
Source Attribution of Human Campylobacteriosis Using Whole-Genome Sequencing Data and Network Analysis

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Set
2.2. Bioinformatics Analysis
2.2.1. Assemblies
2.2.2. cgMLST and wgMLST
2.2.3. SNP
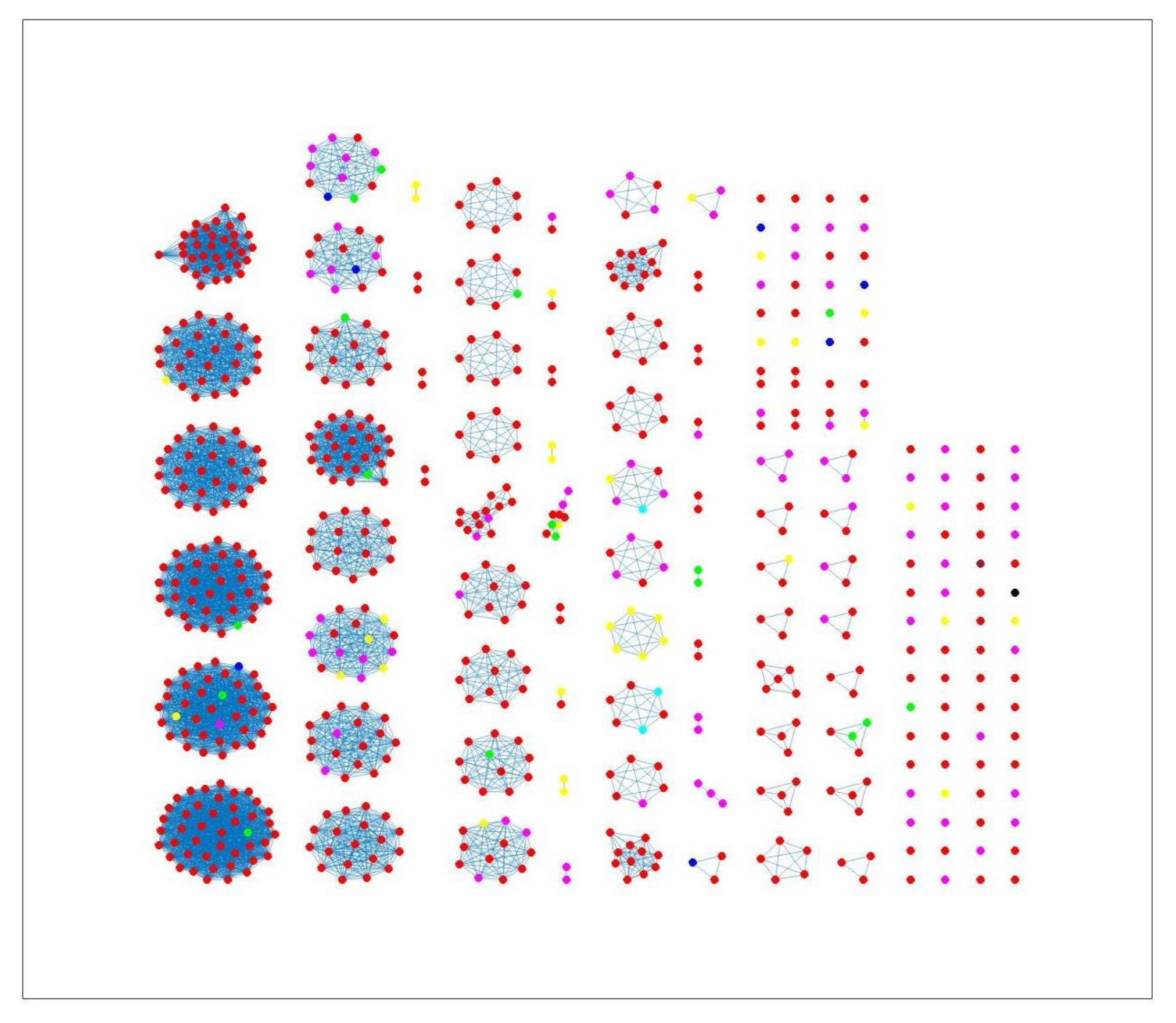
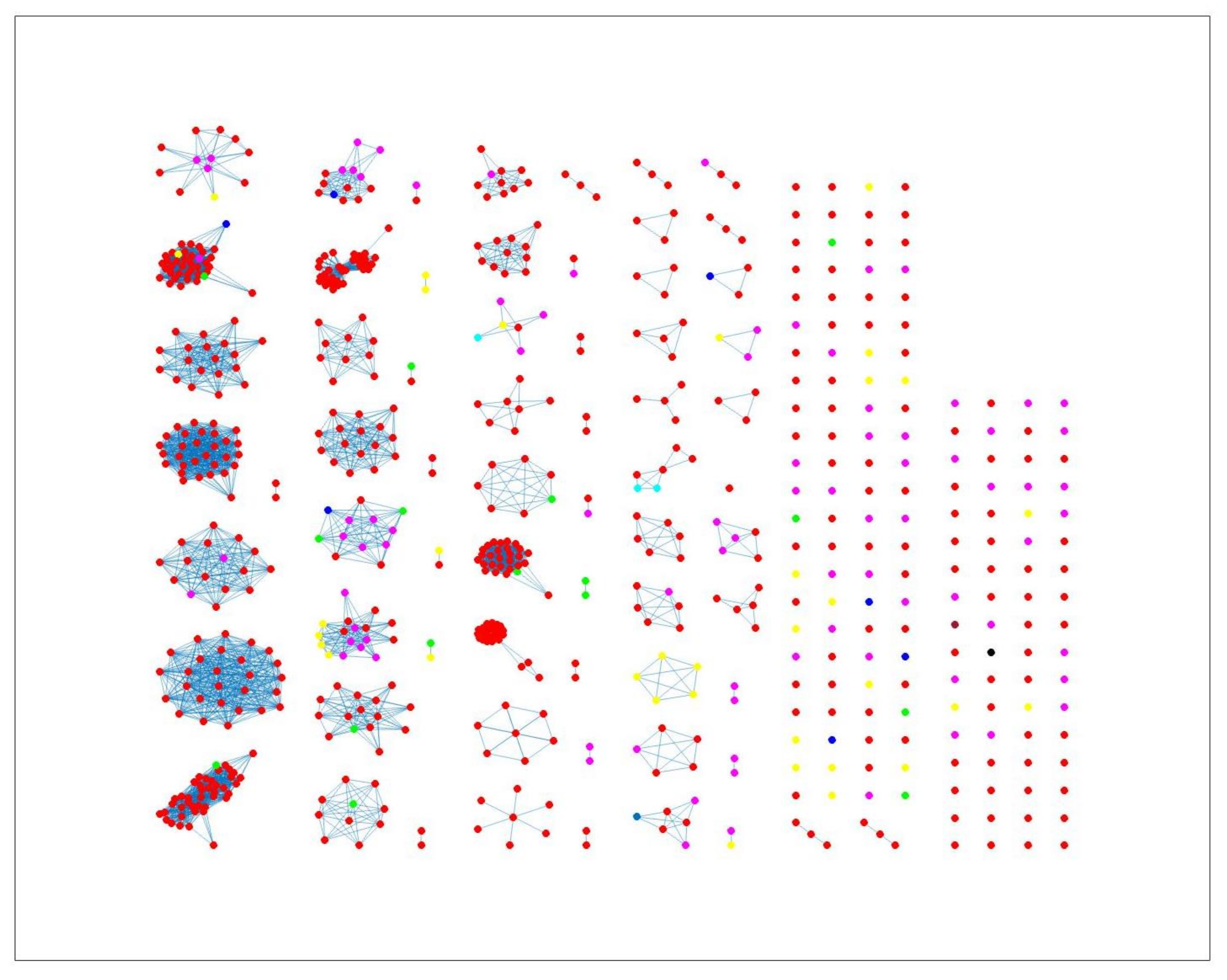
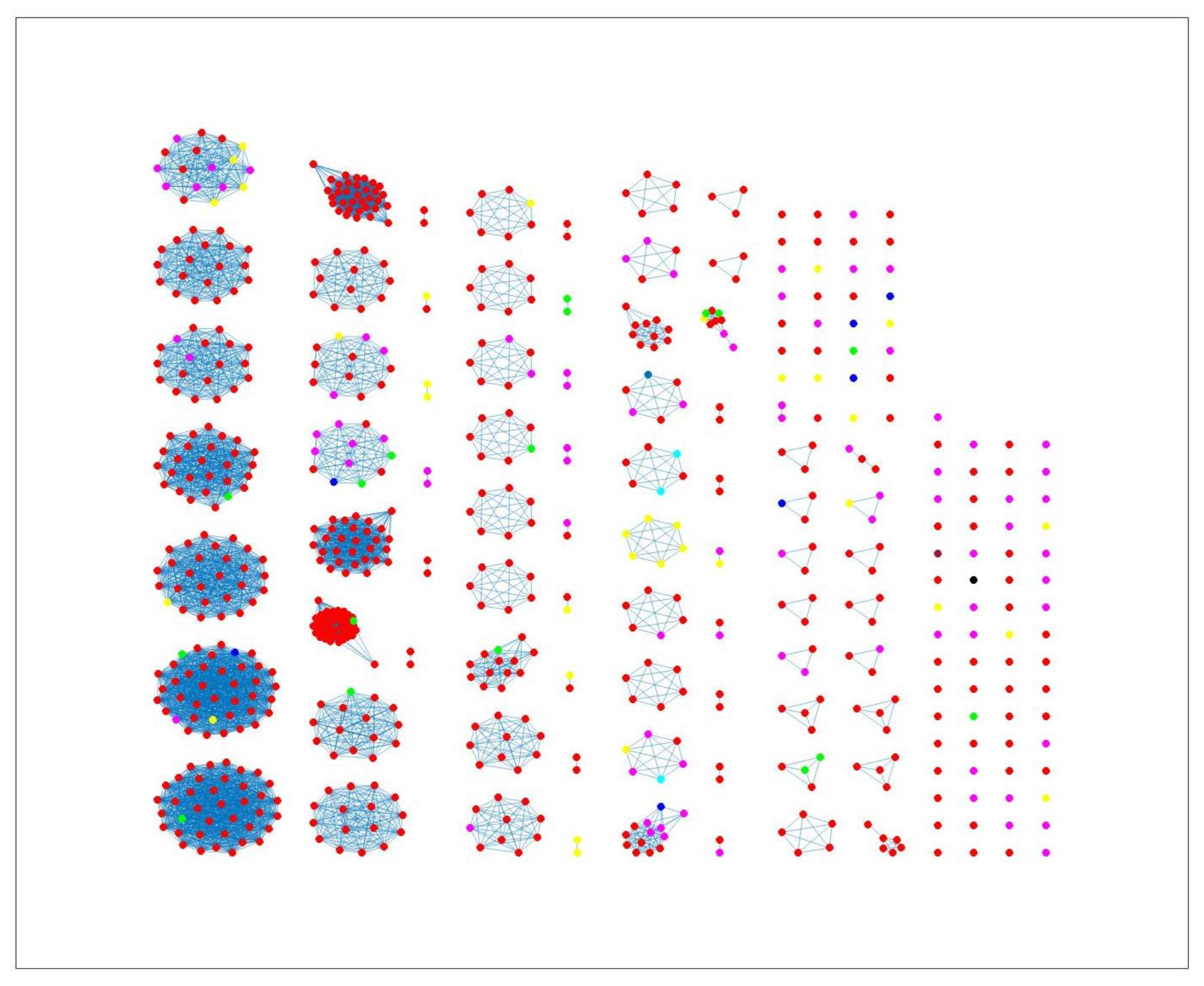
2.3. Network Analysis
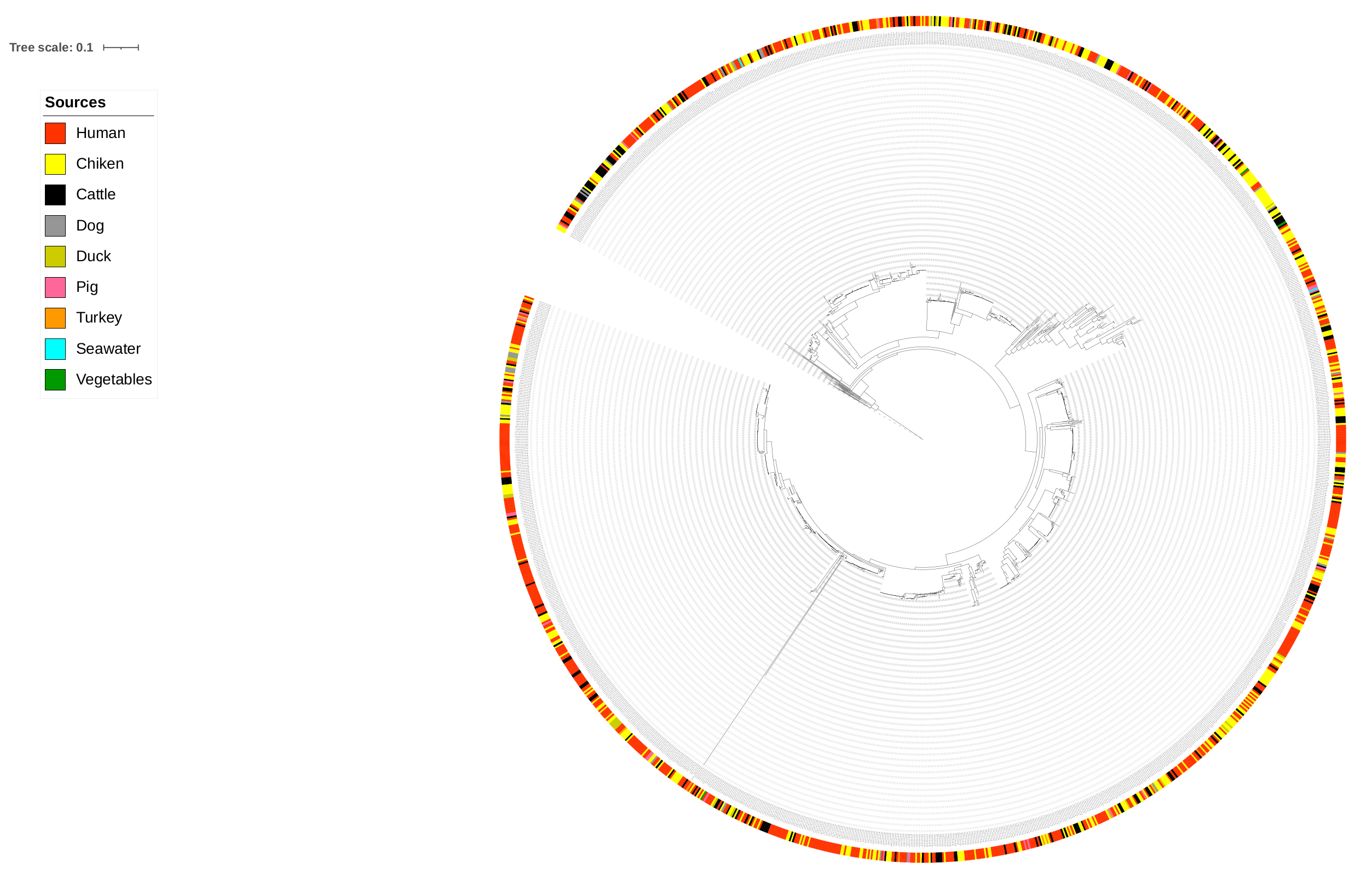
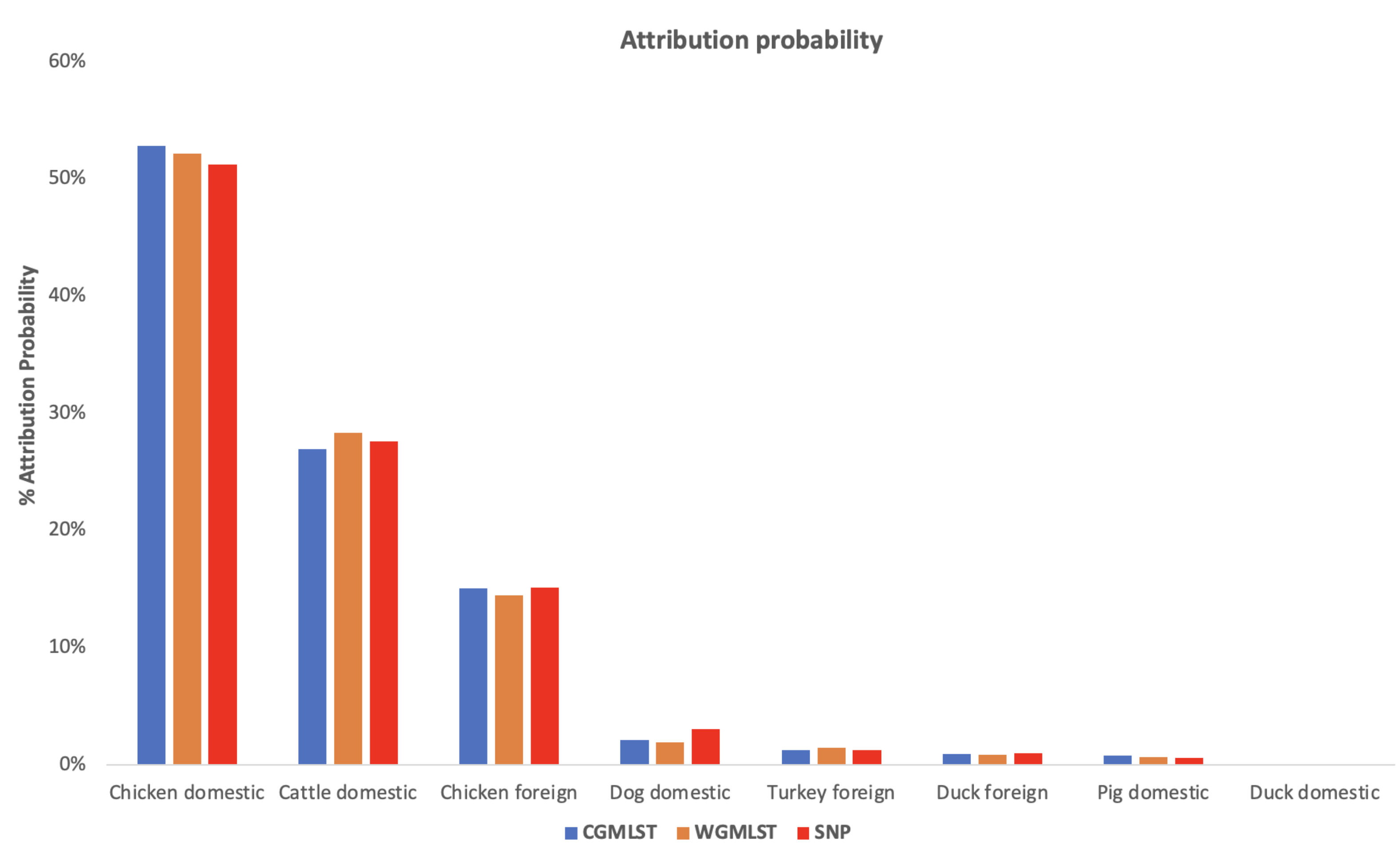
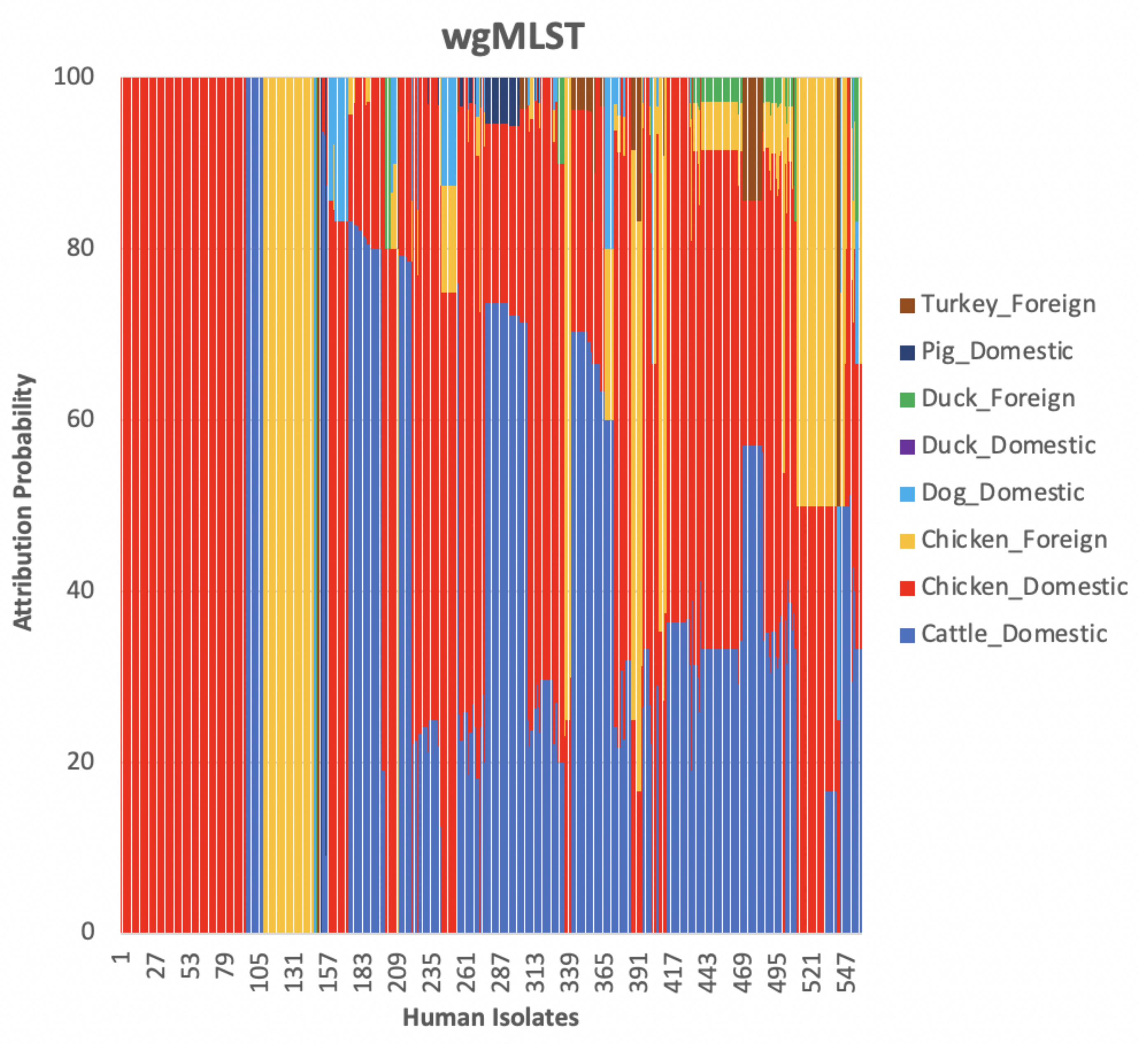
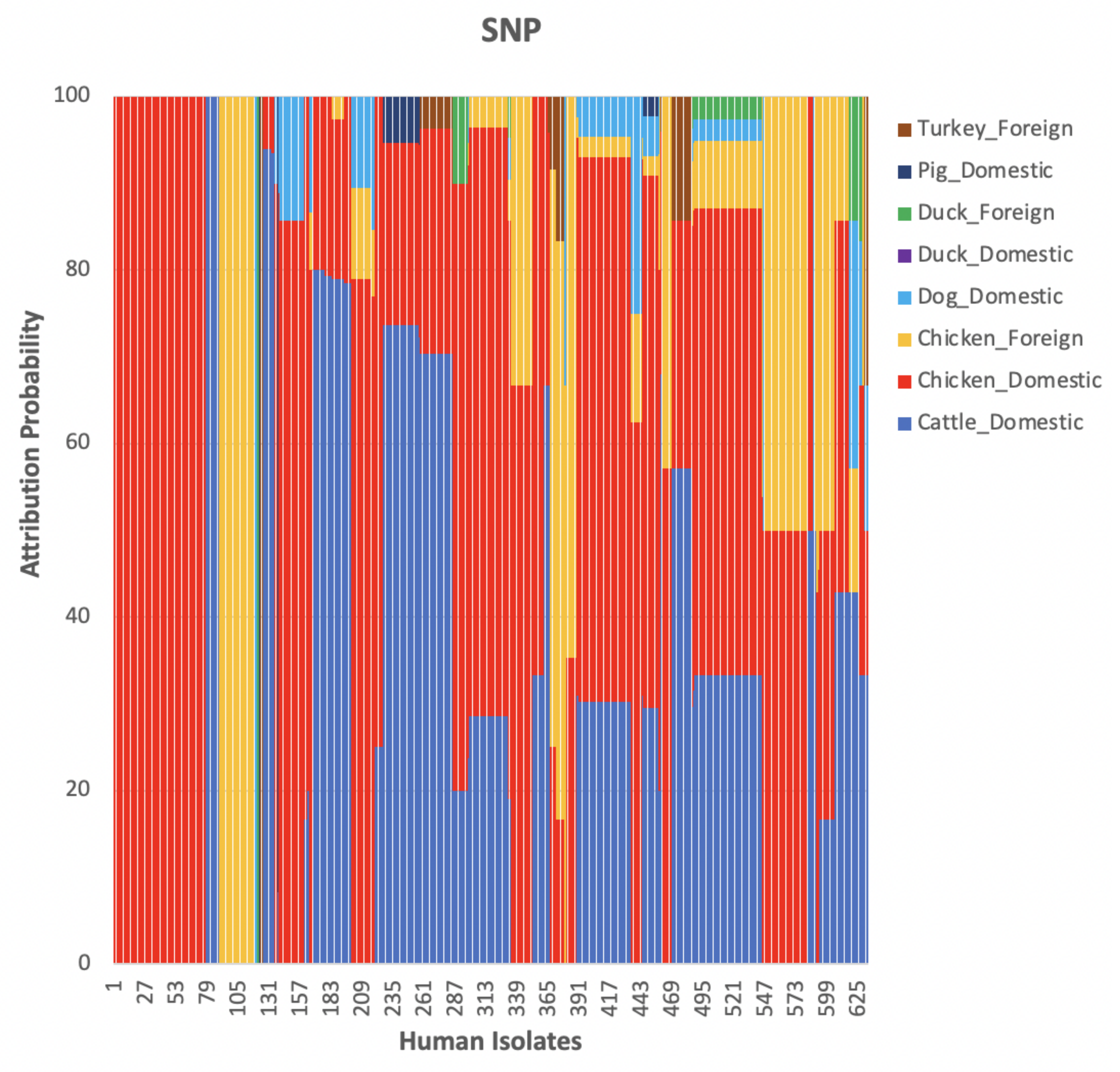
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Campylobacter in Denmark. Available online: https://www.foodsafetynews.com/2020/02/campylobacter-infections-at-record-high-in-denmark/ (accessed on 30 March 2022).
- Wingstrand, A.; Neimann, J.; Engberg, J.; Nielsen, E.M.; Gerner-Smidt, P.; Wegener, H.C.; Mølba, K. Fresh chicken as main risk factor for campylobacteriosis, Denmark. Emerg. Infect. Dis. 2006, 12, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, S.K.; Colles, F.M.; McCARTHY, N.D.; Strachan, N.J.C.; Ogden, I.D.; Forbes, K.J.; Dallas, J.F.; Maiden, M.C.J. Niche segregation and genetic structure of Campylobacter jenuni populations from wild and agricultural host species. Eur. Pubmed Cent. 2011, 20, 3484–3490. [Google Scholar]
- Merlotti, A.; Manfreda, G.; Munck, N.; Hald, T.; Litrup, E.; Nielsen, E.M.; Remondini, D.; Pasquali, F. Network Approach to Source Attribution of Salmonella enterica Serovar Typhimurium and Its Monophasic Variant. Front. Microbiol. 2020, 11, 1205. [Google Scholar] [CrossRef] [PubMed]
- Pires, S.M.; Evers, E.E.; Van Pely, W.; Ayers, T.; Scallan, E.; Angulo, F.J.; Havelaar, A.; Hald, T. Attributing the Human Disease Burden of Foodborne Infections to Specific Sources. Foodborne Pathog. Dis. 2009, 6, 417–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravel, A.; Hurst, M.; Petrica, N.; David, J.; Mutschall, S.K.; Pintar, K.; Taboada, E.N.; Pollari, F. Source attribution of human campylobacteriosis at the point of exposure by combining comparative exposure assessment and subtype comparison based on comparative genomic fingerprinting. PLoS ONE 2017, 12, e0183790. [Google Scholar] [CrossRef] [Green Version]
- Scientific Opinion of the Panel on Biological Hazards on a request from EFSA on Overview of methods for source attribution for human illness from food borne microbiological hazards. Overview of methods for source attribution for human cases of food borne microbiological hazards. EFSA J. 2008, 764, 1–43. [Google Scholar]
- Hald, T.; Vose, D.; Wegener, H.C.; Koupeev, T. Bayesian approach to quantify the contribution of animal-food sources to human salmonellosis. Risk Anal. 2004, 24, 251–265. [Google Scholar] [CrossRef]
- Dingle, K.E.; Colles, F.M.; Ure, R.; Wagenaar, J.A.; Duim, B.; Bolton, F.J.; Fox, A.J.; Wareing, D.R.A.; Maiden, M.C.J. Molecular characterization of Campylobacter jejuni clones: A rational basis for epidemiological investigations. Emerg. Infect. Dis. 2002, 8, 949–955. [Google Scholar] [CrossRef]
- Inns, T.; Ashton, P.M.; Herrera-Leon, S.; Lighthill, J.; Foulkes, S.; Jombart, T.; Rehman, Y.; Fox, A.; Dallman, T.; Pinna, E.D.E.; et al. Prospective use of whole genome sequencing (WGS) detected a multi-country outbreak of Salmonella Enteritidis. Epidemiol. Infect. 2017, 145, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Genestet, C.; Tatai, C.; Berland, J.L.; Claude, J.B.; Westeel, E.; Hodille, E.; Fredenucci, I.; Rasigade, J.P.; Ponsoda, M.; Jacomo, V.; et al. Prospective whole-genome sequencing in tuberculosis outbreak investigation. France, 2017–2018. Emerg. Infect. Dis. 2019, 25, 589–592. [Google Scholar] [CrossRef] [Green Version]
- Schjørring, S.; Lassen, S.G.; Jensen, T.; Moura, A.; Kjeldgaard, J.S.; Müller, L.; Thielke, S.; Leclercq, A.; Maury, M.M.; Tourdjman, M.; et al. Cross-border outbreak of listeriosis caused by cold-smoked salmon, revealed by integrated surveillance and whole genome sequencing (WGS), Denmark and France, 2015 to 2017. Eurosurveillance 2017, 22, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Arning, N.; Sheppard, S.K.; Bayliss, S.; Clifton, D.A.; Wilson, D.J. Machine learning to predict the source of campylobacteriosis using whole genome data. PLoS Genet. 2021, 17, e1009436. [Google Scholar] [CrossRef] [PubMed]
- ECDC. Expert Opinion on Whole Genome Sequencing for Public Health Surveillance; ECDC: Stockholm, Sweden; Solna, Sweden, 2016. [Google Scholar]
- Maiden, M.C.J.; Rensburg, M.J.J.V.; Bray, J.E.; Earle, S.G.; Ford, S.A.; Jolley, K.A.; McCarthy, N.D. MLST revisited: The gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 2013, 11, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Saltykova, A.; Mattheus, W.; Bertrand, S.; Roosens, N.H.C.; Marchal, K.; De Keersmaecker, S.C.J. Detailed Evaluation of Data Analysis Tools for Subtyping of Bacterial Isolates Based on Whole Genome Sequencing: Neisseria meningitidis as a Proof of Concept. Front. Microbiol. 2019, 10, 1–3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, S.; Gu, W.D.; Bakker, H.; Boxrud, D.; Taylor, A.; Roe, C.; Driebe, E.; Engelthaler, D.M.; Allard, M.; et al. Zoonotic Source Attribution of Salmonella enterica Serotype Typhimurium Using Genomic Surveillance Data, United States. Emerg. Infect. Dis. 2019, 25, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Lupolova, N.; Dallman, T.J.; Holden, N.J.; Gally, D.L. Patchy promiscuity: Machine learning applied to predict the host specificity of Salmonella enterica and Escherichia coli. Microb. Genom. 2017, 3, e000135. [Google Scholar] [CrossRef] [Green Version]
- Munck, N.; Njage, P.M.K.; Leekitcharoenphon, P.; Litrup, E.; Hald, T. Application of Whole-Genome Sequences and Machine Learning in Source Attribution of Salmonella Typhimurium. Risk Anal. 2020, 40, 1700–1703. [Google Scholar] [CrossRef]
- Njage, P.M.K.; Leekitcharoenphon, P.; Hansen, L.T.; Hendriksen, R.S.; Faes, C.; Aerts, M.; Hald, T. Quantitative Microbial Risk Assessment Based on Whole Genome Sequencing Data: Case of Listeria monocytogenes. Microorganisms 2020, 8, 1772. [Google Scholar] [CrossRef]
- Njage, P.M.K.; Henry, C.; Leekitcharoenphon, P.; Roussel, S.; Hendriksen, R.S.; Hald, T. Potential of machine learning methods as a tool for predicting risk of illness applying next generation sequencing data: Case of Listeria monocytogenes. Risk Anal. 2019, 39, 1397–1410. [Google Scholar]
- Njage, P.M.K.; Leekitcharoenphon, S.; Hald, T. Machine learning as a tool for microbial risk assessment using next generation sequencing data: Predicting clinical outcomes in shigatoxigenic Escherichia coli. Int. J. Food Microbiol. 2019, 292, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Tanui, C.K.; Karanth, S.; Njage, P.M.K.; Meng, J.; Pradhan, A.K. Machine learning-based predictive modeling to identify genotypic traits associated with Salmonella enterica disease endpoints in isolates from ground chicken. LWT 2022, 154, 112701. [Google Scholar] [CrossRef]
- Bandoy, D.; Weimer, B.C. Biological Machine Learning Combined with Campylobacter Population Genomics Reveals Virulence Gene Allelic Variants Cause Disease. Microorganisms 2020, 8, 549. [Google Scholar] [CrossRef] [PubMed]
- Santo, F.; Darko, H. Community detection in networks: A user guide. Phys. Rep. 2016, 659, 1–44. [Google Scholar]
- Joensen, K.G.; Kiil, K.; Gantzhorn, M.R.; Nauerby, B.; Engberg, J.; Holt, H.M.; Nielsen, H.L.; Petersen, A.M.; Kuhn, K.G.; Sandø, G.; et al. Whole-Genome Sequencing to Detect Numerous Campylobacter jejuni Outbreaks and Match Patient Isolates to Sources, Denmark, 2015–2017. Emerg. Infect. Dis. 2020, 26, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Cody, A.J.; Bray, J.E.; Jolley, K.A.; McCarthy, N.D.; Maiden, M.C.J. Coregenome Multilocus Sequence Typing Scheme for Stable, Comparative Analyses of Campylobacter jejuni and C. coli Human Disease Isolates. J. Clin. Microbiol. 2017, 55, 2086–2097. [Google Scholar] [CrossRef] [Green Version]
- Center for Genomic Epidemiology. Available online: https://www.genomicepidemiology.org/ (accessed on 31 March 2022).
- Cody, A.J.; McCarthy, N.D.; van Rensburg, M.J.; Isinkaye, T.; Bentley, S.D.; Parkhill, J.; Dingle, K.E.; Jolley, K.A.; Maiden, M.C.J. Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing. J. Clin. Microbiol. 2013, 51, 2526–2534. [Google Scholar] [CrossRef] [Green Version]
- Kaas, R.S.; Leekitcharoenphon, P.; Aarestrup, F.M.; Lund, O. Solving the Problem of Comparing Whole Bacterial Genomes across Different Sequencing Platforms. PLoS ONE 2014, 9, 1–6. [Google Scholar]
- Heng, L. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar]
- Heng, L.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar]
- Heng, L.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- Ivica, L.; Bork, P. Interactive Tree Of Life (iTOL) v6: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar]
- Computerome 2.0. Available online: https://www.computerome.dk (accessed on 30 March 2022).
- MATLABR2021b. Available online: https://www.mathworks.com/products/get-matlab.html?s_tid=gn_getml (accessed on 30 March 2022).
- Fruchterman, T.; Reingold, E. Graph drawing by force-directed placement. Soft. Prac. Exp. 1991, 21, 1129–1164. [Google Scholar] [CrossRef]
- Kuhn, M.; Johnson, K. Applied Predictive Modeling, 1st ed.; Springer: New York, NY, USA, 2013; pp. 415–419. [Google Scholar]
- Woodcock, D.J.; Krusche, P.; Strachan, N.J.C.; Forbes, K.J.; Cohan, F.M.; Méric, G.; Sheppard, K.S. Genomic plasticity and rapid host switching can promote the evolution of generalism: A case study in the zoonotic pathogen Campylobacter. Sci. Rep. 2017, 7, 9650. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Performance | cgMLST | wgMLST | SNP |
|---|---|---|---|
| Best threshold | 0.1141 | 0.0105 | 1715 |
| CSC | cgMLST | wgMLST | SNP |
|---|---|---|---|
| Species | 78% | 79% | 69% |
| Year | 61% | 64% | 67% |
| Human Isolates | cgMLST | wgMLST | SNP |
|---|---|---|---|
| Attributed | 632 | 558 | 633 |
| Not attributed | 85 | 159 | 86 |
| True / Pred | Cattle.dk | Chkn.dk | Chkn.for | Dog.dk | Duck.dk | Duck.for | Pig.dk | Turkey.for |
|---|---|---|---|---|---|---|---|---|
| Cattle.dk | 151 | 32 | 1 | 2 | 0 | 1 | 1 | 4 |
| Chkn.dk | 43 | 259 | 30 | 13 | 0 | 4 | 2 | 0 |
| Chkn.for | 2 | 20 | 87 | 0 | 0 | 1 | 0 | 2 |
| Dog.dk | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 |
| Duck.dk | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 |
| Duck.for | 0 | 0 | 0 | 0 | 0 | 17 | 0 | 0 |
| Pig.dk | 0 | 0 | 0 | 0 | 0 | 0 | 29 | 0 |
| Turkey.for | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 |
| True / Pred | Cattle.dk | Chkn.dk | Chkn.for | Dog.dk | Duck.dk | Duck.for | Pig.dk | Turkey.for |
|---|---|---|---|---|---|---|---|---|
| Cattle.dk | 157 | 31 | 2 | 2 | 0 | 1 | 1 | 2 |
| Chkn.dk | 37 | 263 | 24 | 9 | 0 | 2 | 1 | 0 |
| Chkn.for | 2 | 14 | 92 | 0 | 0 | 2 | 0 | 2 |
| Dog.dk | 0 | 0 | 0 | 9 | 0 | 0 | 0 | 0 |
| Duck.dk | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 |
| Duck.for | 0 | 0 | 0 | 0 | 0 | 19 | 0 | 0 |
| Pig.dk | 1 | 0 | 0 | 0 | 0 | 0 | 28 | 0 |
| Turkey.for | 2 | 1 | 0 | 1 | 0 | 0 | 0 | 5 |
| True / Pred | Cattle.dk | Chkn.dk | Chkn.for | Dog.dk | Duck.dk | Duck.for | Pig.dk | Turkey.for |
|---|---|---|---|---|---|---|---|---|
| Cattle.dk | 158 | 36 | 4 | 4 | 0 | 2 | 1 | 4 |
| Chkn.dk | 39 | 253 | 27 | 13 | 0 | 3 | 1 | 0 |
| Chkn.for | 2 | 20 | 89 | 0 | 0 | 1 | 0 | 2 |
| Dog.dk | 0 | 0 | 0 | 5 | 0 | 0 | 0 | 0 |
| Duck.dk | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 |
| Duck.for | 0 | 0 | 0 | 0 | 0 | 18 | 0 | 0 |
| Pig.dk | 1 | 0 | 0 | 0 | 0 | 0 | 28 | 0 |
| Turkey.for | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wainaina, L.; Merlotti, A.; Remondini, D.; Henri, C.; Hald, T.; Njage, P.M.K. Source Attribution of Human Campylobacteriosis Using Whole-Genome Sequencing Data and Network Analysis. Pathogens 2022, 11, 645. https://doi.org/10.3390/pathogens11060645
Wainaina L, Merlotti A, Remondini D, Henri C, Hald T, Njage PMK. Source Attribution of Human Campylobacteriosis Using Whole-Genome Sequencing Data and Network Analysis. Pathogens. 2022; 11(6):645. https://doi.org/10.3390/pathogens11060645
Chicago/Turabian StyleWainaina, Lynda, Alessandra Merlotti, Daniel Remondini, Clementine Henri, Tine Hald, and Patrick Murigu Kamau Njage. 2022. "Source Attribution of Human Campylobacteriosis Using Whole-Genome Sequencing Data and Network Analysis" Pathogens 11, no. 6: 645. https://doi.org/10.3390/pathogens11060645
APA StyleWainaina, L., Merlotti, A., Remondini, D., Henri, C., Hald, T., & Njage, P. M. K. (2022). Source Attribution of Human Campylobacteriosis Using Whole-Genome Sequencing Data and Network Analysis. Pathogens, 11(6), 645. https://doi.org/10.3390/pathogens11060645