


Comparative Genomics Reveal the Utilization Ability of Variable Carbohydrates as Key Genetic Features of Listeria Pathogens in Their Pathogenic Lifestyles

Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Retrieval and Management
2.2. Protein Orthologous Group Analysis
2.3. Phylogenetic Analysis
2.4. Estimation of Core and Pan-Genome Sizes
2.5. Functional Features of the Core and Accessory Genomes
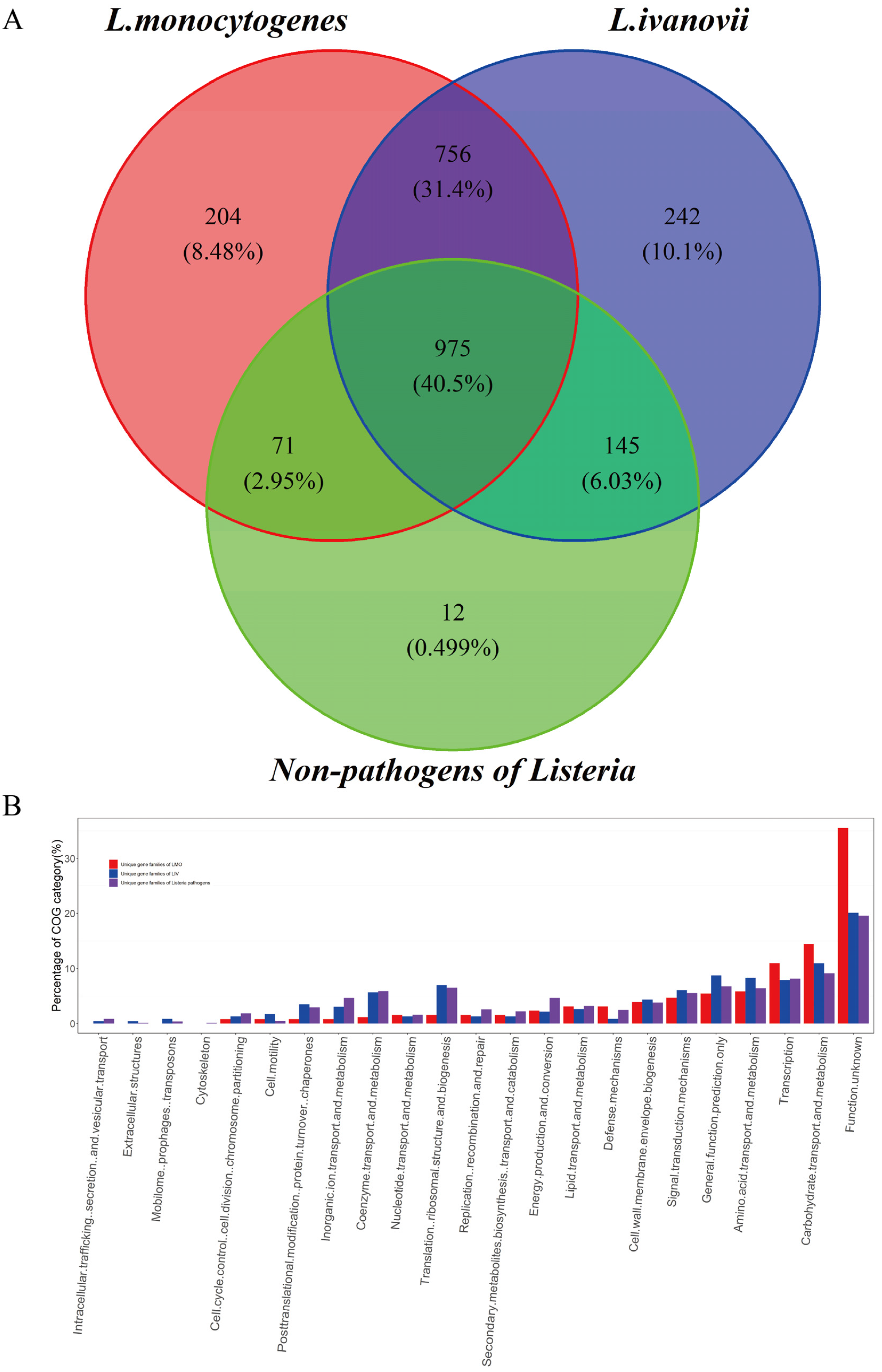
2.6. Unique Gene Analyses of L. monocytogenes and L. ivanovii
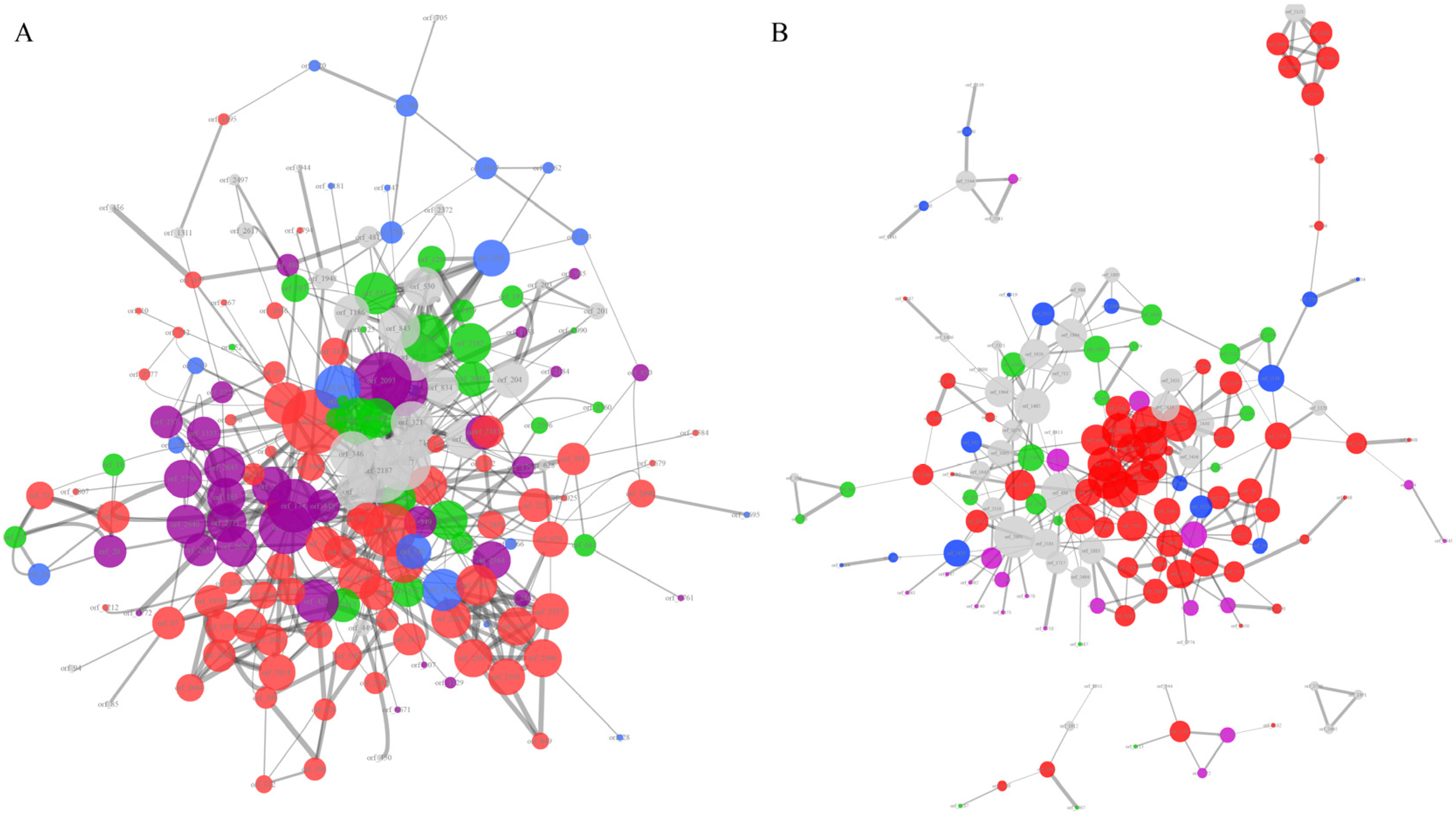
2.7. Protein–Protein Interaction Network Analysis of Unique Protein Sets
2.8. Antibiotic Resistance Gene Investigation of Listeria Genomes
2.9. Identification of CRISPR-Cas Systems of Listeria Genomes
3. Results and Discussion
3.1. Subsection
3.1.1. Genome Statistics and General Features
3.1.2. Homologous Proteome Analysis and Phylogeny of the Listeria Genus
3.1.3. Core and Pan-Genome Sizes of Listeria Species
3.1.4. Functional Features of the Listeria Core and Accessory Genomes
3.1.5. The Transport Systems for Carbohydrates and Ions Enriched in Unique Gene Set of Listeria Pathogens and Closely Related to Their Pathogenesis
3.1.6. Virulence Factor Gene Investigation of the Unique Genes of Listeria Pathogens
3.1.7. Ethanolamine-Metabolic-Related Proteins Enriched in the Protein–Protein Interaction Network of Listeria Pathogens’ Unique Genes
3.1.8. The Distribution of Antibiotics Resistance Genes in the Listeria Species
3.1.9. The Occurrence of Different Types of CRISPR-Cas Systems in Listeria
3.1.10. The Characterization of Plasmids from the Listeria Species
3.2. Formatting of Mathematical Components
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Batt, C.A. LISTERIA|Introduction. In Encyclopedia of Food Microbiology, 2nd ed.; Batt, C.A., Tortorello, M.L., Eds.; Academic Press: Oxford, UK, 2014; pp. 466–469. [Google Scholar]
- Conly, J.M.; Johnston, B.L. Listeria: A persistent food-borne pathogen. Can. J. Infect. Dis. Med. Microbiol. 2008, 19, 327–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, L.; Pichon, M.; Burucoa, C.; Nusser, S.H.A.; Moura, A.; Garcia-Garcera, M.; Lecuit, M. Listeria monocytogenes faecal carriage is common and depends on the gut microbiota. Nat. Commun. 2021, 12, 6826. [Google Scholar] [CrossRef] [PubMed]
- Kreft, J.; Vázquez-Boland, J.A.; Altrock, S.; Dominguez-Bernal, G.; Goebel, W. Pathogenicity islands and other virulence elements in Listeria. Curr. Top. Microbiol. Immunol. 2002, 264, 109–125. [Google Scholar] [PubMed]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef]
- Cossart, P.; Vicente, M.F.; Mengaud, J.; Baquero, F.; Perez-Diaz, J.C.; Berche, P. Listeriolysin O is essential for virulence of Listeria monocytogenes: Direct evidence obtained by gene complementation. Infect. Immun. 1989, 57, 3629–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, G.A.; Marquis, H.; Jones, S.; Johnston, N.C.; Portnoy, D.A.; Goldfine, H. The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread. Infect. Immun. 1995, 63, 4231–4237. [Google Scholar] [CrossRef] [Green Version]
- Vega, Y.; Dickneite, C.; Ripio, M.T.; Böckmann, R.; González-Zorn, B.; Novella, S.; Domínguez-Bernal, G.; Goebel, W.; Vázquez-Boland, J.A. Functional similarities between the Listeria monocytogenes virulence regulator PrfA and cyclic AMP receptor protein: The PrfA* (Gly145Ser) mutation increases binding affinity for target DNA. J. Bacteriol. 1998, 180, 6655–6660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, G.; Ge, L.; Huang, Q.; Chen, C.; Kianian, S.; Roberts, M.F.; Schekman, R.; Portnoy, D.A. Avoidance of autophagy mediated by PlcA or ActA is required for Listeria monocytogenes growth in macrophages. Infect. Immun. 2015, 83, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, D.E.; Agaisse, H. The Metalloprotease Mpl Supports Listeria monocytogenes Dissemination through Resolution of Membrane Protrusions into Vacuoles. Infect. Immun. 2016, 84, 1806–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico pMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods Mol. Biol. 2020, 2075, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Schmartz, G.P.; Hartung, A.; Hirsch, P.; Kern, F.; Fehlmann, T.; Müller, R.; Keller, A. PLSDB: Advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 2022, 50, D273–D278. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Dongen, S. A Cluster Algorithm for Graphs; Centrum Wiskunde & Informatica (CWI): The National Research Institute for Mathematics and Computer Science in the Netherlands: Amsterdam, The Netherlands, 2000; pp. 1–40. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernandez-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Vera Alvarez, R.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.; Chang, H.Y.; Dosztanyi, Z.; Elgebali, S.; Fraser, M. InterPro in 2017—Beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huertacepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, 87–105. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [Green Version]
- Glaser, P.; Frangeul, L.; Buchrieser, C.; Rusniok, C.; Amend, A.; Baquero, F.; Berche, P.; Bloecker, H.; Brandt, P.; Chakraborty, T.; et al. Comparative genomics of Listeria species. Science 2001, 294, 849–852. [Google Scholar] [CrossRef]
- Baroncelli, R.; Amby, D.B.; Zapparata, A.; Sarrocco, S.; Vannacci, G.; Le Floch, G.; Harrison, R.J.; Holub, E.; Sukno, S.A.; Sreenivasaprasad, S.; et al. Gene family expansions and contractions are associated with host range in plant pathogens of the genus Colletotrichum. BMC Genom. 2016, 17, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.W.; Ng, E.Y.W.; Lampidis, R.; Emmerth, M.; Walcher, M.; Kreft, J.; Goebel, W.; Wagner, M.; Schleifer, K.-H. Evolutionary history of the genus Listeria and its virulence genes. Syst. Appl. Microbiol. 2005, 28, 1–18. [Google Scholar] [CrossRef]
- Boerlin, P.; Rocourt, J.; Piffaretti, J.C. Taxonomy of the genus Listeria by using multilocus enzyme electrophoresis. Int. J. Syst. Bacteriol. 1991, 41, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef]
- Lefebure, T.; Stanhope, M.J. Evolution of the core and pan-genome of Streptococcus: Positive selection, recombination, and genome composition. Genome Biol. 2007, 8, R71. [Google Scholar] [CrossRef] [Green Version]
- Siefert, J.L. Defining the Mobilome. In Horizontal Gene Transfer: Genomes in Flux; Gogarten, M.B., Gogarten, J.P., Olendzenski, L.C., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 13–27. [Google Scholar]
- Muchaamba, F.; Eshwar, A.; Stevens, M.; von Ah, U.; Tasara, T. Variable Carbon Source Utilization, Stress Resistance, and Virulence Profiles Among Listeria monocytogenes Strains Responsible for Listeriosis Outbreaks in Switzerland. Front. Microbiol. 2019, 10, 957. [Google Scholar] [CrossRef] [Green Version]
- Deutscher, J.; Aké, F.; Zebre, A.; Cao, T.; Kentache, T.; Pham, Q.; Mokhtari, A.; Joyet, P.; Milohanic, E. Carbohydrate Utilization by Listeria monocytogenes and its Influence on Virulence Gene Expression. In Listeria monocytogenes: Food Sources, Prevalence and Management Strategies; Nova science publishers: Hauppauge, NY, USA, 2014; pp. 49–76. [Google Scholar]
- Milenbachs, A.A.; Brown, D.P.; Moors, M.; Youngman, P. Carbon-source regulation of virulence gene expression in Listeria monocytogenes. Mol. Microbiol. 1997, 23, 1075–1085. [Google Scholar] [CrossRef]
- McLaughlin, H.P.; Hill, C.; Gahan, C.G.M. The impact of iron on Listeria monocytogenes; inside and outside the host. Curr. Opin. Biotechnol. 2011, 22, 194–199. [Google Scholar] [CrossRef]
- Orelle, C.; Mathieu, K.; Jault, J.-M. Multidrug ABC transporters in bacteria. Res. Microbiol. 2019, 170, 381–391. [Google Scholar] [CrossRef]
- Rea, R.B.; Gahan, C.G.M.; Hill, C. Disruption of Putative Regulatory Loci in Listeria monocytogenes Demonstrates a Significant Role for Fur and PerR in Virulence. Infect. Immun. 2004, 72, 717–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledala, N.; Sengupta, M.; Muthaiyan, A.; Wilkinson, B.J.; Jayaswal, R.K. Transcriptomic response of Listeria monocytogenes to iron limitation and Fur mutation. Appl. Environ. Microbiol. 2010, 76, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebreton, A.; Cossart, P. RNA- and protein-mediated control of Listeria monocytogenes virulence gene expression. RNA Biol. 2017, 14, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boujemaa-Paterski, R.; Gouin, E.; Hansen, G.; Samarin, S.; Le Clainche, C.; Didry, D.; Dehoux, P.; Cossart, P.; Kocks, C.; Carlier, M.-F.; et al. Listeria Protein ActA Mimics WASP Family Proteins: It Activates Filament Barbed End Branching by Arp2/3 Complex. Biochemistry 2001, 40, 11390–11404. [Google Scholar] [CrossRef]
- Cabanes, D.; Dussurget, O.; Dehoux, P.; Cossart, P. Auto, a surface associated autolysin of Listeria monocytogenes required for entry into eukaryotic cells and virulence. Mol. Microbiol. 2004, 51, 1601–1614. [Google Scholar] [CrossRef] [PubMed]
- Gouin, E.; Adib-Conquy, M.; Balestrino, D.; Nahori, M.-A.; Villiers, V.; Colland, F.; Dramsi, S.; Dussurget, O.; Cossart, P. The Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the IκB kinase subunit IKKα. Proc. Natl. Acad. Sci. USA 2010, 107, 17333–17338. [Google Scholar] [CrossRef] [Green Version]
- Rajabian, T.; Gavicherla, B.; Heisig, M.; Müller-Altrock, S.; Goebel, W.; Gray-Owen, S.D.; Ireton, K. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat. Cell Biol. 2009, 11, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Chico-Calero, I.; Suárez, M.; González-Zorn, B.; Scortti, M.; Slaghuis, J.; Goebel, W.; Vázquez-Boland, J.A. Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. USA 2002, 99, 431–436. [Google Scholar] [CrossRef] [Green Version]
- González-Zorn, B.; Domínguez-Bernal, G.; Suárez, M.; Ripio, M.T.; Vega, Y.; Novella, S.; Vázquez-Boland, J.A. The smcL gene of Listeria ivanovii encodes a sphingomyelinase C that mediates bacterial escape from the phagocytic vacuole. Mol. Microbiol. 1999, 33, 510–523. [Google Scholar] [CrossRef]
- Mengaud, J.; Ohayon, H.; Gounon, P.; Mège, R.-M.; Cossart, P. E-Cadherin Is the Receptor for Internalin, a Surface Protein Required for Entry of L. monocytogenes into Epithelial Cells. Cell 1996, 84, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Sashinami, H.; Osanai, A.; Asano, Y.; Nakane, A. Autolysin amidase of Listeria monocytogenes promotes efficient colonization of mouse hepatocytes and enhances host immune response. Int. J. Med. Microbiol. 2011, 301, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.; Kakizaki, I.; Nakane, A. Interaction of Listeria monocytogenes autolysin amidase with glycosaminoglycans promotes listerial adhesion to mouse hepatocytes. Biochimie 2012, 94, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Halvorsen, E.M.; Ammendolia, D.A.; Mor-Vaknin, N.; O'Riordan, M.X.D.; Brumell, J.H.; Markovitz, D.M.; Higgins, D.E. Invasion of the Brain by Listeria monocytogenes Is Mediated by InlF and Host Cell Vimentin. mBio 2018, 9, e00160-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malmirchegini, G.R.; Sjodt, M.; Shnitkind, S.; Sawaya, M.R.; Rosinski, J.; Newton, S.M.; Klebba, P.E.; Clubb, R.T. Novel Mechanism of Hemin Capture by Hbp2, the Hemoglobin-binding Hemophore from Listeria monocytogenes. J. Biol. Chem. 2014, 289, 34886–34899. [Google Scholar] [CrossRef] [Green Version]
- Borezée, E.; Pellegrini, E.; Beretti, J.-L.; Berche, P. SvpA, a novel surface virulence-associated protein required for intracellular survival of Listeria monocytogenes. Microbiology 2001, 147, 2913–2923. [Google Scholar] [CrossRef] [Green Version]
- Goldfine, H.; Knob, C.; Alford, D.; Bentz, J. Membrane permeabilization by Listeria monocytogenes phosphatidylinositol-specific phospholipase C is independent of phospholipid hydrolysis and cooperative with listeriolysin O. Proc. Natl. Acad. Sci. USA 1995, 92, 2979–2983. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Boland, J.A.; Kuhn, M.; Berche, P.; Chakraborty, T.; Domínguez-Bernal, G.; Goebel, W.; González-Zorn, B.; Wehland, J.; Kreft, J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001, 14, 584–640. [Google Scholar] [CrossRef] [Green Version]
- Poyart, C.; Abachin, E.; Razafimanantsoa, I.; Berche, P. The zinc metalloprotease of Listeria monocytogenes is required for maturation of phosphatidylcholine phospholipase C: Direct evidence obtained by gene complementation. Infect. Immun. 1993, 61, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Forster, B.M.; Zemansky, J.; Portnoy, D.A.; Marquis, H. Posttranslocation chaperone PrsA2 regulates the maturation and secretion of Listeria monocytogenes proprotein virulence factors. J. Bacteriol. 2011, 193, 5961–5970. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Promadej, N.; Kim, J.W.; Kathariou, S. Teichoic acid glycosylation mediated by gtcA is required for phage adsorption and susceptibility of Listeria monocytogenes serotype 4b. Appl. Environ. Microbiol. 2008, 74, 1653–1655. [Google Scholar] [CrossRef]
- Alves, J.A.; Previato-Mello, M.; Barroso, K.C.M.; Koide, T.; da Silva Neto, J.F. The MarR family regulator OsbR controls oxidative stress response, anaerobic nitrate respiration, and biofilm formation in Chromobacterium violaceum. BMC Microbiol. 2021, 21, 304. [Google Scholar] [CrossRef] [PubMed]
- O'Neil, H.S.; Marquis, H. Listeria monocytogenes flagella are used for motility, not as adhesins, to increase host cell invasion. Infect. Immun. 2006, 74, 6675–6681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemon, K.P.; Higgins, D.E.; Kolter, R. Flagellar motility is critical for Listeria monocytogenes biofilm formation. J. Bacteriol. 2007, 189, 4418–4424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Z.; Boeren, S.; Bhandula, V.; Light, S.H.; Smid, E.J.; Notebaart, R.A.; Abee, T. Bacterial Microcompartments Coupled with Extracellular Electron Transfer Drive the Anaerobic Utilization of Ethanolamine in Listeria monocytogenes. mSystems 2021, 6, e01349-20. [Google Scholar] [CrossRef] [PubMed]
- Garsin, D.A. Ethanolamine utilization in bacterial pathogens: Roles and regulation. Nat. Rev. Microbiol. 2010, 8, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Joseph, B.; Przybilla, K.; Stühler, C.; Schauer, K.; Slaghuis, J.; Fuchs, T.M.; Goebel, W. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J. Bacteriol. 2006, 188, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Toledoarana, A.; Dussurget, O.; Nikitas, G.; Sesto, N.; Guetrevillet, H.; Balestrino, D.; Loh, E.; Gripenland, J.; Tiensuu, T.; Vaitkevicius, K. The Listeria transcriptional landscape from saprophytism to virulence. Nature 2009, 459, 950–956. [Google Scholar] [CrossRef]
- Anast, J.M.; Schmitz-Esser, S. The transcriptome of Listeria monocytogenes during co-cultivation with cheese rind bacteria suggests adaptation by induction of ethanolamine and 1,2-propanediol catabolism pathway genes. PLoS ONE 2020, 15, e0233945. [Google Scholar] [CrossRef]
- Tang, S.; Orsi, R.H.; den Bakker, H.C.; Wiedmann, M.; Boor, K.J.; Bergholz, T.M. Transcriptomic Analysis of the Adaptation of Listeria monocytogenes to Growth on Vacuum-Packed Cold Smoked Salmon. Appl. Environ. Microbiol. 2015, 81, 6812–6824. [Google Scholar] [CrossRef] [Green Version]
- Olaimat, A.N.; Al-Holy, M.A.; Shahbaz, H.M.; Al-Nabulsi, A.A.; Abu Ghoush, M.H.; Osaili, T.M.; Ayyash, M.M.; Holley, R.A. Emergence of Antibiotic Resistance in Listeria monocytogenes Isolated from Food Products: A Comprehensive Review. Compr Rev Food Sci Food Saf 2018, 17, 1277–1292. [Google Scholar] [CrossRef]
- Walsh, D.; Duffy, G.; Sheridan, J.J.; Blair, I.S.; McDowell, D.A. Antibiotic resistance among Listeria, including Listeria monocytogenes, in retail foods. J. Appl. Microbiol. 2001, 90, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, E.; Gerbaud, G.; Jacquet, C.; Rocourt, J.; Courvalin, P. Incidence of antibiotic resistance in Listeria species. J. Infect. Dis. 1995, 172, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Louwen, R.; Staals, R.H.; Endtz, H.P.; van Baarlen, P.; van der Oost, J. The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 2014, 78, 74–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, T.R.; Napier, B.A.; Schroeder, M.R.; Louwen, R.; Zhao, J.; Chin, C.Y.; Ratner, H.K.; Llewellyn, A.C.; Jones, C.L.; Laroui, H.; et al. A CRISPR-Cas system enhances envelope integrity mediating antibiotic resistance and inflammasome evasion. Proc. Natl. Acad. Sci. USA 2014, 111, 11163–11168. [Google Scholar] [CrossRef] [Green Version]
- Poyart-Salmeron, C.; Carlier, C.; Trieu-Cuot, P.; Courtieu, A.L.; Courvalin, P. Transferable plasmid-mediated antibiotic resistance in Listeria monocytogenes. Lancet 1990, 335, 1422–1426. [Google Scholar] [CrossRef]
- Chmielowska, C.; Korsak, D.; Chapkauskaitse, E.; Decewicz, P.; Lasek, R.; Szuplewska, M.; Bartosik, D. Plasmidome of Listeria spp.-The repA-Family Business. Int. J. Mol. Sci. 2021, 22, 10320. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Number of Genomes | Genome Size (bp) 1 | GC Content (%) 1 | Number of Scaffolds | Number of Plasmids | CDS 1,2 | rRNA 1 | tRNA 1 | Isolation Source |
|---|---|---|---|---|---|---|---|---|---|
| L.grayi | 4 | 2,600,550 (2,598,321, 2,751,104) | 41.52 (41.47, 41.73) | 1–4 | 0–1 | 2609 (2561, 2982) | 12 (7, 18) | 58 (52, 65) | Homo sapiens, Plant |
| L. innocua | 8 | 3,019,705 (2,889,727, 3,115,943) | 37.38 (34.65, 37.48) | 1 | 0–1 | 2934 (2622, 3130) | 18 (0, 18) | 67 (24, 67) | Food, Meat, Environment |
| L. ivanovii | 10 | 2,924,216 (2,893,311, 3,133,385) | 37.11 (34.59, 37.15) | 1 | 0 | 2808 (2682, 2924) | 18 (1, 18) | 67 (27, 67) | Food, Sheep |
| L. marthii | 3 | 2,884,551 (2,840,389, 2,947,729) | 38.62 (36.79, 38.74) | 1 | 0 | 2759 (2597, 2863) | 18 (2, 18) | 67 (28, 67) | Environment |
| L. monocytogenes | 275 | 2,964,472 (2,776,517, 3,243,301) | 37.98 (37.74, 38.29) | 1 | 0–1 | 2906 (2730, 3323) | 18 (4, 20) | 67 (49, 72) | Cow, Chicken, Food, Homo sapiens, Pig, Goat, Meat, Horse, Rabbit, Environment |
| L. seeligeri | 5 | 2,927,941 (2,797,636, 3,140,155) | 37.30 (35.17, 37.38) | 1 | 0–1 | 2779 (2590, 2846) | 8 (1, 18) | 63 (24, 67) | Environment |
| L. welshimeri | 2 | 2,814,134 (2,814,130, 2,814,137) | 36.35 (36.35, 36.35) | 1 | 0 | 2,780 (2778, 2781) | 18 (18, 18) | 66 (66, 66) | Food |
| Total | 295 | 2,959,738 (2,598,321, 3,243,301) | 37.97 (34.59, 41.73) | 1–4 | 0–1 | 2902 (2561, 3323) | 18 (0, 20) | 67 (24, 72) | - |
| Type of Transporter | Type of Substrate | Protein Name | ORFs 1 (Referenced to L. monocytogenes EGD-e_169963) | ORFs (Referenced to L. ivanovii 1638) |
|---|---|---|---|---|
| Phosphotransferase system | cellobiose | CelC, CelA, CelB | orf_1724, orf_2698, orf_915, orf_2723, orf_916 | orf_1158, orf_242, orf_2009, orf_186, orf_2008 |
| fructose | FrwB | orf_2144, orf_397, orf_425, orf_633 | orf_2291, orf_722 | |
| FrwC | orf_398, orf_426, orf_632 | NE 2 | ||
| mannose/fructose/N-acetylgalactosamine | AgaB, ManY, ManZ, ManX | orf_2009, orf_22, orf_2008, orf_783, orf_96, orf_2007, orf_24, orf_782, orf_2004, orf_785 | orf_2656, orf_869, orf_2151, orf_2780, orf_870, orf_2152, orf_871, orf_2149, orf_874 | |
| sorbitol | SrlB, SrlE, SrlA, | orf_542, orf_543, orf_544 | orf_2404, orf_2403, orf_2402 | |
| galactitol | SgaB | orf_1979, orf_2663 | NE | |
| mannitol | MtlA | orf_2816 | NE | |
| ABC-type transport system | sugar transport | MalK, UgpA | orf_2132, orf_2856, orf_861 | orf_2613, orf_2068, orf_735, orf_75 |
| polysaccharide | LplB | orf_2016 | orf_862 | |
| glycerol-3-phosphate | UgpB | orf_2014, orf_2857, orf_860 | orf_864 | |
| UgpE | orf_2015, orf_2855 | NE | ||
| multidrug transport | MdlB, CcmA, YadH | orf_106, orf_1135, orf_2766, orf_607, orf_608, orf_2235, orf_980, orf_987, orf_981, orf_988 | orf_143, orf_1807, orf_2332, orf_2333, orf_2767, orf_1934, orf_2207, orf_635, orf_1932, orf_1933, orf_2206 | |
| Mn2+/Zn2+/Fe3+/Co2+/Mo2+ | PotA, CbiM, CbiN, FepB, FepD, NlpA, ZnuC, ZnuB, ModA, ModB, ZnuA, FepC | orf_1040,orf_1207, orf_1208, orf_2192, orf_1964, orf_281, orf_1453, orf_1855, orf_1452, orf_1854, orf_1042, orf_1041, orf_1676, orf_2190 | orf_1727, orf_1726, orf_678, orf_915, orf_2606, orf_1028, orf_1450, orf_1029, orf_1451,orf_1878, orf_1879, orf_1206, orf_680 | |
| antimicrobial peptide, polar amino acid, methionine, lipoprotein, | SalY, GlnQ, AbcC, LolD, MetP, LolC | orf1_192, orf1_2259, orf1_2358, orf1_1511, orf1_279, orf1_1227 | orf1_2529, orf1_2703, orf1_611, orf1_2607, orf1_1393, orf1_2608, orf1_1700 | |
| Others | EcfA2, DdpA, Uup, CcmA, CydD | orf1_133, orf1_1210, orf1_2731, orf1_920, orf1_745, orf1_1134 | orf1_2004, orf1_2751, orf1_1724, orf1_2188, orf1_178, orf1_1808 |
| Function Category | Gene Name | UGLMO 1 | UFLIV 2 | UGLP(LMO) 1 | UGLP(LIV) 2 | Gene’s Description of VFDB |
|---|---|---|---|---|---|---|
| Regulation | prfA | orf1_197 | Listeriolysin-positive regulatory protein | |||
| Motility | actA | orf1_201 | Actin-assembly-inducing protein precursor | |||
| Invasion | aut | orf_1077 | Autolysin | |||
| inlA | orf_431 | orf_2509 | Internalin A | |||
| inlB | orf_432 | orf_2506 | Internalin B | |||
| inlC | orf_348 | Internalin C | ||||
| Immune modulation | cps4I | orf_2549 | orf_401 | Capsular polysaccharide biosynthesis protein Cps4I | ||
| gndA | orf_1382 | orf_1532 | NADP-dependent phosphogluconate dehydrogenase | |||
| hasC | orf_1079 | orf_1839 | UTP–glucose-1-phosphate uridylyltransferase | |||
| Nutritional/metabolic factor | mgtB | orf_2703 | orf_239 | Mg2+ transport protein | ||
| hbp1/svpA | orf_2194 | orf_676 | Haemoglobin-binding protein 1 | |||
| hbp2 | orf_2193 | orf_677 | Hypothetical protein | |||
| isdE | orf_2192 | orf_678 | Iron-regulated surface-determinant protein E | |||
| hpt | orf1_2093 | Hexose phosphate transport protein | ||||
| Exoenzyme | smcL | orf1_1675 | Sphingomyelinase-c | |||
| mpl | orf_200 | orf_2695 | Zinc metalloproteinase precursor | |||
| Exotoxin | cylR2 | orf1_2557 | Cytolysin regulator R2 | |||
| plcB | orf_202 | orf_2693 | Phospholipase C | |||
| hly | orf_199 | orf_2696 | Listeriolysin O precursor | |||
| plcA | orf_198 | orf_2697 | Phosphatidylinositol-specific phospholipase c | |||
| Adherence | tufA | orf_2666 | orf_278 | Elongation factor Tu | ||
| ami | orf_2570 | orf_1680 | Autolysin amidase, adhesin | |||
| inlF | orf_407 | orf_474 | Internalin F | |||
| Post-translational modification | gtcA | orf_2561 | orf_385 | Wall teichoic acid glycosylation protein | ||
| prsA2 | orf_2227 | orf_643 | Post-translocation chaperone |
| Species | Number of Each Cas-CRISPR System Type | |||||||
|---|---|---|---|---|---|---|---|---|
| I-B_n1 | I-B_n2 | II-A_n1 | II-A_n2 | II-C_n1 | III-B_n1 | VI-A_n2 | Total | |
| L. grayi | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| L. innocua | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 4 |
| L. ivanovii | 2 | 0 | 0 | 2 | 2 | 0 | 0 | 6 |
| L. marthii | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 |
| L. monocytogenes | 38 | 3 | 51 | 10 | 0 | 1 | 0 | 103 |
| L. seeligeri | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 5 |
| L. welshimeri | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 44 | 3 | 56 | 12 | 2 | 1 | 1 | 119 |
| Species | Number of Each Plasmid Type | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LMIVRS16815 | M640p00130 | M643p00680 | pLGUG1 | pLI100 | pLIS3 | pLIS5 | pLM33 | pLM5578 | pLMIV | pLMUKDL7 | Total | |
| L. grayi | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
| L. innocua | 0 | 2 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 5 |
| L. monocytogenes | 1 | 6 | 3 | 4 | 0 | 2 | 0 | 26 | 6 | 2 | 1 | 51 |
| L. seeligeri | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Total | 1 | 8 | 3 | 7 | 1 | 2 | 1 | 28 | 6 | 2 | 1 | 60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Q.; Zhu, X.; Long, Q.; Yi, X.; Yang, A.; Long, X.; Cao, D. Comparative Genomics Reveal the Utilization Ability of Variable Carbohydrates as Key Genetic Features of Listeria Pathogens in Their Pathogenic Lifestyles. Pathogens 2022, 11, 1430. https://doi.org/10.3390/pathogens11121430
Lu Q, Zhu X, Long Q, Yi X, Yang A, Long X, Cao D. Comparative Genomics Reveal the Utilization Ability of Variable Carbohydrates as Key Genetic Features of Listeria Pathogens in Their Pathogenic Lifestyles. Pathogens. 2022; 11(12):1430. https://doi.org/10.3390/pathogens11121430
Chicago/Turabian StyleLu, Qunfeng, Xiaoying Zhu, Qinqin Long, Xueli Yi, Anni Yang, Xidai Long, and Demin Cao. 2022. "Comparative Genomics Reveal the Utilization Ability of Variable Carbohydrates as Key Genetic Features of Listeria Pathogens in Their Pathogenic Lifestyles" Pathogens 11, no. 12: 1430. https://doi.org/10.3390/pathogens11121430
APA StyleLu, Q., Zhu, X., Long, Q., Yi, X., Yang, A., Long, X., & Cao, D. (2022). Comparative Genomics Reveal the Utilization Ability of Variable Carbohydrates as Key Genetic Features of Listeria Pathogens in Their Pathogenic Lifestyles. Pathogens, 11(12), 1430. https://doi.org/10.3390/pathogens11121430