
Genetic Diversity of Circumsporozoite Surface Protein of Plasmodium vivax from the Central Highlands, Vietnam

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Blood Samples
2.2. Amplification and Sequence Analysis of Vietnamese pvcsp
2.3. Analyses of Genetic Diversity and Natural Selection of pvcsp
2.4. Comparative Analyses of Genetic Diversity and Natural Selection in Global P. vivax Population
3. Results
3.1. Allelic Diversity of Vietnamese pvcsp
3.2. Polymorphism of the N-Terminal Non-Repeat Region in Vietnamese pvcsp
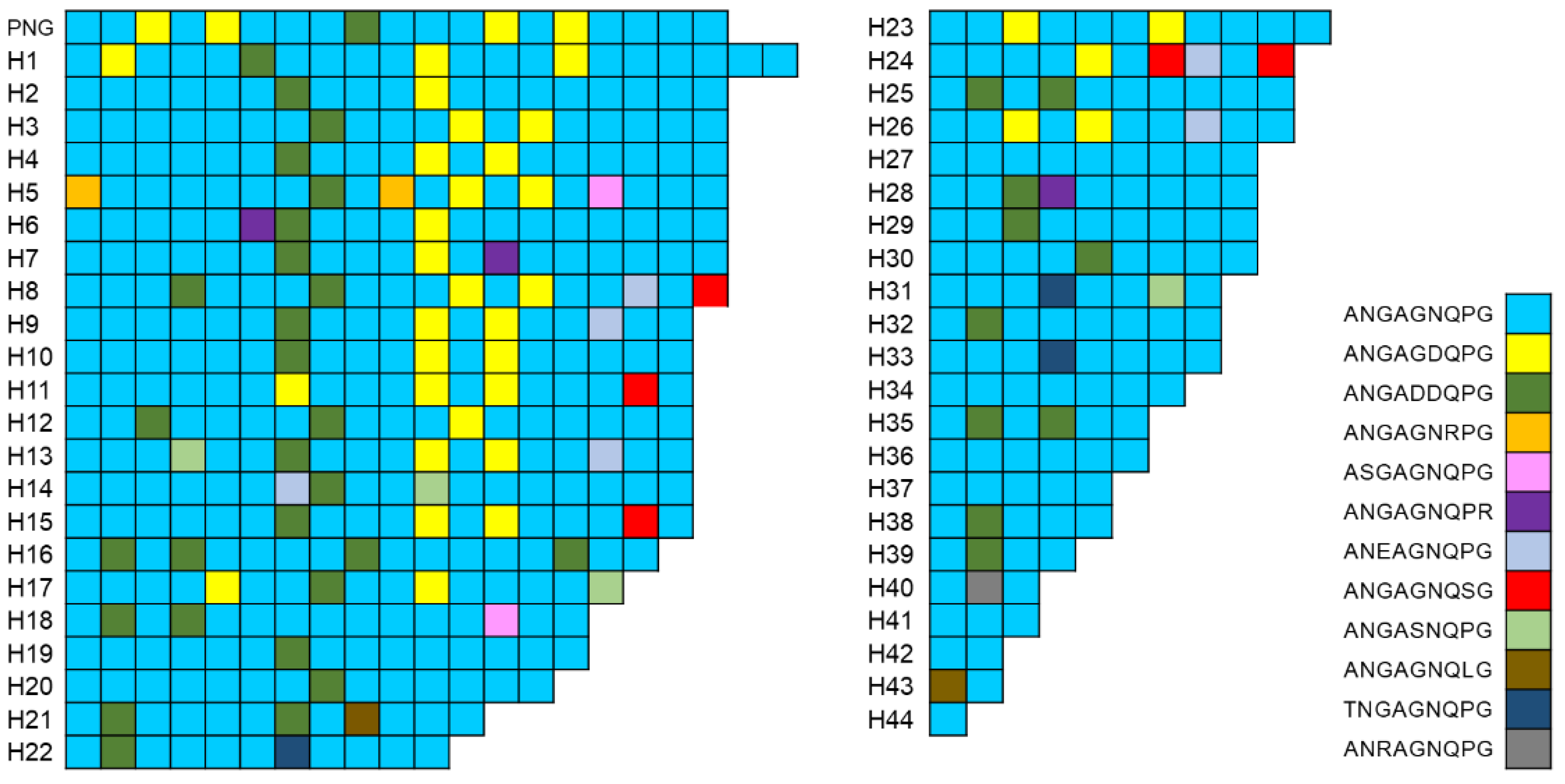
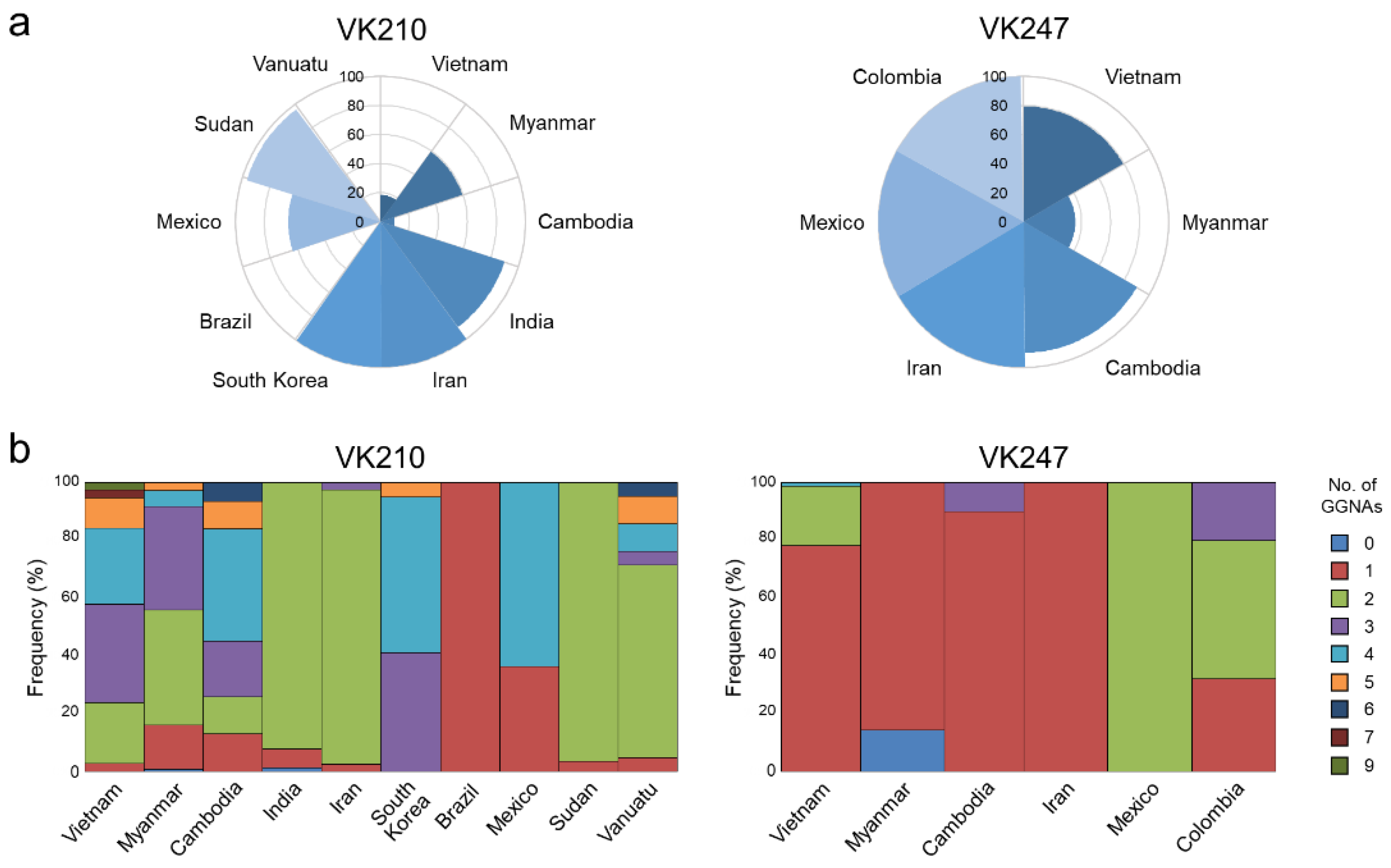
3.3. Polymorphic Pattern of the CRR in Vietnamese pvcsp
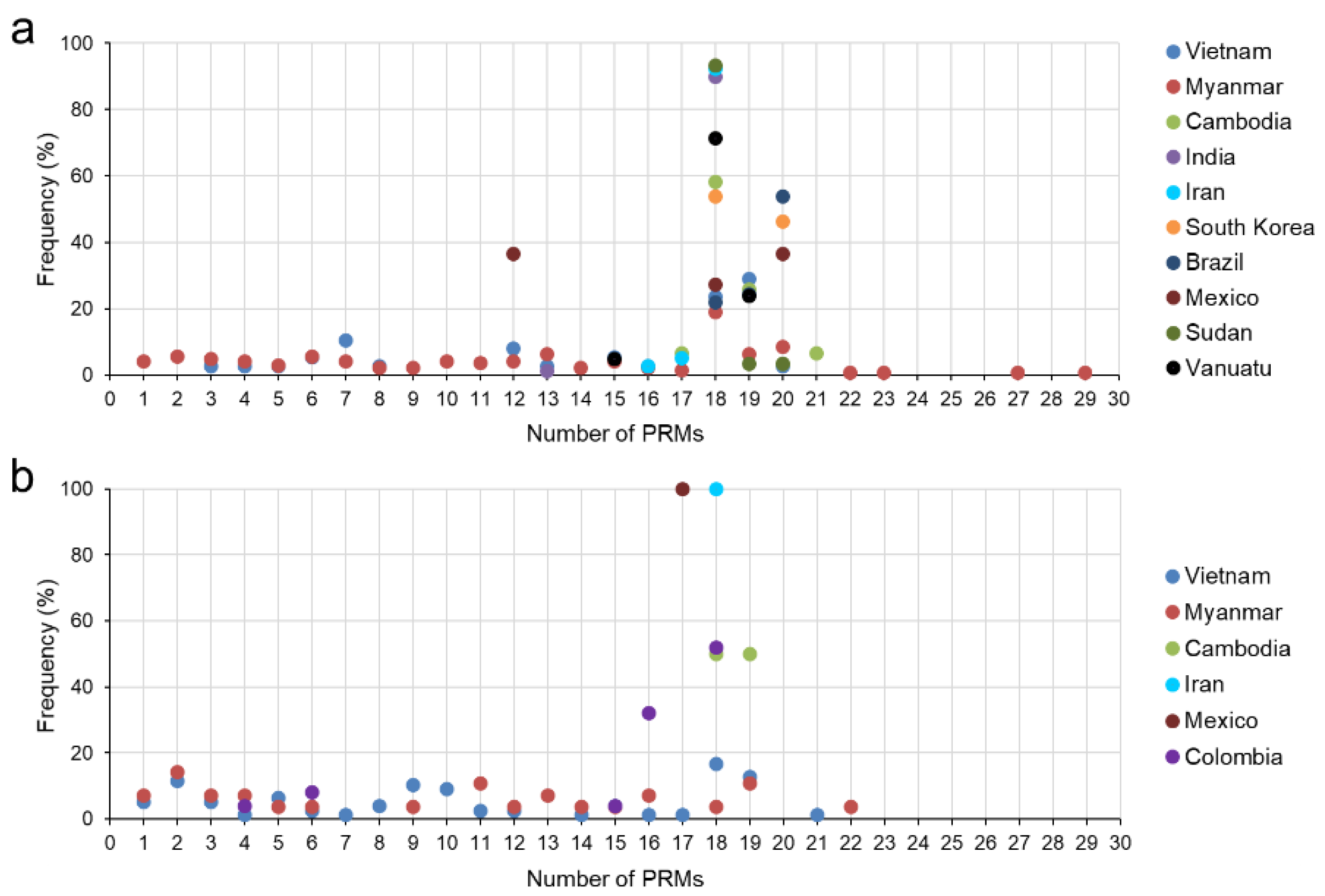
3.4. Size Polymorphism in the CRR of the Global pvcsp
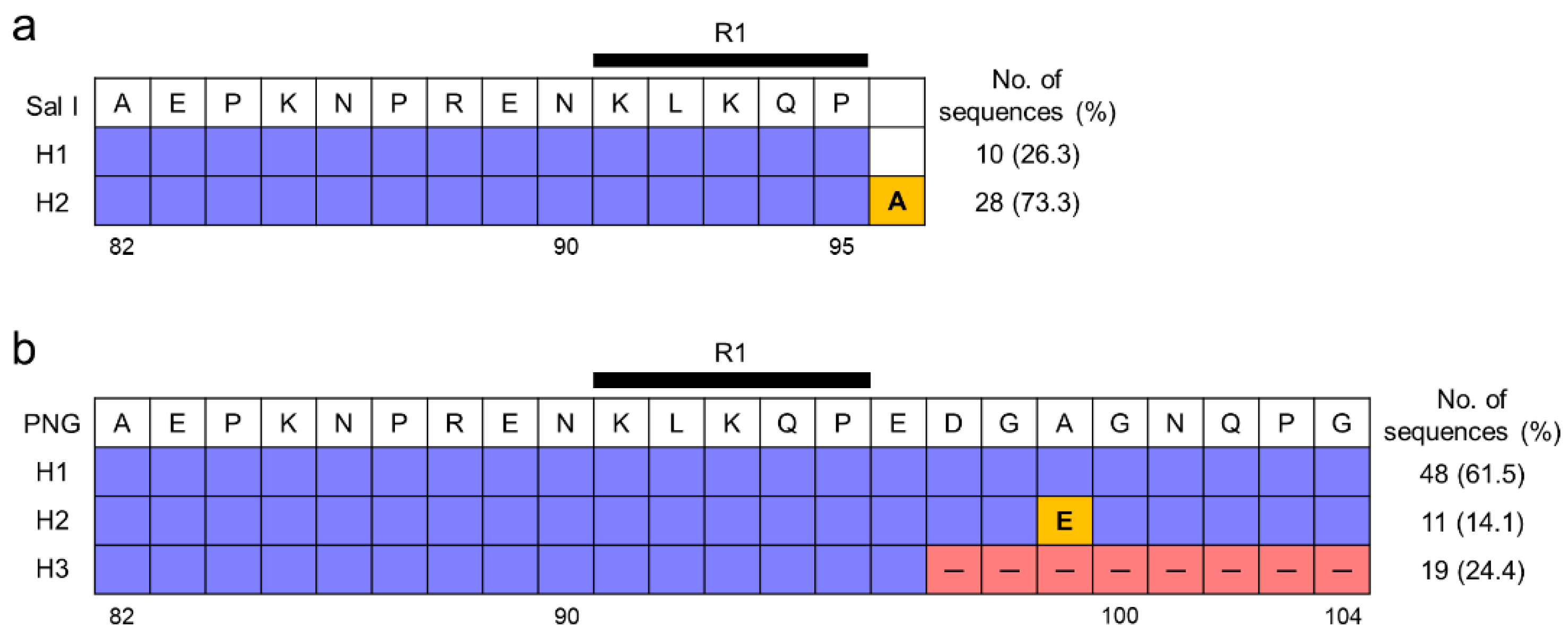
3.5. Polymorphism of the C-Terminal Non-Repeat Region in Vietnamese pvcsp
3.6. Nucleotide Diversity and Natural Selection in N- and C-Terminal Non-Repeat Regions of Vietnamese and Global pvcsp VK210 Variants
3.7. Nucleotide Diversity and Natural Selection in N- and C-Terminal Non-Repeat Regions of Vietnamese and Global pvcsp VK247 Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Caraballo, H.; King, K. Emergency department management of mosquito-borne illness: Malaria, dengue, and West Nile virus. Emerg. Med. Pract. 2014, 16, 1–23. [Google Scholar] [PubMed]
- Cox, F.E.G. History of human parasitology. Clin. Microbiol. Rev. 2002, 15, 595–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. World Malaria Report 2021; WHO: Geneva, Switzerland, 2021; ISBN 9789240040496. [Google Scholar]
- White, N.J.; Imwong, M. Relapse. Adv. Parasitol. 2012, 80, 113–150. [Google Scholar] [PubMed]
- Mueller, I.; Galinski, M.R.; Baird, J.K.; Carlton, J.M.; Kochar, D.K.; Alonso, P.L.; del Portillo, H.A. Key gaps in the knowledge of Plasmodium vivax, a neglected human malaria parasite. Lancet Infect. Dis. 2009, 9, 555–566. [Google Scholar] [CrossRef]
- Rogerson, S.J.; Carter, R. Severe vivax malaria: Newly recognised or rediscovered? PLoS Med. 2008, 5, e136. [Google Scholar] [CrossRef] [Green Version]
- Anstey, N.M.; Russell, B.; Yeo, T.W.; Price, R.N. The pathophysiology of vivax malaria. Trends Parasitol. 2009, 25, 220–227. [Google Scholar] [CrossRef]
- Rahimi, B.A.; Thakkinstian, A.; White, N.J.; Sirivichayakul, C.; Dondorp, A.M.; Chokejindachai, W. Severe vivax malaria: A systematic review and meta-analysis of clinical studies since 1900. Malar. J. 2014, 13, 481. [Google Scholar] [CrossRef] [Green Version]
- Baird, J.K. Chloroquine resistance in Plasmodium vivax. Antimicrob. Agents Chemother. 2004, 48, 4075–4083. [Google Scholar] [CrossRef] [Green Version]
- Price, R.N.; Auburn, S.; Marfurt, J.; Cheng, Q. Phenotypic and genotypic characterisation of drug-resistant Plasmodium vivax. Trends Parasitol. 2012, 28, 522–529. [Google Scholar] [CrossRef] [Green Version]
- Rathore, D.; Sacci, J.B.; De La Vega, P.; McCutchan, T.F. Binding and invasion of liver cells by Plasmodium falciparum sporozoites. Essential involvement of the amino terminus of circumsporozoite protein. J. Biol. Chem. 2002, 277, 7092–7098. [Google Scholar] [CrossRef]
- Coppi, A.; Natarajan, R.; Pradel, G.; Bennett, B.L.; James, E.R.; Roggero, M.A.; Corradin, G.; Persson, C.; Tewari, R.; Sinnis, P. The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J. Exp. Med. 2011, 208, 341–356. [Google Scholar] [CrossRef]
- Arnot, D.E.; Barnwell, J.W.; Tam, J.P.; Nussenzweig, V.; Nussenzweig, R.S.; Enea, V. Circumsporozoite protein of Plasmodium vivax: Gene cloning and characterization of the immunodominant epitope. Science 1985, 230, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, R.; Wirtz, R.A.; Lanar, D.E.; Sattabongkot, J.; Hall, T.; Waters, A.P.; Prasittisuk, C. Circumsporozoite protein heterogeneity in the human malaria parasite Plasmodium vivax. Science 1989, 245, 973–976. [Google Scholar] [CrossRef]
- Qari, S.H.; Shi, Y.P.; Goldman, I.F.; Udhaykumar, V.; Collins, W.E.; Lal, A.A.; Alpers, M.P. Identification of Plasmodium vivax-like human malaria parasite. Lancet 1993, 341, 780–783. [Google Scholar] [CrossRef]
- González-Cerón, L.; Martinez-Barnetche, J.; Montero-Solís, C.; Santillán, F.; Soto, A.M.; Rodríguez, M.H.; Espinosa, B.J.; Chávez, O.A. Molecular epidemiology of Plasmodium vivax in Latin America: Polymorphism and evolutionary relationships of the circumsporozoite gene. Malar. J. 2013, 12, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Võ, T.C.; Lê, H.G.; Kang, J.-M.; Moe, M.; Naw, H.; Myint, M.K.; Lee, J.; Sohn, W.-M.; Kim, T.-S.; Na, B.-K. Genetic polymorphism and natural selection of circumsporozoite protein in Myanmar Plasmodium vivax. Malar. J. 2020, 19, 303. [Google Scholar] [CrossRef] [PubMed]
- Yadava, A.; Sattabongkot, J.; Washington, M.A.; Ware, L.A.; Majam, V.; Zheng, H.; Kumar, S.; Ockenhouse, C.F. A novel chimeric Plasmodium vivax circumsporozoite protein induces biologically functional antibodies that recognize both VK210 and VK247 sporozoites. Infect. Immun. 2007, 75, 1177–1185. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.W.; Yadava, A.; Tosh, D.; Sattabongkot, J.; Komisar, J.; Ware, L.A.; McCarthy, W.F.; Cowden, J.J.; Regules, J.; Spring, M.D.; et al. Phase 1/2a Trial of Plasmodium vivax Malaria Vaccine Candidate VMP001/AS01B in Malaria-Naive Adults: Safety, Immunogenicity, and Efficacy. PLoS Negl. Trop. Dis. 2016, 10, e0004423. [Google Scholar] [CrossRef] [PubMed]
- Võ, T.C.; Lê, H.G.; Kang, J.M.; Naw, H.; Fan, C.K.; Trinh, N.T.M.; Quang, H.H.; Na, B.K. Molecular surveillance of malaria in the Central Highlands, Vietnam. Parasitol. Int. 2021, 83, 102374. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef] [PubMed]
- Parobek, C.M.; Bailey, J.A.; Hathaway, N.J.; Socheat, D.; Rogers, W.O.; Juliano, J.J. Differing Patterns of Selection and Geospatial Genetic Diversity within Two Leading Plasmodium vivax Candidate Vaccine Antigens. PLoS Negl. Trop. Dis. 2014, 8, e2796. [Google Scholar] [CrossRef] [Green Version]
- Santos-Ciminera, P.D.; das Alecrim, M.G.C.; Roberts, D.R.; Quinnan, G.V., Jr. Molecular epidemiology of Plasmodium vivax in the State of Amazonas, Brazil. Acta Trop. 2007, 102, 38–46. [Google Scholar] [CrossRef]
- Talha, A.A.; Pirahmadi, S.; Mehrizi, A.A.; Djadid, N.D.; Nour, B.Y.M.; Zakeri, S. Molecular genetic analysis of Plasmodium vivax isolates from Eastern and Central Sudan using pvcsp and pvmsp-3α genes as molecular markers. Infect. Genet. Evol. 2015, 32, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, A.; Chaves, L.F.; Taleo, G.; Kalkoa, M.; Isozumi, R.; Wickremasinghe, R.; Perlmann, H.; Takeo, S.; Tsuboi, T.; Tachibana, S.I.; et al. Characteristic age distribution of Plasmodium vivax infections after malaria elimination on Aneityum Island, Vanuatu. Infect. Immun. 2014, 82, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Maneerattanasak, S.; Gosi, P.; Krudsood, S.; Tongshoob, J.; Lanteri, C.A.; Snounou, G.; Khusmith, S. Genetic diversity among Plasmodium vivax isolates along the Thai-Myanmar border of Thailand. Malar. J. 2016, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Kibria, M.G.; Elahi, R.; Mohon, A.N.; Khan, W.A.; Haque, R.; Alam, M.S. Genetic diversity of Plasmodium vivax in clinical isolates from Bangladesh. Malar. J. 2015, 14, 267. [Google Scholar] [CrossRef]
- Manandhar, S.; Bhusal, C.L.; Ghimire, U.; Singh, S.P.; Karmacharya, D.B.; Dixit, S.M. A study on relapse/re-infection rate of Plasmodium vivax malaria and identification of the predominant genotypes of P. vivax in two endemic districts of Nepal. Malar. J. 2013, 12, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakeri, S.; Safi, N.; Afsharpad, M.; Butt, W.; Ghasemi, F.; Mehrizi, A.A.; Atta, H.; Zamani, G.; Djadid, N.D. Genetic structure of Plasmodium vivax isolates from two malaria endemic areas in Afghanistan. Acta Trop. 2010, 113, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Henry-Halldin, C.N.; Sepe, D.; Susapu, M.; McNamara, D.T.; Bockarie, M.; King, C.L.; Zimmerman, P.A. High-throughput molecular diagnosis of circumsporozoite variants VK210 and VK247 detects complex Plasmodium vivax infections in malaria endemic populations in Papua New Guinea. Infect. Genet. Evol. 2011, 11, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.M.; Lê, H.G.; Võ, T.C.; Naw, H.; Yoo, W.G.; Sohn, W.M.; Trinh, N.T.M.; Quang, H.H.; Na, B.K. Genetic polymorphism and natural selection of apical membrane antigen-1 in Plasmodium falciparum isolates from Vietnam. Genes 2021, 12, 1903. [Google Scholar] [CrossRef] [PubMed]
- Quang, N.D.; Hoa, P.T.P.; Tuan, M.S.; Viet, N.X.; Jalloh, A.; Matsuoka, H. Polymorphism at the apical membrane antigen 1 gene (AMA1) of the malaria parasite Plasmodium falciparum in a Vietnamese population. Biochem. Genet. 2009, 47, 370–383. [Google Scholar] [CrossRef]
- Van Long, B.; Allen, G.; Brauny, M.; Linh, L.T.K.; Pallerla, S.R.; Huyen, T.T.T.; Van Tong, H.; Toan, N.L.; Quyet, D.; Son, H.A.; et al. Molecular surveillance and temporal monitoring of malaria parasites in focal Vietnamese provinces. Malar. J. 2020, 19, 458. [Google Scholar] [CrossRef]
- Van Hong, N.; Delgado-Ratto, C.; Thanh, P.V.; Van den Eede, P.; Guetens, P.; Binh, N.T.H.; Phuc, B.Q.; Duong, T.T.; Van Geertruyden, J.P.; D’Alessandro, U.; et al. Population Genetics of Plasmodium vivax in Four Rural Communities in Central Vietnam. PLoS Negl. Trop. Dis. 2016, 10, e0004434. [Google Scholar] [CrossRef]
- Hoffmann, E.H.E.; Ribolla, P.E.M.; Ferreira, M.U. Genetic relatedness of Plasmodium falciparum isolates and the origin of allelic diversity at the merozoite surface protein-1 (MSP-1) locus in Brazil and Vietnam. Malar. J. 2003, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.U.; Liu, Q.; Zhou, M.; Kimura, M.; Kaneko, O.; Van Thien, H.; Isomura, S.; Tanabe, K.; Kawamoto, F. Stable patterns of allelic diversity at the Merozoite Surface Protein-1 locus of Plasmodium falciparum in clinical isolates from Southern Vietnam. J. Eukaryot. Microbiol. 1998, 45, 131–136. [Google Scholar] [CrossRef]
- Cui, L.; Mascorro, C.N.; Fan, Q.; Rzomp, K.A.; Khuntirat, B.; Zhou, G.; Chen, H.; Yan, G.; Sattabongkot, J. Genetic diversity and multiple infections of Plasmodium vivax malaria in western Thailand. Am. J. Trop. Med. Hyg. 2003, 68, 613–619. [Google Scholar] [CrossRef]
- Rodriguez, M.H.; Gonzalez-Ceron, L.; Hernandez, J.E.; Nettel, J.A.; Villarreal, C.; Kain, K.C.; Wirtz, R.A. Different prevalences of Plasmodium vivax phenotypes VK210 and VK247 associated with the distribution of Anopheles albimanus and Anopheles pseudopunctipennis in Mexico. Am. J. Trop. Med. Hyg. 2000, 62, 122–127. [Google Scholar] [CrossRef]
- Manguin, S.; Boete, C. Global Impact of Mosquito Biodiversity, Human Vector-Borne Diseases and Environmental Change. In The Importance of Biological Interactions in the Study of Biodiversity; IntechOpen: London, UK, 2011; pp. 27–58. [Google Scholar]
- Sinka, M.E.; Bangs, M.J.; Manguin, S.; Chareonviriyaphap, T.; Patil, A.P.; Temperley, W.H.; Gething, P.W.; Elyazar, I.R.; Kabaria, C.W.; Harbach, R.E.; et al. The dominant anopheles vectors of human malaria in the Asia-Pacific region: Occurrence data, distribution maps and bionomic précis. Parasites Vectors 2011, 4, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwonkerd, W.; Ritthison, W.; Ngo, C.T.; Tainchum, K.; Bangs, M.J.; Chareonviriyaphap, T. Vector Biology and Malaria Transmission in Southeast Asia. In Anopheles mosquitoes—New Insights into Malaria Vectors; IntechOpen: London, UK, 2013. [Google Scholar]
- Nguyen, T.Q.; Nguyen, M.D.; Pham, V.X.; Ro, H.M.; Edstein, M.D.; Chow, W.K.; Martin, N.J.; Hertz, J.C.; Motoki, M.T. Entomological survey in two communes with residual malaria transmission in Gia Lai Province in the central highlands of Vietnam. Malar. J. 2021, 20, 403. [Google Scholar] [CrossRef]
- Hernández-Martínez, M.Á.; Escalante, A.A.; Arévalo-Herrera, M.; Herrera, S. Antigenic diversity of the Plasmodium vivax circumsporozoite protein in parasite isolates of Western Colombia. Am. J. Trop. Med. Hyg. 2011, 84, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenet, S.M.; Tapia, L.L.; Escalante, A.A.; Durand, S.; Lucas, C.; Bacon, D.J. Genetic diversity and population structure of genes encoding vaccine candidate antigens of Plasmodium vivax. Malar. J. 2012, 11, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, P.; Shakibaei, M.; Patankar, M.S.; Clavijo, P.; Beavis, R.C.; Clark, G.F.; Frevert, U. The malaria circumsporozoite protein: Interaction of the conserved regions I and II-plus with heparin-like oligosaccharides in heparan sulfate. Exp. Parasitol. 1997, 85, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Patil, A.; Orjuela-Sánchez, P.; da Silva-Nunes, M.; Ferreira, M.U. Evolutionary dynamics of the immunodominant repeats of the Plasmodium vivax malaria-vaccine candidate circumsporozoite protein (CSP). Infect. Genet. Evol. 2010, 10, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.R.; Imwong, M.; Nandy, A.; Chotivanich, K.; Nontprasert, A.; Tonomsing, N.; Maji, A.; Addy, M.; Day, N.P.J.; White, N.J.; et al. Genetic diversity of Plasmodium vivax in Kolkata, India. Malar. J. 2006, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.L. The evolution of amino acid repeat arrays in Plasmodium and other organisms. J. Mol. Evol. 2004, 59, 528–535. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Country | n | K | S | H | Hd ± SD | π ± SD | dN–dS | Tajima’s Dp value | Fu & Li’s Dp value | Fu & Li’s Fp value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| N-terminal | Vietnam | 38 | 0.102 | 1 | 2 | 0.102 ± 0.065 | 0.0024 ± 0.0016 | −0.0120 | −0.8255a | 0.5688a | 0.2028a |
| Myanmar# | 143 | 0.098 | 5 | 7 | 0.096 ± 0.034 | 0.0024 ± 0.0009 | 0.0008 | −1.9359c | −3.9086b | −3.8409b | |
| Cambodia# | 31 | 0.125 | 1 | 2 | 0.125 ± 0.077 | 0.0030 ± 0.0018 | 0.0039 | −0.7737a | 0.5907a | 0.2450a | |
| India# | 79 | 1.972 | 23 | 28 | 0.661 ± 0.063 | 0.0470 ± 0.0074 | −0.0909 | −2.0261c | −1.3952a | −1.9514e | |
| Iran# | 39 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| South Korea# | 39 | 0.051 | 1 | 2 | 0.051 ± 0.048 | 0.0012 ± 0.0011 | 0.0016 | −1.1264a | −1.7662a | −1.8293a | |
| Brazil# | 41 | 0.049 | 1 | 2 | 0.049 ± 0.046 | 0.0012 ± 0.0011 | −0.0059 | −1.1219a | −1.7816a | −1.8406a | |
| Mexico# | 11 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Sudan# | 30 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Vanuatu# | 21 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| C-terminal | Vietnam | 38 | 0.713 | 2 | 3 | 0.383 ± 0.081 | 0.0095 ± 0.0020 | −0.0450 | 0.9518a | 0.7771a | 0.9581a |
| Myanmar# | 143 | 0.209 | 10 | 11 | 0.186 ± 0.044 | 0.0035 ± 0.0009 | −0.0035 | −2.2251d | −3.5089b | −3.6329b | |
| Cambodia# | 31 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| India# | 79 | 0.276 | 8 | 6 | 0.123 ± 0.051 | 0.0043 ± 0.0020 | −0.0149 | −2.1953d | −4.4419b | −4.3524b | |
| Iran# | 39 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| South Korea# | 39 | 0.103 | 2 | 3 | 0.101 ± 0.065 | 0.0008 ± 0.0005 | −0.0037 | −1.4889a | −2.4148e | −2.4864e | |
| Brazil# | 41 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mexico# | 11 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Sudan# | 30 | 0.067 | 1 | 2 | 0.067 ± 0.061 | 0.0009 ± 0.0008 | −0.0041 | −1.1470a | −1.6821a | −1.7655a | |
| Vanuatu# | 21 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Region | Country | n | K | S | H | Hd ± SD | π ± SD | dN–dS | Tajima’s Dp Value | Fu & Li’s Dp Value | Fu & Li’s Fp Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| N-terminal | Vietnam | 79 | 0.370 | 1 | 2 | 0.370 ± 0.050 | 0.0082 ± 0.0011 | 0.0110 | 0.9975a | 0.5078a | 0.7588a |
| Myanmar# | 28 | 0.405 | 1 | 3 | 0.405 ± 0.094 | 0.0090 ± 0.0021 | 0.0117 | –0.4445a | −0.7114a | −0.7369a | |
| Cambodia# | 10 | 0.200 | 1 | 2 | 0.200 ± 0.154 | 0.0029 ± 0.0022 | 0.0038 | −1.1117a | −1.2434a | −1.3467a | |
| Iran# | 11 | 1.309 | 3 | 2 | 0.436 ± 0.133 | 0.0190 ± 0.0058 | −0.0543 | 0.9518a | 1.1271a | 1.2185a | |
| Mexico# | 8 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Colombia# | 25 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| C-terminal | Vietnam | 79 | 1.357 | 8 | 9 | 0.531 ± 0.063 | 0.0133 ± 0.0019 | −0.0290 | −0.4083a | 1.2744a | 0.8376a |
| Myanmar# | 28 | 0.495 | 6 | 6 | 0.331 ± 0.114 | 0.0068 ± 0.0027 | −0.0150 | −1.9719b | −2.5946b | −2.8039b | |
| Cambodia# | 10 | 0.400 | 2 | 2 | 0.200 ± 0.154 | 0.0039 ± 0.0030 | −0.0172 | −1.4009a | −1.5866a | −1.7190a | |
| Iran# | 11 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mexico# | 8 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Colombia# | 25 | 0.500 | 2 | 3 | 0.440 ± 0.095 | 0.0039 ± 0.0010 | 0.0017 | −0.1215a | −0.6754a | −0.6012a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Võ, T.C.; Trinh, N.T.M.; Lê, H.G.; Kang, J.-M.; Yoo, W.G.; Quang, H.H.; Na, B.-K. Genetic Diversity of Circumsporozoite Surface Protein of Plasmodium vivax from the Central Highlands, Vietnam. Pathogens 2022, 11, 1158. https://doi.org/10.3390/pathogens11101158
Võ TC, Trinh NTM, Lê HG, Kang J-M, Yoo WG, Quang HH, Na B-K. Genetic Diversity of Circumsporozoite Surface Protein of Plasmodium vivax from the Central Highlands, Vietnam. Pathogens. 2022; 11(10):1158. https://doi.org/10.3390/pathogens11101158
Chicago/Turabian StyleVõ, Tuấn Cường, Nguyen Thi Minh Trinh, Hương Giang Lê, Jung-Mi Kang, Won Gi Yoo, Huynh Hong Quang, and Byoung-Kuk Na. 2022. "Genetic Diversity of Circumsporozoite Surface Protein of Plasmodium vivax from the Central Highlands, Vietnam" Pathogens 11, no. 10: 1158. https://doi.org/10.3390/pathogens11101158