Serves as a Potential Marker of Metastasis. Tumour Biol. 2014, 35, 8645–8652. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Perez-Perez, G.I.; Kleanthous, H.; Cover, T.L.; Peek, R.M.; Chyou, P.H.; Stemmermann, G.N.; Nomura, A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995, 55, 2111–2115. [Google Scholar]
- Covacci, A.; Censini, S.; Bugnoli, M.; Petracca, R.; Burroni, D.; Macchia, G.; Massone, A.; Papini, E.; Xiang, Z.; Figura, N. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. USA 1993, 90, 5791–5795. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Taylor, J.D.; Wyatt, J.I.; Heatley, R.V.; Shallcross, T.M.; Tompkins, D.S.; Rathbone, B.J. Mucosal IgA recognition of Helicobacter pylori 120 kda protein, peptic ulceration, and gastric pathology. Lancet 1991, 338, 332–335. [Google Scholar] [CrossRef]
- Gwack, J.; Shin, A.; Kim, C.S.; Ko, K.P.; Kim, Y.; Jun, J.K.; Bae, J.; Park, S.K.; Hong, Y.C.; Kang, D.; et al. CagA-producing Helicobacter pylori and increased risk of gastric cancer: A nested case-control study in Korea. Br. J. Cancer 2006, 95, 639–641. [Google Scholar] [CrossRef]
- Valle, J.; Gisbert, J.P. Helicobacter pylori Infection and precancerous lesions of the stomach. Hepato-Gastroenterol. 2001, 48, 1548–1551. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Month. | Groups | 1st Order Bile Duct | 2nd Order Bile Duct | Worm | |||
|---|---|---|---|---|---|---|---|
| Surface | Peri-Nucleus | Crypt | Tegument | Gut | |||
| 1 | OV/HP+ | ++ | + | 20% (1/5) | - | + | + |
| OV/HP- | +++ | + | 0% (0/5) | - | + | - | |
| HP+ | - | - | 0% (0/5) | - | NA | NA | |
| HP- | - | - | 0% (0/5) | - | NA | NA | |
| 3 | OV/HP+ | + | +++ | 20% (1/5) | - | + | + |
| OV/HP- | + | +++ | 0% (0/5) | - | + | - | |
| HP+ | - | - | 0% (0/5) | - | NA | NA | |
| HP- | - | - | 0% (0/5) | - | NA | NA | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suyapoh, W.; Tirnitz-Parker, J.E.E.; Tangkawattana, S.; Suttiprapa, S.; Sripa, B. Biliary Migration, Colonization, and Pathogenesis of O. viverrini Co-Infected with CagA+ Helicobacter pylori. Pathogens 2021, 10, 1089. https://doi.org/10.3390/pathogens10091089
Suyapoh W, Tirnitz-Parker JEE, Tangkawattana S, Suttiprapa S, Sripa B. Biliary Migration, Colonization, and Pathogenesis of O. viverrini Co-Infected with CagA+ Helicobacter pylori. Pathogens. 2021; 10(9):1089. https://doi.org/10.3390/pathogens10091089
Chicago/Turabian StyleSuyapoh, Watcharapol, Janina E. E. Tirnitz-Parker, Sirikachorn Tangkawattana, Sutas Suttiprapa, and Banchob Sripa. 2021. "Biliary Migration, Colonization, and Pathogenesis of O. viverrini Co-Infected with CagA+ Helicobacter pylori" Pathogens 10, no. 9: 1089. https://doi.org/10.3390/pathogens10091089
APA StyleSuyapoh, W., Tirnitz-Parker, J. E. E., Tangkawattana, S., Suttiprapa, S., & Sripa, B. (2021). Biliary Migration, Colonization, and Pathogenesis of O. viverrini Co-Infected with CagA+ Helicobacter pylori. Pathogens, 10(9), 1089. https://doi.org/10.3390/pathogens10091089