
Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Availability
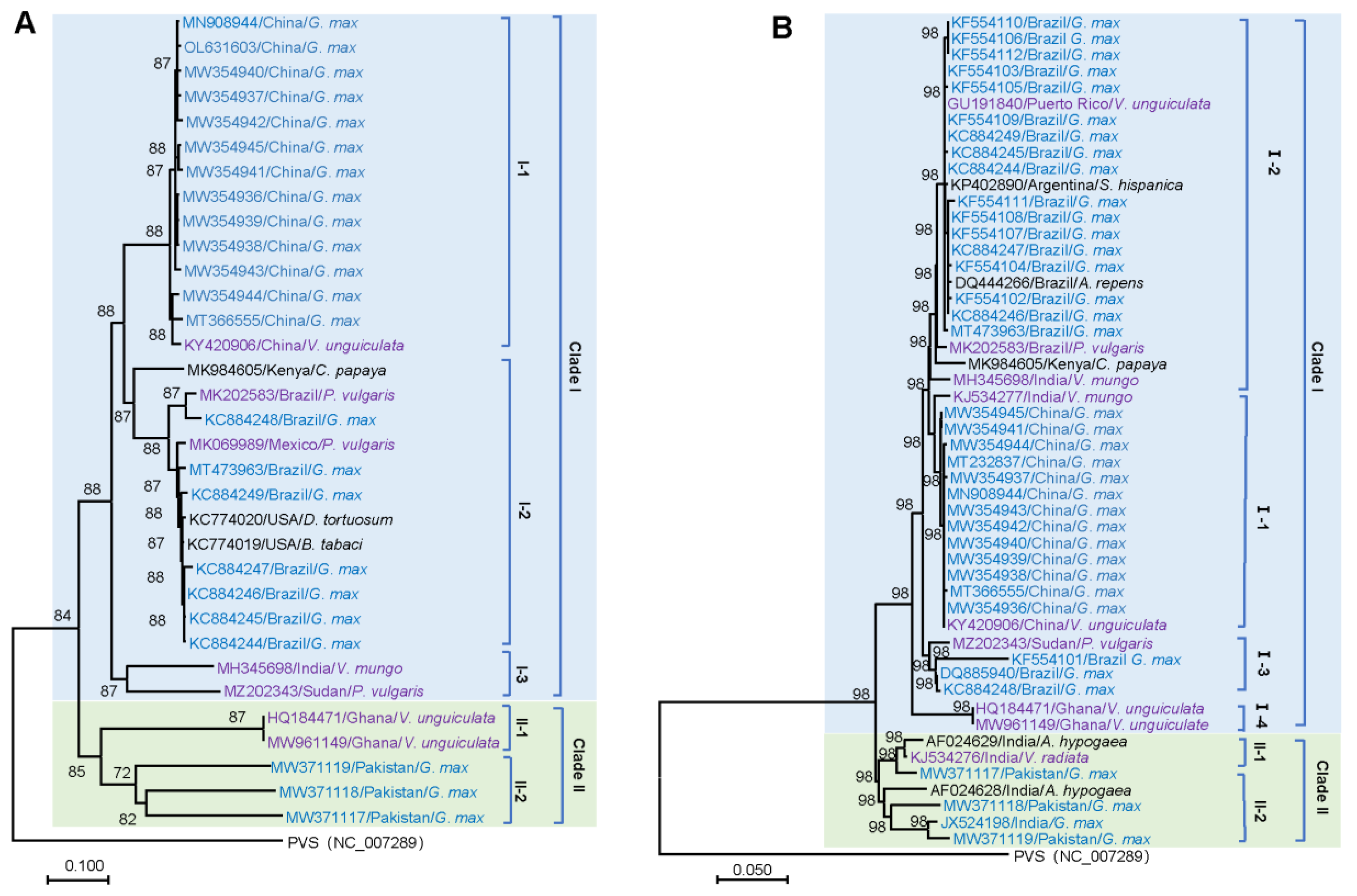
2.2. Phylogenetic Analysis
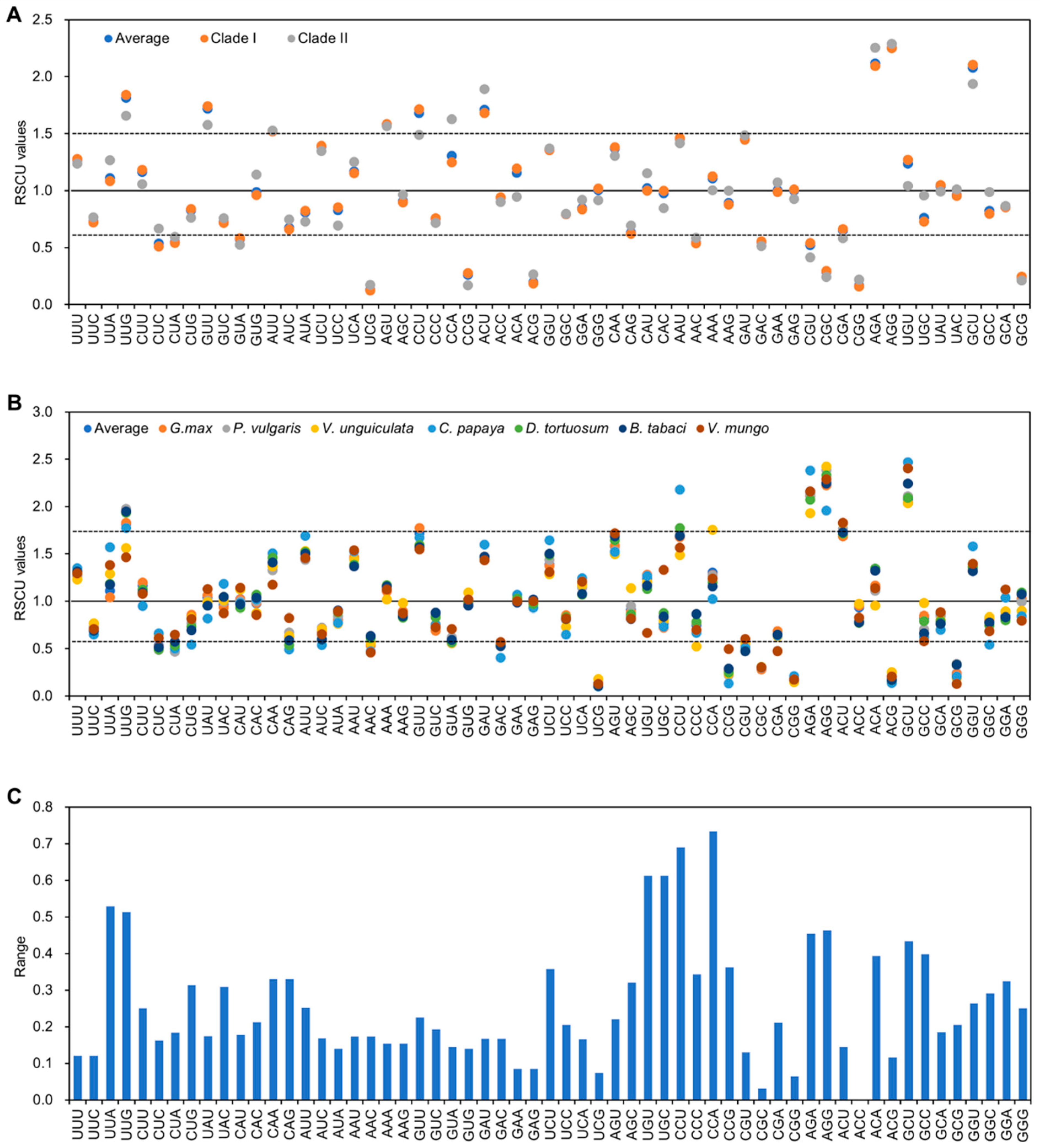
2.3. Calculation of the Relative Synonymous Codon Usage (RSCU)
2.4. Nucleotide Composition Analysis
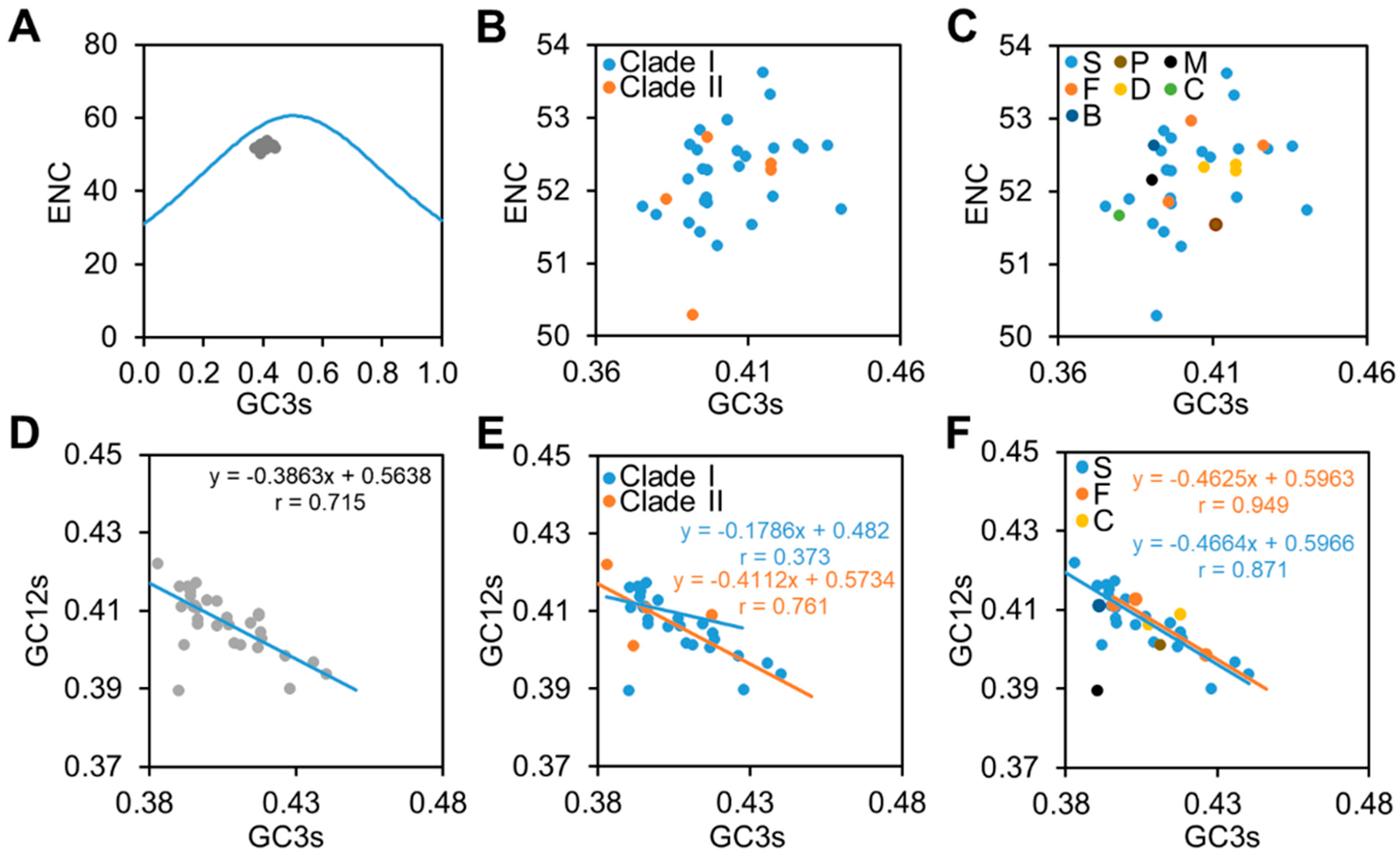
2.5. Analysis of Effective Number of Codons (ENC)
2.6. Neutrality Plot Analysis
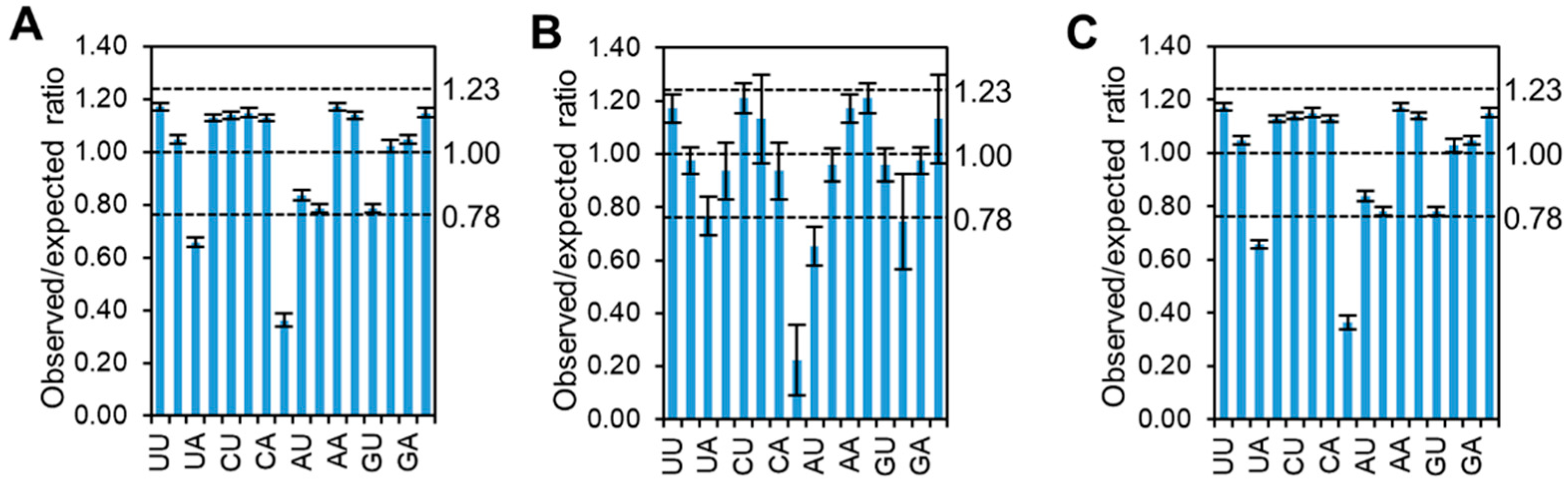
2.7. Dinucleotide Odds Ratio
3. Results
3.1. Phylogenetic Analysis
3.2. Nucleotide Composition Analysis
3.3. Natural Selection Is the Major Force Influencing the Codon Usage of CpMMV
3.4. RSCU Patterns of CpMMV
3.5. Dinucleotide Frequency Affects the Codon Pattern of CpMMV
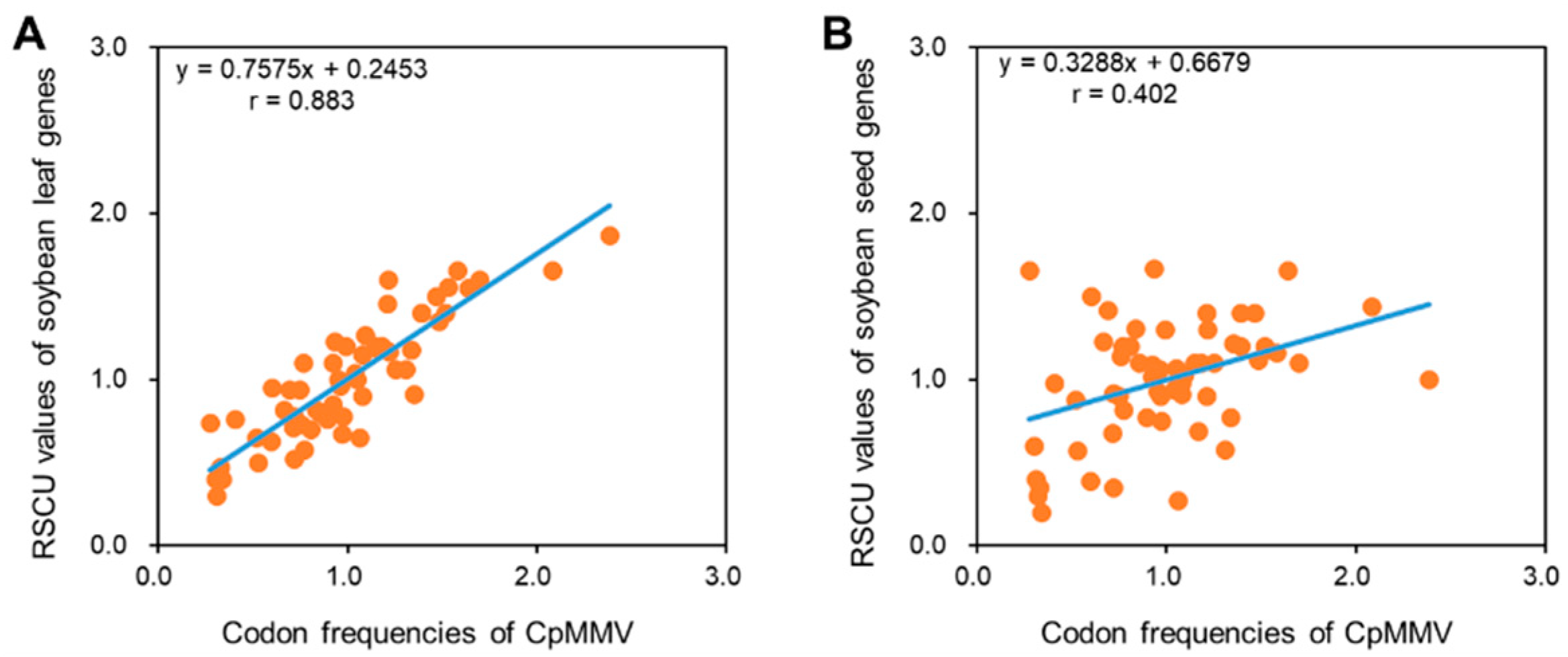
3.6. Codon Adaptation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jeyanandarajah, P.; Brunt, A.A. The natural occurrence, transmission, properties and possible affinities of cowpea mild mottle virus. J. Phytopathol. 1993, 137, 148–156. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, R.; Xiang, H.; Abouelnasr, H.; Li, D.; Yu, J.; McBeath, J.H.; Han, C. Discovery and characterization of a novel carlavirus infecting potatoes in China. PLoS ONE 2013, 8, e69255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senshu, H.; Yamaji, Y.; Minato, N.; Shiraishi, T.; Maejima, K.; Hashimoto, M.; Miura, C.; Neriya, Y.; Namba, S. A dual strategy for the suppression of host antiviral silencing: Two distinct suppressors for viral replication and viral movement encoded by Potato virus M. J. Virol. 2011, 85, 10269–10278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, N.; Komatsu, K.; Ayukawa, Y.; Matsuo, Y.; Hashimoto, M.; Netsu, O.; Teraoka, T.; Yamaji, Y.; Namba, S.; Arie, T. N-terminal region of cysteine-rich protein (CRP) in carlaviruses is involved in the determination of symptom types. Mol. Plant Pathol. 2018, 19, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Zanardo, L.G.; Carvalho, C.M. Cowpea mild mottle virus (Carlavirus, Betaflexiviridae): A review. Trop. Plant Pathol. 2017, 42, 417–430. [Google Scholar] [CrossRef]
- Brunt, A.A.; Kenten, R.H. Cowpea mild mottle, a newly recognized virus infecting cowpeas (Vigna unguiculata) in Ghana. Ann. Appl. Biol. 1973, 74, 67–74. [Google Scholar] [CrossRef]
- Iwaki, M.; Thongeearkom, P.; Prommin, M.; Honda, Y.; Hibi, T. Whitefly transmission and some properties of cowpea mild mottle virus on soybean in Thailand. Plant Dis. 1982, 66, 365–368. [Google Scholar] [CrossRef]
- Iizuka, N.; Rajeshwari, R.; Reddy, D.V.R.; Goto, T.; Muniyappa, V.; Bharathan, N.; Ghanekar, A.M. Natural occurrence of a strain of cowpea mild mottle virus on groundnut (Arachis hypogaea) in India. J. Phytopathol. 1984, 109, 245–253. [Google Scholar] [CrossRef]
- Antignus, Y.; Cohen, S. Purification and some properties of a new strain of cowpea mild mottle virus in Israel. Ann. Appl. Biol. 1987, 110, 563–569. [Google Scholar] [CrossRef]
- Mansour, A.; Al-Musa, A.; Vetten, H.J.; Lesemann, D.E. Properties of a cowpea mild mottle virus (CpMMV) isolate from eggplant in Jordan and evidence for biological and serological differences between CPMMV isolates from Leguminons and Solanaceous hosts. J. Phytopathol. 1998, 146, 539–547. [Google Scholar] [CrossRef]
- Pardina, P.E.R.; Arneodo, J.D.; Truol, G.A.; Herrera, P.S.; Laguna, I.G. First report of cowpea mild mottle virus in bean crops in Argentina. Australas. Plant Pathol. 2004, 33, 129–130. [Google Scholar]
- Brito, M.; Fernández-Rodríguez, T.; Garrido, M.J.; Mejías, A.; Romano, M.; Marys, E. First report of cowpea mild mottle carlavirus on yardlong bean (Vigna unguiculata subsp. sesquipedalis) in Venezuela. Viruses 2012, 4, 3804–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.A.; Chien, L.Y.; Tsai, C.F.; Lin, Y.Y.; Cheng, Y.H. First report of cowpea mild mottle virus in cowpea and french bean in Taiwan. Plant Dis. 2013, 97, 1001. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, L.G.; Silva, F.N.; Lima, A.T.M.; Milanesi, D.F.; Castilho-Urquiza, G.P.; Almeida, A.M.R.; Zerbini, F.M.; Carvalho, C.M. Molecular variability of cowpea mild mottle virus infecting soybean in Brazil. Arch. Virol. 2014, 159, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Lamas, N.S.; Matos, V.O.R.L.; Alves-Freitas, D.M.T.; Melo, F.L.; Costa, A.F.; Faria, J.C.; Ribeiro, S.G. Occurrence of cowpea mild mottle virus in common bean and associated weeds in Northeastern Brazil. Plant Dis. 2017, 101, 1828. [Google Scholar] [CrossRef]
- Wei, Z.Y.; Wu, G.W.; Ye, Z.X.; Jiang, C.; Mao, C.Y.; Zhang, H.H.; Miao, R.P.; Yan, F.; Li, J.M.; Chen, J.P.; et al. First report of cowpea mild mottle virus infecting soybean in China. Plant Dis. 2020, 104, 2534. [Google Scholar] [CrossRef]
- Wei, Z.; Mao, C.; Jiang, C.; Zhang, H.; Chen, J.; Sun, Z. Identification of a new genetic clade of cowpea mild mottle virus and characterization of its interaction with soybean mosaic virus in co-infected soybean. Front. Microbiol. 2021, 12, 650773. [Google Scholar] [CrossRef]
- Zanardo, L.G.; Silva, F.N.; Bicalho, A.A.C.; Urquiza, G.P.C.; Lima, A.T.M.; Almeida, A.M.R.; Zerbini, F.M.; Carvalho, C.M. Molecular and biological characterization of cowpea mild mottle virus isolates infecting soybean in Brazil and evidence of recombination. Plant Pathol. 2014, 63, 456–465. [Google Scholar] [CrossRef]
- Zanardo, L.; Trindade, T.; Mar, T.; Barbosa, T.; Milanesi, D.; Alves, M.; Lima, R.; Zerbini, F.; Janssen, A.; Mizubuti, E.; et al. Experimental evolution of cowpea mild mottle virus reveals recombination-driven reduction in virulence accompanied by increases in diversity and viral fitness. Virus Res. 2021, 303, 198389. [Google Scholar] [CrossRef]
- McInerney, J.O. GCUA: General codon usage analysis. Bioinformatics 1998, 14, 372–373. [Google Scholar] [CrossRef]
- Cheng, X.F.; Wu, X.Y.; Wang, H.; Sun, Y.; Qian, Y.; Luo, L. High codon adaptation in citrus tristeza virus to its citrus host. Virol. J. 2012, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Chen, X.; Ushijima, H.; Frey, T.K. Analysis of base and codon usage by rubella virus. Arch. Virol. 2012, 157, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.M.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L.M. Codon usage bias and the evolution of influenza A viruses. Codon usage biases of influenza virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Guo, Y.; Luo, L.; Wang, Y.P.; Dong, Z.M.; Sun, S.H.; Qiu, L.J. Analysis of nuclear gene codon bias on soybean genome and transcriptome. Zuo Wu Xue Bao 2011, 37, 965–974. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, N. Translation-coupled violation of parity rule 2 in human genes is not the cause of heterogeneity of the DNA G + C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Cheng, X.F.; Virk, N.; Chen, W.; Ji, S.; Ji, S.; Sun, Y.; Wu, X. CpG usage in RNA viruses: Data and hypotheses. PLoS ONE 2013, 8, e74109. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, Y.; Cheng, S.; Li, S.; Gong, Y.; Zhang, Z.; Lu, Q.; Hu, R.; Liu, Y.; Zhang, D. Genomic sequences measure and molecular characteristics of cowpea mild mottle virus of Hainan isolate. China Veg. 2019, 6, 35–38. [Google Scholar]
- Rima, B.K.; McFerran, N.V. Dinucleotide and stop codon frequencies in single-stranded RNA viruses. J. Gen. Virol. 1997, 78, 2859–2870. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.C.; Grover, A.; Kahl, G. Mining microsatellites in eukaryotic genomes. Trends Biotechnol. 2007, 25, 490–498. [Google Scholar] [CrossRef]
- Karlin, S.; Burge, C. Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 1995, 11, 283–290. [Google Scholar] [PubMed]
- Karlin, S.; Mrázek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlin, S.; Doerfler, W.; Cardon, L.R. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 1994, 68, 2889–2897. [Google Scholar] [CrossRef] [Green Version]
- González de Prádena, A.; Sánchez Jimenez, A.; San León, D.; Simmonds, P.; García, J.A.; Valli, A.A. Plant virus genome is shaped by specific dinucleotide restrictions that influence viral infection. mBio 2020, 11, e02818–e02819. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.; Fros, J.; Bertran, A.; Sechan, F.; Odon, V.; Torrance, L.; Kormelink, R.; Simmonds, P. A functional investigation of the suppression of CpG and UpA dinucleotide frequencies in plant RNA virus genomes. Sci. Rep. 2019, 9, 18359. [Google Scholar] [CrossRef]
- Tulloch, F.; Atkinson, N.J.; Evans, D.J.; Ryan, M.D.; Simmonds, P. RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. Elife 2014, 3, e04531. [Google Scholar] [CrossRef] [Green Version]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Odon, V.; Fros, J.J.; Goonawardane, N.; Dietrich, I.; Ibrahim, A.; Alshaikhahmed, K.; Nguyen, D.; Simmonds, P. The role of ZAP and OAS3/RNAseL pathways in the attenuation of an RNA virus with elevated frequencies of CpG and UpA dinucleotides. Nucleic Acids Res. 2019, 47, 8061–8083. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef]
- Tian, L.; Shen, X.; Murphy, R.W.; Shen, Y. The adaptation of codon usage of +ssRNA viruses to their hosts. Infect. Genet. Evol. 2018, 63, 175–179. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Gan, H.; Liang, X. Analysis of synonymous codon usage bias in potato virus M and its adaption to hosts. Viruses 2019, 11, 752. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | GenBank Accession No | Country | Isolate Name | Host |
|---|---|---|---|---|
| 1 | AF024628 | India | CPMMV-H | Arachis hypogaea |
| 2 | AF024629 | India | CPMMV-M | A. hypogaea |
| 3 | DQ444266 | Brazil | Arachis | A. repens |
| 4 | DQ885940 | Brazil | Barreiras | Glycine max |
| 5 | GU191840 | Puerto Rico | CpMMVPR | Vigna unguiculata |
| 6 | HQ184471 | Ghana | Ghana | V. unguiculata |
| 7 | JX524198 | India | D1 | G. max |
| 8 | KC774019 | USA | Whiteflies_2007 | Bemisia tabaci |
| 9 | KC774020 | USA | Florida_Bean_2011 | Desmodium tortuosum |
| 10 | KC884244 | Brazil | CPMMV:BR:MG:09:2 | G. max |
| 11 | KC884245 | Brazil | CPMMV:BR:MG:09:3 | G. max |
| 12 | KC884246 | Brazil | CPMMV:BR:MT:02:1 | G. max |
| 13 | KC884247 | Brazil | CPMMV:BR:BA:02 | G. max |
| 14 | KC884248 | Brazil | CPMMV:BR:GO:01:1 | G. max |
| 15 | KC884249 | Brazil | CPMMV:BR:GO:10:5 | G. max |
| 16 | KF554101 | Brazil | BR:GO:10:4 | G. max |
| 17 | KF554102 | Brazil | BR:MA:02 | G. max |
| 18 | KF554103 | Brazil | CPMMV:BR:MG:09:1 | G. max |
| 19 | KF554104 | Brazil | CPMMV:BR:MG:09:4 | G. max |
| 20 | KF554105 | Brazil | CPMMV:BR:MG:09:5 | G. max |
| 21 | KF554106 | Brazil | CPMMV:BR:MG:09:6 | G. max |
| 22 | KF554107 | Brazil | BR:MG:09:7 | G. max |
| 23 | KF554108 | Brazil | BR:MG:09:11 | G. max |
| 24 | KF554109 | Brazil | BR:MG:09:12 | G. max |
| 25 | KF554110 | Brazil | BR:MG:09:15 | G. max |
| 26 | KF554111 | Brazil | BR:MG:09:16 | G. max |
| 27 | KF554112 | Brazil | BR:PA:02 | G. max |
| 28 | KJ534276 | India | MUNGBEAN1 | V. radiata |
| 29 | KJ534277 | India | URDBEAN1 | V. mungo |
| 30 | KP402890 | Argentina | MA01 | Salvia hispanica |
| 31 | KY420906 | China | Hainan1 | V. unguiculata |
| 32 | MH345698 | India | CpMMV-Urd-Kanpur | V. mungo |
| 33 | MK069989 | Mexico | CN2 | Phaseolus vulgaris |
| 34 | MK202583 | Brazil | CPMMV:BR:GO:14 | P. vulgaris |
| 35 | MK984605 | Kenya | KE-Kit_01 | Carica papaya |
| 36 | MN908944 | China | Anhui_SZ_DN1383 | G. max |
| 37 | MT232837 | China | cpmmv-anhui-sz | G. max |
| 38 | MT366555 | China | CPMMV-JS | G. max |
| 39 | MT473963 | Brazil | Casa Branca_BR | G. max |
| 40 | MW354936 | China | CPMMV_HN_LH | G. max |
| 41 | MW354937 | China | CPMMV_SD_JX | G. max |
| 42 | MW354938 | China | CPMMV_HN_XC | G. max |
| 43 | MW354939 | China | CPMMV_HB_JZ | G. max |
| 44 | MW354940 | China | CPMMV_HN_SQ | G. max |
| 45 | MW354941 | China | CPMMV_SD_JN | G. max |
| 46 | MW354942 | China | CPMMV_JL_GZL | G. max |
| 47 | MW354943 | China | CPMMV_JL_CC | G. max |
| 48 | MW354944 | China | CPMMV_JS_NJ | G. max |
| 49 | MW354945 | China | CPMMV_AH_FY | G. max |
| 50 | MW371117 | Pakistan | PK1 | G. max |
| 51 | MW371118 | Pakistan | PK2 | G. max |
| 52 | MW371119 | Pakistan | PK3 | G. max |
| 53 | MW961149 | Ghana | DSMZ PV-0090 | V. unguiculate |
| 54 | MZ202343 | Sudan | DSMZ PV-0907 | P. vulgaris |
| 55 | OL631603 | China | AH-FY | G. max |
| Catalogs | Overall | Clade-I | Clade-II | Soybean | Cowpea | French Bean | Mung Bean | Papaya |
|---|---|---|---|---|---|---|---|---|
| A | 0.29 | 0.29 | 0.30 | 0.29 | 0.30 | 0.29 | 0.30 | 0.30 |
| U | 0.30 | 0.30 | 0.29 | 0.30 | 0.29 | 0.30 | 0.30 | 0.31 |
| C | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.18 | 0.17 |
| G | 0.23 | 0.23 | 0.23 | 0.23 | 0.23 | 0.23 | 0.23 | 0.22 |
| GC | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.40 | 0.39 |
| GC1 | 0.41 | 0.41 | 0.42 | 0.42 | 0.41 | 0.42 | 0.39 | 0.39 |
| GC2 | 0.40 | 0.40 | 0.40 | 0.40 | 0.41 | 0.39 | 0.41 | 0.39 |
| GC12 | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.41 | 0.40 | 0.39 |
| GC3 | 0.40 | 0.40 | 0.40 | 0.40 | 0.41 | 0.41 | 0.41 | 0.39 |
| A3 | 0.35 | 0.34 | 0.36 | 0.35 | 0.35 | 0.34 | 0.34 | 0.36 |
| U3 | 0.41 | 0.41 | 0.40 | 0.41 | 0.39 | 0.41 | 0.41 | 0.41 |
| C3 | 0.24 | 0.24 | 0.24 | 0.24 | 0.24 | 0.25 | 0.24 | 0.22 |
| G3 | 0.28 | 0.28 | 0.27 | 0.28 | 0.28 | 0.28 | 0.29 | 0.28 |
| ENC | 52.23 | 52.28 | 51.91 | 52.22 | 52.33 | 52.49 | 51.54 | 52.16 |
| AA | Codon | Overall | AA | Codon | Overall | AA | Codon | Overall |
|---|---|---|---|---|---|---|---|---|
| Phe | UUU | 1.27 | Pro | CCU | 1.68 | Glu | GAA | 1.00 |
| UUC | 0.73 | CCC | 0.75 | GAG | 1.00 | |||
| Leu | UUA | 1.11 | CCA | 1.31 | Arg | CGU | 0.52 | |
| UUG | 1.81 | CCG | 0.26 | CGC | 0.29 | |||
| CUU | 1.16 | Thr | ACU | 1.71 | CGA | 0.65 | ||
| CUC | 0.53 | ACC | 0.94 | CGG | 0.17 | |||
| CUA | 0.55 | ACA | 1.16 | AGA | 2.12 | |||
| CUG | 0.83 | ACG | 0.20 | AGG | 2.25 | |||
| Val | GUU | 1.72 | Gly | GGU | 1.36 | Cys | UGU | 1.24 |
| GUC | 0.72 | GGC | 0.79 | UGC | 0.76 | |||
| GUA | 0.57 | GGA | 0.85 | Tyr | UAU | 1.04 | ||
| GUG | 0.99 | GGG | 1.00 | UAC | 0.96 | |||
| Ser | UCU | 1.39 | Gln | CAA | 1.37 | Ala | GCU | 2.08 |
| UCC | 0.83 | CAG | 0.63 | GCC | 0.83 | |||
| UCA | 1.17 | His | CAU | 1.02 | GCA | 0.86 | ||
| UCG | 0.13 | CAC | 0.98 | GCG | 0.24 | |||
| AGU | 1.58 | Asn | AAU | 1.46 | Ile | AUU | 1.52 | |
| AGC | 0.91 | AAC | 0.54 | AUC | 0.67 | |||
| Lys | AAA | 1.11 | Asp | GAU | 1.45 | AUA | 0.81 | |
| AAG | 0.89 | GAC | 0.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Liu, Y.; Wu, X.; Cheng, X.; Wu, X. Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution. Pathogens 2022, 11, 419. https://doi.org/10.3390/pathogens11040419
Yang S, Liu Y, Wu X, Cheng X, Wu X. Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution. Pathogens. 2022; 11(4):419. https://doi.org/10.3390/pathogens11040419
Chicago/Turabian StyleYang, Siqi, Ye Liu, Xiaoyun Wu, Xiaofei Cheng, and Xiaoxia Wu. 2022. "Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution" Pathogens 11, no. 4: 419. https://doi.org/10.3390/pathogens11040419
APA StyleYang, S., Liu, Y., Wu, X., Cheng, X., & Wu, X. (2022). Synonymous Codon Pattern of Cowpea Mild Mottle Virus Sheds Light on Its Host Adaptation and Genome Evolution. Pathogens, 11(4), 419. https://doi.org/10.3390/pathogens11040419