Abstract
Background: Neuronal apoptosis is a major contributor to Alzheimer’s disease (AD). Periodontitis is a significant risk factor for AD. The periodontal pathogens Porphyromonas gingivalis and Treponema denticola have been shown to initiate the hallmark pathologies and behavioral symptoms of AD. Studies have found that T. denticola infection induced Tau hyperphosphorylation and amyloid β accumulation in the hippocampi of mice. Aβ accumulation is closely associated with neuronal apoptosis. However, the roles of T. denticola in neuronal apoptosis remain unclear and its roles in AD pathology need further study. Objective: This study aimed to investigate whether oral infection with T. denticola induced alveolar bone loss and neuronal apoptosis in mice. Methods: C57BL/6 mice were orally administered with T. denticola, Micro-CT was employed to assess the alveolar bone resorption. Western blotting, quantitative PCR, and TUNEL staining were utilized to detect the apoptosis-associated changes in mouse hippocampi. N2a were co-cultured with T. denticola to verify in vivo results. Results: Mice infected with T. denticola exhibited more alveolar bone loss compared with the control mice. T. denticola oral infection induced neuronal apoptosis in hippocampi of mice. Consistent results of the apoptosis-associated protein expression were observed in N2a cells treated with T. denticola and Aβ1–42 in vitro. However, the Aβ inhibitor reversed these results, suggesting that Aβ1–42 mediates T. denticola infection-induced neuronal apoptosis. Conclusions: This study found that oral infected T. denticola caused alveolar bone loss, and induced neuronal apoptosis by promoting Aβ accumulation in mice, providing evidence for the link between periodontitis and AD.
1. Introduction
Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder characterized by memory and cognitive decline, disorientation, and personality changes [1]. It is a chronic disease with a high incidence of 10–50% in people over age 65, affects 43.8 million people worldwide, and is the fifth leading cause of death in the world [2,3]. Senile plaques composed of Aβ, neurofibrillary tangles (NFTs) formed by hyperphosphorylated tau, and progressive loss of synapses and neurons [4] are pathological hallmarks of AD. The causes of AD are unclear. However, patients with AD exhibit neuroinflammation associated with infection, including microglial cell activation and altered inflammatory factor profiles, implying that infection may be an etiology of AD [5].
Chronic periodontitis is a common and widespread oral infectious disease [6]. Periodontal pathogens and their virulence factors, such as Porphyromonas gingivalis (P. gingivalis) and Treponema denticola (T. denticola), can enter distant organs through the circulatory system via erosive and swollen periodontal tissue, resulting in the onset and progression of a variety of systemic diseases [7]. Chronic periodontitis has been identified as a major risk factor for AD [8,9,10]. Epidemiological investigations have shown that the degree of cognitive impairment in patients with severe periodontitis is three times greater than that of patients with mild periodontitis or without periodontitis [11]. In the elderly with normal cognitive function, alveolar bone resorption is positively correlated with Aβ deposition in brain tissues [12]. Studies have shown that P. gingivalis can enter the brain tissue of AD patients, and induce Aβ accumulation [13,14], tau hyperphosphorylation, neuroinflammation, and neuron loss [15,16]. Furthermore, as a predominant spirochete in the subgingival plaque of the gingival crevice and periodontal pocket, a possible link between T. denticola and AD has been reported [17]. One study demonstrated that spirochetes were found in 91 percent of 495 brain and blood samples from AD patients, but 0 percent of 185 samples from controls [18]. T. denticola has also been detected in the postmortem brain tissues of AD patients using PCR [19]. Our previous research demonstrated that oral infections of T. denticola could promote AD pathology in the hippocampi of mice, including an increase in Aβ burden [20], tau hyperphosphorylation, and neuroinflammation [21]. However, further studies are needed to determine the relationship between T. denticola and AD.
Neuronal apoptosis is a major contributor to neurodegenerative disorders such as AD, Parkinson’s disease, and amyotrophic lateral sclerosis [22]. Johnson reported on the potential role of neuronal apoptosis in AD in 1994 [23]. Numerous studies have found the relationship between Aβ and neuronal apoptosis in AD [24,25]. Aβ accumulation, in particular, contributes to neurodegeneration by activating pro-apoptotic proteins to induce mitochondrial dysfunction [26]. Zhao and Huang et al. discovered that intracellular Aβ aggregation occurred in the early stages of AD, and Aβ aggregation in the cytoplasm might cause structural damage in synapses, functional abnormalities, and neuronal apoptosis [27]. Kawahara reported that Aβ oligomers caused neuronal cell death in the later stage of AD [28]. The most common subtypes of Aβ, Aβ1–40, and Aβ1–42 are neurotoxic and play key roles in neuronal apoptosis and cognitive impairment in AD [29]. T. denticola infection induced Aβ1–40 and Aβ1–42 accumulation in mouse hippocampi in our previous study [20]. Therefore, we hypothesized that T. denticola infection might induce neuronal apoptosis and accelerate the pathological progression of AD by increasing the Aβ burden.
In this study, a mouse model was used to investigate whether oral infection with T. denticola induced alveolar bone resorption and neuronal apoptosis in the hippocampi. Mouse neuroblastoma N2a cells were incubated with T. denticola suspension in vitro to validate the pro-apoptotic effect and underlying mechanisms.
2. Results
2.1. T. denticola Induced Alveolar Bone Resorption and Neuronal Apoptosis in the Mouse Hippocampi
T. denticola and P. gingivalis were detected in the saliva of all mice administered the specific bacteria orally, indicating that the bacteria colonized successfully (Figure S2). ABL was measured as the distance from the cementoenamel junction (CEJ) of the second maxillary molar to the alveolar bone crest (ABC) in three-dimensional reconstruction images. The Micro-CT images revealed that mice infected with T. denticola and P. gingivalis exhibited significantly more ABL than control mice (Figure 1A). TUNEL staining results revealed that hippocampi of mice in the T. denticola infection group and the P. gingivalis infection group exhibited significantly higher apoptosis rates than those in the blank control group (Figure 1B). The expression of cleaved caspase-3 was determined by western blotting. As shown in Figure 1C, the level of cleaved caspase-3 was significantly higher in the hippocampi of the T. denticola and the P. gingivalis infection groups compared with the blank control group.
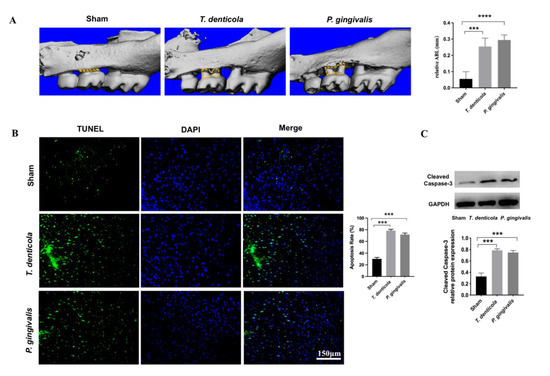
Figure 1.
Oral infection with T. denticola induced alveolar bone resorption and neuronal apoptosis in the hippocampi of mice. (A) Morphometric evaluation of total horizontal ABL in mice. The yellow dotted line indicates the area between the CEJ of the maxillary molar and the ABC; (B) The apoptosis rate in the hippocampi of mice was examined using TUNEL staining, n = 4 mice per group. (C) The protein levels of cleaved caspase-3 in the mouse hippocampus were examined by western blotting, n = 3 mice per group. Results are presented as the mean ± SD, ***: p < 0.001, ****: p < 0.0001.
2.2. T. denticola Oral Infection Regulated the Expressions of Apoptosis-Associated Genes and Proteins
The hippocampi of T. denticola- or P. gingivalis- infected mice had higher levels of BCL2-like 11 (Bim) and BCL2-associated X protein (Bax), and lower levels of B cell leukemia/lymphoma 2 (Bcl2) and Bclw compared with the sham group (Figure 2A). Furthermore, Bclw showed the greatest change. Gene expression levels of Jnk2, Jnk3, and Smac were examined, and the results revealed that the expression levels of Jnk2 and Smac were significantly up-regulated (Figure 2A). Moreover, BCL-W protein was significantly down-regulated, whereas SAPK/JNK and SMAC were significantly up-regulated in the hippocampi of the T. denticola- or P. gingivalis-infected groups (Figure 2B).
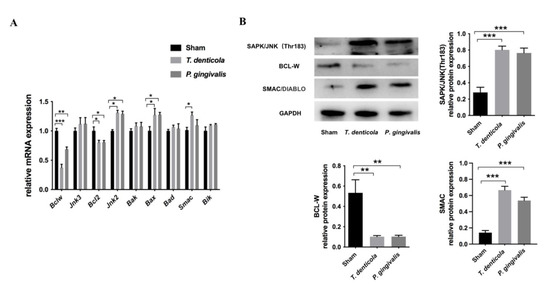
Figure 2.
T. denticola oral infection regulated the expression levels of apoptosis-associated genes and proteins. (A) The gene expression levels of Bclw, Bcl2, Bik, Bax, Bak, Bad, Jnk2, Jnk3, and Smac in the hippocampi of mice were measured using Quantitative real-time PCR (qRT-PCR). The values are shown as the mean ± SD of three independent experiments, n = 4 mice per group. (B) The protein levels of BCL-W, SAPK/JNK, and SMAC in the mice hippocampi were examined by western blotting, and the results were quantitatively analyzed. Results are presented as the mean ± SD, n = 3 mice per group *: p < 0.05, **: p < 0.01, ***: p < 0.001.
2.3. Amyloid-β1–42 Mediated T. denticola Infection-Induced Neuronal Apoptosis
To test if T. denticola directly promoted neuronal apoptosis, N2a cells were cultured with T. denticola. The results showed that apoptosis and the protein level of cleaved caspase-3 were significantly higher in N2a cells co-cultured with T. denticola than those in the control group, which was consistent with the results in vivo (Figure 3A,B). To further explore the potential mechanisms, N2a cells were stimulated with Aβ1–40, and Aβ1–42. The apoptosis rates were significantly increased in the Aβ1–42- and T. denticola-treated groups, whereas there was no statistical difference between the Aβ1–40 group and the control group (Figure 3A). The levels of cleaved caspase-3 of Aβ1–42- and T. denticola-treated groups were significantly higher than in the control group (Figure 3B). The expression levels of SAPK/JNK and SMAC proteins were increased, while the expression of BCL-W protein was decreased in both the Aβ1–42- and T. denticola-treated groups (Figure 3C). The expression levels of cleaved caspase-3 and BCL-W changed after Aβ1–40 treatment, while the expression levels of SAPK/JNK and SMAC did not differ between the Aβ1–40 and control groups (Figure 3B,C).
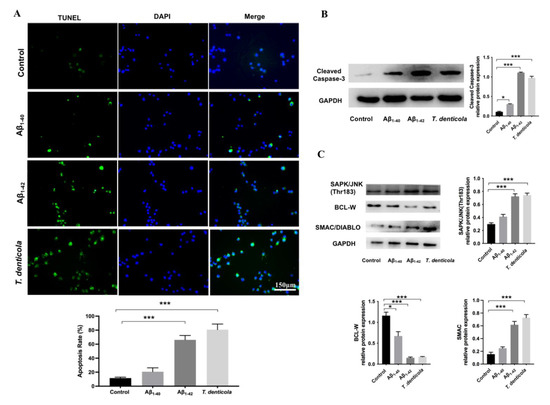
Figure 3.
T. denticola and Aβ1–42 induced apoptosis in N2a cells via a similar mechanism. (A) N2a cells were co-cultured with T. denticola, Aβ1–40, and Aβ1–42, and the apoptosis rate was examined using TUNEL staining. (B) The protein levels of cleaved caspase-3 in N2a cells in each group were examined by western blotting, and the results were quantitatively analyzed. (C) The protein levels of SAPK/JNK, BCL-W, and SMAC in N2a cells in each group were examined by western blotting, and the results were quantitatively analyzed. Results are presented as the mean ± SD, *: p < 0.05, ***: p < 0.001, n = 3 per experiment.
Moreover, the N2a cells were pretreated with an Aβ inhibitor KMI1303, then cocultured with T. denticola. The apoptosis rate was significantly decreased following KMI1303 treatment (Figure 4A). Compared with the T. denticola-treated group, the levels of cleaved caspase-3, SMAC, and SAPK/JNK were significantly decreased, and the level of BCL-W, was significantly increased in N2a cells pretreated with KMI1303 (Figure 4B). All the results suggest that Aβ1–42 mediates T. denticola infection-induced neuronal apoptosis.
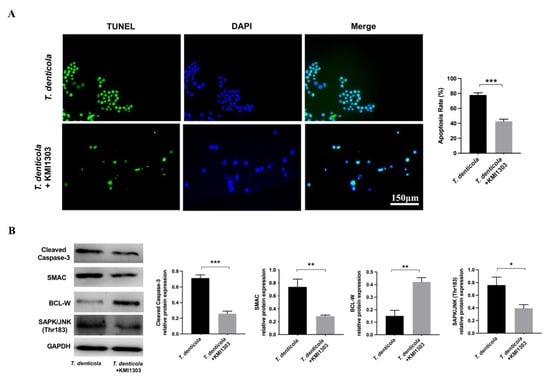
Figure 4.
Amyloid-β mediated the effect of T. denticola infection on neuronal apoptosis. (A) N2a cells were pretreated with KMI1303, then co-cultured with T. denticola, and the apoptosis rate was examined using TUNEL staining. (B) The protein levels of cleaved caspase-3, BCL-W, SAPK/JNK, and SMAC in N2a cells in each group were assessed by western blotting, and the results were quantitatively analyzed. Results are presented as the mean ± SD, *: p < 0.05, **: p < 0.01, ***: p < 0.001, n = 3 per experiment.
3. Discussion
Chronic periodontitis has been identified as a significant risk factor for AD [12]. Infection with periodontal pathogens, such as P. gingivalis and T. denticola, promotes AD pathogenesis in the hippocampi of mice, including Aβ accumulation, tau hyperphosphorylation, and inflammatory responses [13,14,16]. Noguchi and Moore reported that T. denticola could infect the cerebral cortex and cause atrophic dementia, cortical atrophy, and local amyloidosis as early as 1913 [30]. Increasing numbers of studies have suggested that T. denticola may contribute to AD pathogenesis [17,31,32,33]. Our previous study found that oral T. denticola infection caused bacterial invasion in the brain and increased the Aβ burden in the hippocampi of mice. Aβ accumulation is closely associated with neuronal apoptosis, which is a key factor in the pathological process of AD [22]. In this study, we aimed to explore how T. denticola induces neuronal apoptosis in AD.
We found an increased neuronal apoptosis rate following T. denticola exposure in vivo and in vitro. Based on our previous study, P. gingivalis infection was used as a positive control for apoptosis in mouse hippocampi. Our findings were consistent with previous research that found apoptotic changes in AD brain tissue slices, with apoptosis rates in patients with AD up to 50 times higher compared to those in the control group [34,35]. The caspase family of proteins is a primary apoptosis effector, among which cleaved caspase-3 is a marker of early apoptosis [36,37,38]. Therefore, we measured the expression of cleaved caspase-3 both in vivo and in vitro. The results showed that the levels of cleaved caspase-3 were significantly higher in the hippocampi of mice infected with T. denticola and in N2a cells stimulated with T. denticola. These findings indicated that T. denticola oral infection could induce neuronal apoptosis.
To investigate the potential mechanism by which T. denticola oral infection triggered apoptosis in the hippocampi of mice, we further looked at the mRNA levels of Bcl2 and its family members, Jnk2, Jnk3, and Smac, which are important in regulating the mitochondrial pathway in apoptosis. The BCL-2 protein family regulates cellular life and death signals and mediates intrinsic apoptotic pathways [39]. Studies have reported that aberrant expression of BCL-2 family proteins is closely related to AD [40]. Pro-survival (e.g., BCL-2, BCL-XL, BCL-W) and pro-death (e.g., BAK, BAX, BID, BIM, BIK) proteins are members of the BCL-2 family. We found that Bcl-2 and Bclw were down-regulated and Bax and Bim were up-regulated, with Bclw showing the most significant change. BCL-W is an anti-apoptotic protein that protects against AD pathology in vivo and in vitro [41]. In addition, BCL-W has been shown to protect neurons from Aβ-induced neuronal apoptosis by inhibiting the mitochondrial release of SMAC [42]. Consistent with these findings, our study provided further evidence of the role of BCL-W in neuronal apoptosis.
SAPK/JNK, a distant member of the mitogen-activated protein kinase (MAPK) superfamily, has previously been shown to trigger neuronal degeneration and death in different brain pathological conditions and diseases [43,44]. JNK has three isoforms (JNK1, 2, and 3) that can play different roles in the regulation of apoptosis [45]. JNK activation has been linked to transcriptional regulation of BCL-2 family members and plays a critical role in neuron death, senile plaque formation, and tau phosphorylation in AD [46,47,48]. Moreover, Yao et al. reported that Aβ reduced BCL-W protein levels via a JNK-dependent mechanism [46]. A previous study found that SMAC/DIABLO, a mitochondrial protein that may promote apoptosis by neutralizing one or more members of the IAP family of apoptosis inhibitory proteins [49]. SMAC was released from the mitochondrial inner membrane space into the cytoplasm to bind and activate caspases under pro-apoptotic conditions and in response to mitochondrial outer membrane permeabilization [50]. However, BCL-W inhibited the release of SMAC from mitochondria and prevented apoptosis [51]. Studies have suggested that downregulation of BCL-W and subsequent SMAC release might be key components in the pathway of Aβ-induced neuronal apoptosis [46]. Consistent with these findings, we discovered that in response to T. denticola infection, BCL-W was down-regulated, while SAPK/JNK and SMAC were up-regulated, implying that T. denticola may induce neuronal apoptosis via activation of the JNK pathway, down-regulation of the anti-apoptotic protein BCL-W, and release of the pro-apoptotic protein SMAC. However, further studies are needed to address the role of T. denticola infection in neuronal apoptosis in AD.
Aβ aggregation is a key step in the pathological process of AD. Previous research has shown that Aβ accumulated primarily extracellularly in brain tissue [52]. An increasing number of studies have found evidence for intracellular accumulation of Aβ [53,54,55]. In our previous study, we found that T. denticola can enter the mouse hippocampus and directly induce intra- and extracellular Aβ1–40 and Aβ1–42 accumulation in the hippocampus [20]. As Aβ is known to be neurotoxic and may mediate neuronal apoptosis through endogenous pathways, we hypothesized that T. denticola infection and Aβ might promote neuronal apoptosis through similar mechanisms. In the present study, Aβ1–42- and T. denticola-treated N2a cells showed increased apoptosis, up-regulation of cleaved caspase-3, SAPK/JNK, and SMAC, and down-regulation of BCL-W. Moreover, an Aβ inhibitor reversed the T. denticola-induced N2a cell apoptosis. KMI1303 is an inhibitor of β-secretase known to inhibit T. denticola-inducing Aβ production. In our previous study, we confirmed that the expression of Aβ1–42 in the KMI1303-treated group is significantly lower than that in the coculture group [20]. These results suggested that Aβ1–42 was involved in the effect of T. denticola infection on neuronal apoptosis. Aβ aggregation was found to be the initiating factor in neuronal degeneration in mice, and intracerebral injection of Aβ1–42 induced neuronal damage and caspase cleavage in the hippocampi of rats [56,57,58]. Mitochondrial apoptotic pathways that trigger mitochondrial dysfunction and DNA damage in Aβ-exposed cells are likely involved in the Aβ neuronal toxicity cascade [59]. Furthermore, Aβ induced apoptosis by modulating the expression of apoptosis-related genes, such as the BCL-2 family of proteins [60]. Longpre et al. observed JNK activation in Aβ-treated neurons and inhibition of JNK activation significantly attenuated Aβ-induced neuronal toxicity [61]. These results suggested that Aβ1–42 might mediate the effect of T. denticola on neuronal apoptosis by reducing BCL-W expression via JNK activation, resulting in SMAC release (Figure 5).
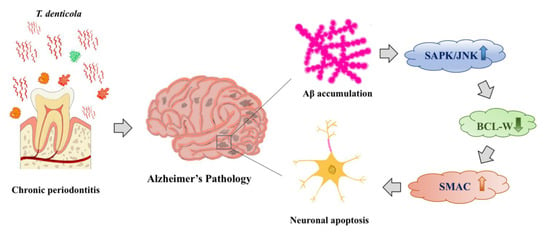
Figure 5.
Schematic overview of T. denticola-induced neuronal apoptosis. T. denticola infection induces Aβ accumulation in hippocampi, Aβ induces neuronal apoptosis via activation of the JNK pathway, down-regulation of the anti-apoptotic protein BCL-W, and release of the pro-apoptotic protein SMAC.
It is worth noting that previous studies have provided evidences that P. gingivalis plays a critical role in the pathogenesis of AD by inducing Aβ accumulation, Tau hyperphosphorylation, neuronal apoptosis, and neuroinflammation in mice [13,14,15,16]. Therefore, we set P. gingivalis-infection group as positive control, and the results showed that T. denticola- and P. gingivalis-infection groups had no significant difference in neuronal apoptosis, thus we did not set a P. gingivalis-infection in vitro experiments and mainly focused on the impact and mechanism of T. denticola infection in AD.
Further studies are needed to determine how oral T. denticola treatment could induce intracranial infection and neuro-amyloidosis. Several studies have revealed the relationship between chronic periodontitis or periodontal pathogens and blood-brain barrier (BBB) damage [62,63,64]. Pathogens and/or their virulence factors entering the brain facilitate BBB disruption in AD, which may be an early feature of the disease [65]. Future research needs to explore whether T. denticola infection disrupts the integrity of the hippocampal BBB and whether virulence factors of T. denticola can enter the hippocampus and play a role in the pathogenesis of AD.
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
T. denticola ATCC 35405 was cultured in a new oral spirochete (NOS) medium in an anaerobic system as described previously [20]. P. gingivalis ATCC 33277 was cultured in a brain-heart infusion medium (OXOID, Basingstoke, UK) supplemented with defibrinated sheep blood, hemin (0.5 mg/mL), and menadione (10 mg/mL) in an anaerobic system (Gene Science, Cambridge, MA, USA) [15]. Bacterial were collected and washed twice with PBS. Bacterial concentrations were measured using a spectrophotometer at an optical density of 600 nm, then diluted with PBS containing 3% carboxymethyl cellulose (CMC) to a concentration of 109 CFU/mL.
4.2. Mouse Treatment
All animal experiments were conducted at the State Key Laboratory of Oral Diseases and were approved by the Research Ethics Committee of West China Hospital of Stomatology (WCHSIRB-D-2019-013). Fifteen 8-week-old male C57BL/6 mice (20–22 g) were purchased from the Animal Experiment Center of Sichuan University. All the animal studies were reported following the ARRIVE guidelines. Mice were housed in individually ventilated cages in a standard environment (24–26 °C room temperature, 55% ± 10% humidity) under specific-pathogen-free conditions on a 12-h light/dark cycle with free access to water and food. They were randomly divided into three groups: sham-infection (blank control), T. denticola-infection (experimental group), and P. gingivalis-infection (positive control). (n = 5 in each group). G*Power 3.1 software (Düsseldorf, Germany) was used to calculate the sample size [66], which was based on the data from our previous study. According to the difference between two independent groups (t-test), the sample size was calculated based on the expression levels of Aβ1–40 in the hippocampi of two groups (Mean = 305.2, SD = 7.395/Mean = 553.8, SD = 100.2) with an alpha level of 0.05 (type II error) and a power of 95% (type I error). The sample size was calculated to be 4. Considering the possibility of unexpected death during the experiment, 5 mice were included in each group.
To suppress endogenous oral microorganisms, all mice were given 1 mg/mL of kanamycin in drinking water for 3 days before the first oral administration of periodontal bacteria. The experimental and positive control groups were orally administered the appropriate bacterium (109 CFU/mL/50 µL) for 24 weeks at a frequency of three times per week [14,67], while the blank control group received an equal volume of PBS with 3% CMC solution. The mice were anesthetized with pentobarbital sodium euthanasia one week after the final treatment, and their hearts were quickly perfused with chilled PBS (0.1 M, pH 7.3) (Figure S1A). The hippocampi were dissected, and the left hemispheres were fixed with 4% paraformaldehyde and the right hemispheres were stored at −80 °C.
4.3. PCR
A DNeasy Blood & Tissue Kit (Qiagen, Los Angeles, CA, USA) was used to extract genomic DNA from saliva. DNA amplification was performed with a PCR amplification kit (Takara, Tokyo, Japan). Briefly, the PCR mixture was made up of 12.5 μL Taq PCR Master Mix, 100 ng (100 ng/μL) DNA sample, 1 μL forward primer, 1 μL reverse primer, and 9.5 μL sterilized ddH2O. The reaction was carried out at 94 °C for 4 min, 40 cycles of 94 °C for 30 s, 55 °C for 5 s and 72 °C for 30 s/60 s; and 72 °C for 2 min. The amplified DNA products were electrophoresed on a 2% agarose gel at 100 V for 30 min. To determine whether the samples contained T. denticola ATCC 35405 or P. gingivalis ATCC 33277, the specific bands of the samples were compared to those in the positive group. The PCR primer sequences were as follows: (1) T. denticola: forward, 5′-TAATACCGAATGTGCTCATTTACAT-3′; reverse, 5′-CTGCCATATCTCTATGTCATTGCTCTT-3′; product, 860 bp; (2) P. gingivalis: forward, 5′-AGGCAGCTTGCCATACTGCG-3′; reverse, 5′-ACTGTTAGCAACTACCGATGT-3′; product, 405 bp (Sangon Biotech Co., Ltd., Shanghai, China).
4.4. Measurement of Alveolar Bone Loss
The maxilla was extracted and fixed in 4% paraformaldehyde for 48 h. Scanning was done with a Micro-CT instrument (Scanco Medical, Zurich, Switzerland) under the following conditions: samples were placed to make the long axis of the tooth parallel to the scanning ray, current 145 mA, voltage 55 kVp, resolution 12 μm, integration time 300 ms. Materialise Mimics software was used to perform the three-dimensional reconstruction. The distance from the CEJ to the ABC of the second maxillary molar was measured at three different sites (the distal root, root furcation groove, and mesial root) on the lingual side with ImageJ software. The alveolar bone level of the maxilla was calculated by averaging the values from these three sites.
4.5. Cell Culture and Treatment
N2a cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum in a humidified (5% CO2, 37 °C) incubator. Following confirmation of cell status, cells were plated in 6-well or 24-well tissue culture plates. The experimental groups were co-incubated with T. denticola (multiplicity of infection (MOI) 1:100) or 25 μM Aβ1–40, 25 μM Aβ1–42 for 2 h, while the control group was incubated with DMEM containing 10% FBS. The medium was then replaced with an equal volume of fresh medium, and the cells were cultured at 37 °C for 12 h (Figure S1B).
To investigate the role of Aβ in T. denticola-induced apoptosis, N2a cells in the experimental groups were pretreated with 1 μM KMI1303 (Bioss, Beijing, China) for 4 h before being incubated with the T. denticola for 2 h. The medium was then replaced with an equal volume of fresh medium, and the cells were cultured at 37 °C for 12 h (Figure S1C).
4.6. TUNEL Staining
TUNEL staining was carried out as described previously with some modifications using a TUNEL kit (Roche, Basel, Switzerland) [68]. Briefly, the left hippocampus was paraffinized and sectioned, then deparaffinized in xylene, rehydrated using an ethanol gradient (100%, 95%, 90%, 80%, and 70%; 3 min per rehydration step), washed in PBS (5 min), and incubated with 20 μg/mL proteinase K in 10 mM Tris-HCl, pH 7.4–7.8 at 37 °C for 20 min in a humidified chamber. TUNEL staining was used to detect DNA fragmentation [69]. The slides were incubated with a TUNEL reaction mixture in a humidified chamber at 37 °C for 60 min before being rinsed with PBS three times. The slides were incubated with converter-POD in a humidified chamber at 37 °C for 30 min, then with PBS three times. One hundred microliters of DAB (5 μL 20× diaminobenzidine 3 + 1 μL 30% H2O2 + 94 μL PBS) substrate was added to the slides and allowed to react at 15–25 °C for 10 min. The slides were rinsed with PBS and counterstained with hematoxylin. After washing with running water, the slides were dehydrated, cleared, and sealed with neutral balsam. Images were captured using a fluorescence microscope.
4.7. Western Blotting
Western blotting was performed as described in a previous study with some modifications [20]. Protein extracts from cells or hippocampi were prepared in a modified RIPA buffer supplemented with protease inhibitors (200612, Signalway Antibody, Greenbelt, MD, USA). The BCA method was used to determine the concentration of protein. The protein extracts were boiled after being diluted in SDS-PAGE protein loading buffer (5×) (Beyotime, Shanghai, China) at a ratio of 4:1. Following separation, the proteins were transferred to polyvinylidene difluoride membranes and blocked in TBST buffer (20 mM Tris–HCl, pH 7.4, 137 mM NaCl, and 0.1% Tween-20) with 5% non-fat milk at 37 °C for 1 h, and incubated at 4 °C with primary rabbit polyclonal antibodies (cleaved caspase-3, 49500, 1:500, Signalway Antibody; BCL-W, 40641, 1:1000, Signalway Antibody; SAPK/JNK (pThr183), 11249, 1:500, Signalway Antibody; SMAC/DIABLO, 39330, 1:500, Signalway Antibody; GAPDH, 21612, 1:3000, Signalway Antibody) overnight. After extensive rinsing, the membranes were incubated with the appropriate HRP-conjugated secondary antibody, then visualized using Super ECL Plus reagents. The gray values of the protein bands were quantified by the optical density using ImageJ software (1.41v, US National Institutes of Health, Bethesda, MD, USA).
4.8. qRT-PCR
The procedure was carried out in accordance with the MIQE guidelines [70]. Total RNA was extracted from mouse hippocampi or cells using an RNApure total RNA fast isolation kit (BioTeke, Beijing, China), then reverse-transcribed using an Evo M-MLV RT kit with gDNA Clean for qPCR II (Accurate Biology, Changsha, China) according to the manufacturer’s instructions. The resulting cDNA served as a template for quantitative PCR analysis using gene-specific primers (TSINGKE, Beijing, China). Real-time quantitative polymerase chain reactions were performed with TB Green Premix Ex Taq II (TAKARA, Tokyo, Japan) using an Applied Biosystems QuantStudio 6 Flex Real-Time PCR System. The cycling conditions were as follows: initial denaturation at 95 °C for 30 s, then 40 cycles of 95 °C for 5 s, and 60 °C for 30 s, followed by 95 °C for 15 s, 60 °C for 1 min, then 95 °C for 15 s. The fluorescence intensity was monitored at the end of each amplification step. Quantitative measurements of the target gene levels were normalized to GAPDH and the results were expressed as fold changes of the threshold cycle (Ct) value relative to control using the 2−ΔΔCt method. Primers and amplicon size were shown in Table 1.

Table 1.
Specific primer pairs used in qRT-PCR.
4.9. Statistical Analysis
Data were presented as the mean ± standard deviation (SD) and analyzed using SPSS 16.0 statistical software (SPSS Inc., Chicago, IL, USA). The student’s t-test was used to analyze statistical differences. Differences were considered significantly different if the p-value was <0.05.
5. Conclusions
In conclusion, this study showed that T. denticola oral infection could induce alveolar bone loss and neuronal apoptosis in mice. The potential mechanism might be related to the intrinsic mitochondrial pathway mediated by Aβ. These findings provided novel insights into the important role of T. denticola in AD pathogenesis and suggested that prevention and treatment of periodontitis may be beneficial for preventing and slowing the progression of AD.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/pathogens11101150/s1, Figure S1: Study design; Figure S2: Bacteria detection (determined by PCR) in the saliva of mice.
Author Contributions
Conceptualization, L.W., X.S., Z.T., L.J., H.Z., X.C. and H.W.; methodology, L.W., X.S., L.J., H.Z., X.C. and H.W.; validation, L.W., X.S.; formal analysis, L.W., X.S. and Z.T.; Project administration, L.W., X.S., X.C. and H.W.; writing—original draft preparation, L.W.; writing—review and editing, L.W., X.S. and X.C.; supervision, X.C. and H.W.; funding acquisition, X.C. and H.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by Science and Technology Program for Overseas Students in Sichuan Province (2021-29-1 to H.W.), Sichuan Province Science and Technology Support Program (2021YFSY0011 to H.W., 2022NSFSC1359 to X.C.), and Research Funding from West China Hospital of Stomatology Sichuan University (RCDWJS2021-16 to X.C.).
Institutional Review Board Statement
The animal study protocol was approved by the Research Ethics Committee of West China Hospital of Stomatology (WCHSIRB-D-2019-013).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author (X.C. and H.W.).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hardy, J. A hundred years of Alzheimer’s disease research. Neuron 2006, 52, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.B.D.D. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar]
- Hodson, R. Alzheimer’s disease. Nature 2018, 559, S1. [Google Scholar] [CrossRef]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.B. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. Periodontitis: Facts, fallacies and the future. Periodontol 2000 2017, 75, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Ren, J.; Yu, H.; Yu, W.; Zhou, Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun. Ageing 2018, 15, 6. [Google Scholar] [CrossRef]
- Ide, M.; Harris, M.; Stevens, A.; Sussams, R.; Hopkins, V.; Culliford, D.; Fuller, J.; Ibbett, P.; Raybould, R.; Thomas, R.; et al. Periodontitis and Cognitive Decline in Alzheimer’s Disease. PLoS ONE 2016, 11, e0151081. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Dasanayake, A.P.; Brys, M.; Glodzik-Sobanska, L.; de Leon, M.J. Inflammation and Alzheimer’s disease: Possible role of periodontal diseases. Alzheimers Dement. 2008, 4, 242–250. [Google Scholar] [CrossRef]
- Kamer, A.R.; Craig, R.G.; Niederman, R.; Fortea, J.; de Leon, M.J. Periodontal disease as a possible cause for Alzheimer’s disease. Periodontol 2000 2020, 83, 242–271. [Google Scholar] [CrossRef]
- Iwasaki, M.; Kimura, Y.; Ogawa, H.; Yamaga, T.; Ansai, T.; Wada, T.; Sakamoto, R.; Ishimoto, Y.; Fujisawa, M.; Okumiya, K.; et al. Periodontitis, periodontal inflammation, and mild cognitive impairment: A 5-year cohort study. J. Periodontal Res. 2019, 54, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Kamer, A.R.; Pirraglia, E.; Tsui, W.; Rusinek, H.; Vallabhajosula, S.; Mosconi, L.; Yi, L.; McHugh, P.; Craig, R.G.; Svetcov, S.; et al. Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol. Aging 2015, 36, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marcyzk, A.; Konradi, A.; Nguyn, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed]
- Ilievski, V.; Zuchowska, P.K.; Green, S.J.; Toth, P.T.; Ragozzino, M.E.; Le, K.; Aljewari, H.W.; O’Brien-Simpson, N.; Reynolds, E.C.; Watanabe, K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS ONE 2018, 13, e0204941. [Google Scholar] [CrossRef]
- Tang, Z.; Liang, D.; Cheng, M.; Su, X.; Liu, R.; Zhang, Y.; Wu, H. Effects of Porphyromonas gingivalis and Its Underlying Mechanisms on Alzheimer-Like Tau Hyperphosphorylation in Sprague-Dawley Rats. J. Mol. Neurosci. 2021, 71, 89–100. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y.; . Guo, J.; Sun, J.; Sun, Q. Salvianolic Acid B improves cognitive impairment by inhibiting neuroinflammation and decreasing Abeta level in Porphyromonas gingivalis-infected mice. Aging 2020, 12, 10117–10128. [Google Scholar] [CrossRef]
- Miklossy, J. Alzheimer’s disease—A spirochetosis? Neuroreport 1993, 4, 841–848. [Google Scholar] [CrossRef]
- Miklossy, J. Alzheimer’s disease—A neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J. Neuroinflamm. 2011, 8, 90. [Google Scholar] [CrossRef]
- Riviere, G.R.; Riviere, K.H.; Smith, K.S. Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol. Immunol. 2002, 17, 113–118. [Google Scholar] [CrossRef]
- Su, X.; Tang, Z.; Lu, Z.; Liu, Y.; He, W.; Jiang, J.; Zhang, Y.; Wu, H. Oral Treponema denticola Infection Induces Aβ1–40 and Aβ1–42 Accumulation in the Hippocampus of C57BL/6 Mice. J. Mol. Neurosci. 2021, 71, 1506–1514. [Google Scholar] [CrossRef]
- Tang, Z.; Cheng, X.; Su, X.; Wu, L.; Cai, Q.; Wu, H. Treponema denticola Induces Alzheimer-Like Tau Hyperphosphorylation by Activating Hippocampal Neuroinflammation in Mice. J. Dent. Res. 2022, 101, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.B.; Bonni, A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 2004, 72, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.M., Jr. Possible role of neuronal apoptosis in Alzheimer’s disease. Neurobiol. Aging 1994, 15, S187–S189. [Google Scholar] [CrossRef]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-beta is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimers Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef]
- Wirths, O.; Zampar, S. Emerging roles of N- and C-terminally truncated Aβ species in Alzheimer’s disease. Expert Opin. Ther. Targets 2019, 23, 991–1004. [Google Scholar] [CrossRef]
- Liu, T.; Wang, F.; LePochat, P.; Woo, J.-A.A.; Bukhari, M.Z.; Hong, K.W.; Trotter, C.; Kang, D.E. Cofilin-mediated Neuronal Apoptosis via p53 Translocation and PLD1 Regulation. Sci. Rep. 2017, 7, 11532. [Google Scholar] [CrossRef]
- Huang, J.-K.; Ma, P.-L.; Ji, S.-Y.; Zhao, X.-L.; Tan, J.-X.; Sun, X.-J.; Huang, F.-D. Age-dependent alterations in the presynaptic active zone in a Drosophila model of Alzheimer’s disease. Neurobiol. Dis. 2013, 51, 161–167. [Google Scholar] [CrossRef]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets—protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar]
- Lesné, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-β protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef]
- Noguchi, H.; Moore, J.W. A Demonstration of Treponema Pallidum in the Brain in Cases of General Paralysis. J. Exp. Med. 1913, 17, 232–238. [Google Scholar] [CrossRef]
- Gatz, M.; Mortimer, J.A.; Fratiglioni, L.; Johansson, B.; Berg, S.; Reynolds, C.A.; Pedersen, N.L. Potentially modifiable risk factors for dementia in identical twins. Alzheimers Dement. 2006, 2, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kornman, K.S. Interleukin 1 genetics, inflammatory mechanisms, and nutrigenetic opportunities to modulate diseases of aging. Am. J. Clin. Nutr. 2006, 83, 475S–483S. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.B. A Novel Approach to the Treatment and Prevention of Alzheimer’s Disease Based on the Pathology and Microbiology. J. Alzheimers Dis. 2021, 84, 61–67. [Google Scholar] [CrossRef]
- Su, J.H.; Anderson, A.J.; Cummings, B.J.; Cotman, C.W. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 1994, 5, 2529–2533. [Google Scholar] [CrossRef]
- Lucassen, P.J.; Chung, W.C.; Kamphorst, W.; Swaab, D.F. DNA damage distribution in the human brain as shown by in situ end labeling; area-specific differences in aging and Alzheimer disease in the absence of apoptotic morphology. J. Neuropathol. Exp. Neurol. 1997, 56, 887–900. [Google Scholar] [CrossRef]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef]
- Hugon, J.; Terro, F.; Esclaire, F.; Yardin, C. Markers of apoptosis and models of programmed cell death in Alzheimer’s disease. J. Neural. Transm. Suppl. 2000, 59, 125–131. [Google Scholar] [PubMed]
- Masliah, E.; Mallory, M.; Alford, M.; Tanaka, S.; Hansen, L.A. Caspase dependent DNA fragmentation might be associated with excitotoxicity in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1998, 57, 1041–1052. [Google Scholar] [CrossRef]
- Volkmann, N.; Marassi, F.M.; Newmeyer, D.D.; Hanein, D. The rheostat in the membrane: BCL-2 family proteins and apoptosis. Cell Death Differ 2014, 21, 206–215. [Google Scholar] [CrossRef]
- Zaitoun, I.S.; Wintheiser, C.M.; Jamali, N.; Wang, S.; Suscha, A.; Darjatmoko, S.R.; Schlek, K.; Hanna, B.A.; Lindner, V.; Sheibani, N.; et al. Bcl-2 Expression in Pericytes and Astrocytes Impacts Vascular Development and Homeostasis. Sci. Rep. 2019, 9, 9700. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, Y.; Ogawa, O.; Lee, H.; Raina, A.K.; Siedlek, S.L.; Harris, P.L.R.; Fujioka, H.; Shimohama, S.; Tabaton, M.; et al. Neuroprotective properties of Bcl-w in Alzheimer disease. J. Neurochem. 2004, 89, 1233–1240. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. BCL-w: Apoptotic and non-apoptotic role in health and disease. Cell Death Dis 2020, 11, 260. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Centeno, C.; Tomasi, S.; Forloni, G.; Bonny, C.; Vercelli, A.; Borsello, T. Time-course of c-Jun N-terminal kinase activation after cerebral ischemia and effect of D-JNKI1 on c-Jun and caspase-3 activation. Neuroscience 2007, 150, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Spigolon, G.; Veronesi, C.; Bonny, C.; Vercelli, A. c-Jun N-terminal kinase signaling pathway in excitotoxic cell death following kainic acid-induced status epilepticus. Eur. J. Neurosci. 2010, 31, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Dibling, B. The true face of JNK activation in apoptosis. Aging Cell 2002, 1, 112–116. [Google Scholar] [CrossRef]
- Yao, M.; Nguyen, T.V.; Pike, C.J. Beta-amyloid-induced neuronal apoptosis involves c-Jun N-terminal kinase-dependent downregulation of Bcl-w. J. Neurosci. 2005, 25, 1149–1158. [Google Scholar] [CrossRef]
- Marques, C.A.; Keil, U.; Bonert, A.; Steiner, B.; Haass, C.; Muller, W.E.; Eckert, A. Neurotoxic mechanisms caused by the Alzheimer’s disease-linked Swedish amyloid precursor protein mutation: Oxidative stress, caspases, and the JNK pathway. J. Biol. Chem. 2003, 278, 28294–28302. [Google Scholar] [CrossRef]
- Sahara, N.; Murayama, M.; Lee, B.; Park, J.-M.; Lagalwar, S.; Binder, L.I.; Takashima, A. Active c-jun N-terminal kinase induces caspase cleavage of tau and additional phosphorylation by GSK-3beta is required for tau aggregation. Eur. J. Neurosci. 2008, 27, 2897–2906. [Google Scholar] [CrossRef]
- Brenner, D.; Mak, T.W. Mitochondrial cell death effectors. Curr. Opin. Cell Biol. 2009, 21, 871–877. [Google Scholar] [CrossRef]
- Adrain, C.; Creagh, E.M.; Martin, S.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef]
- Hu, X.L.; Olsson, T.; Johansson, I.M.; Brännström, T.; Wester, P. Dynamic changes of the anti- and pro-apoptotic proteins Bcl-w, Bcl-2, and Bax with Smac/Diablo mitochondrial release after photothrombotic ring stroke in rats. Eur. J. Neurosci. 2004, 20, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, T.; Bieger, S.C.; Bruhl, B.; Tienari, P.J.; Ida, N.; Allsop, D.; Roberts, G.W.; Masters, C.L.; Dotti, C.G.; Unsicker, K.; et al. Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat. Med. 1997, 3, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Bathini, P.; Foucras, S.; Dupanloup, I.; Imeri, H.; Perna, A.; Berreux, J.-L.; Doucey, M.-A.; Annoni, J.-M.; Alberi, L.A. Classifying dementia progression using microbial profiling of saliva. Alzheimers Dement. 2020, 12, e12000. [Google Scholar] [CrossRef] [PubMed]
- Tzanoulinou, S.; Brandi, R.; Arisi, I.; D’Onofrio, M.; Urfer, S.M.; Sandi, C.; Constam, D.; Capsoni, S. Pathogen-free husbandry conditions alleviate behavioral deficits and neurodegeneration in AD10 anti-NGF mice. J. Alzheimers Dis. 2014, 38, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Loeb, M.B.; Molloy, D.W.; Smieja, M.; Standish, T.; Goldsmith, C.H.; Mahony, J.; Smith, S.; Borrie, M.; Decoteau, E.; Davidson, W.; et al. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 2004, 52, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Sun, C.; Zheng, M.; Liu, S.; Shi, R. Amentoflavone suppresses amyloid beta1-42 neurotoxicity in Alzheimer’s disease through the inhibition of pyroptosis. Life Sci. 2019, 239, 117043. [Google Scholar] [CrossRef]
- Miguel-Hidalgo, J.J.; Vecino, B.; Fernández-Novoa, L.; Álvarez, A.; Cacabelos, R. Neuroprotective role of S12024 against neurodegeneration in the rat dentate gyrus. Eur Neuropsychopharmacol 1998, 8, 203–208. [Google Scholar] [CrossRef]
- Viola, K.L.; Klein, W.L. Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. [Google Scholar] [CrossRef]
- Xu, S.; Zhong, M.; Zhang, L.; Wang, Y.; Zhou, Z.; Hao, Y.; Zhang, W.; Yang, X.; Wei, A.; Pei, L.; et al. Overexpression of Tfam protects mitochondria against beta-amyloid-induced oxidative damage in SH-SY5Y cells. FEBS J. 2009, 276, 3800–3809. [Google Scholar] [CrossRef]
- Wang, X.T.; Pei, D.S.; Xu, J.; Guan, Q.-H.; Sun, Y.-F.; Liu, X.-M.; Zhang, G.-Y. Opposing effects of Bad phosphorylation at two distinct sites by Akt1 and JNK1/2 on ischemic brain injury. Cell Signal 2007, 19, 1844–1856. [Google Scholar] [CrossRef]
- Longpre, F.; Garneau, P.; Christen, Y.; Ramassamy, C. Protection by EGb 761 against beta-amyloid-induced neurotoxicity: Involvement of NF-kappaB, SIRT1, and MAPKs pathways and inhibition of amyloid fibril formation. Free Radic. Biol. Med. 2006, 41, 1781–1794. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Nagano, K.; Sugiura, S.; Hagiwara, M.; Tanigawa, N.; Abiko, Y.; Yoshimura, F.; Furuichi, Y.; Matsushita, K. E-selectin mediates Porphyromonas gingivalis adherence to human endothelial cells. Infect. Immun. 2012, 80, 2570–2576. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Cheng, L.; Liu, D.; Wang, J.; Zhang, X.; Shu, R.; Liang, J. Role of p38 mitogen-activated protein kinase pathway in Porphyromonas gingivalis lipopolysaccharide-induced VCAM-1 expression in human aortic endothelial cells. J. Periodontol. 2012, 83, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Liu, Y.; Huang, W.; Qing, H.; Kadowaki, T.; Kashiwazaki, H.; Ni, J.; Wu, Z. Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid beta accumulation after Porphyromonas gingivalis infection. J. Neurochem. 2021, 158, 724–736. [Google Scholar] [CrossRef]
- Kouki, M.A.; Pritchard, A.B.; Alder, J.E.; Crean, S. Do Periodontal Pathogens or Associated Virulence Factors Have a Deleterious Effect on the Blood-Brain Barrier, Contributing to Alzheimer’s Disease? J. Alzheimers Dis. 2022, 85, 957–973. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.-G. Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef]
- Rivera, M.F.; Chukkapalli, S.S.; Velsko, I.M.; Lee, J.-Y.; Bhattacharyya, I.; Dolce, C.; Toro, E.J.; Holliday, S.; Kesavalu, L. Bis-enoxacin blocks rat alveolar bone resorption from experimental periodontitis. PLoS ONE 2014, 9, e92119. [Google Scholar] [CrossRef]
- Ribeiro, S.; Sharma, R.; Gupta, S.; Cakar, Z.; de Geyter, C.; Agarwal, A. Inter- and intra-laboratory standardization of TUNEL assay for assessment of sperm DNA fragmentation. Andrology 2017, 5, 477–485. [Google Scholar] [CrossRef]
- Gavrieli, Y.; Sherman, Y.; Ben-Sasson, S.A. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992, 119, 493–501. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).