
Antagonism of Protein Kinase R by Large DNA Viruses


Abstract
:1. Introduction
2. Poxviral Antagonists of Host Defense Pathways
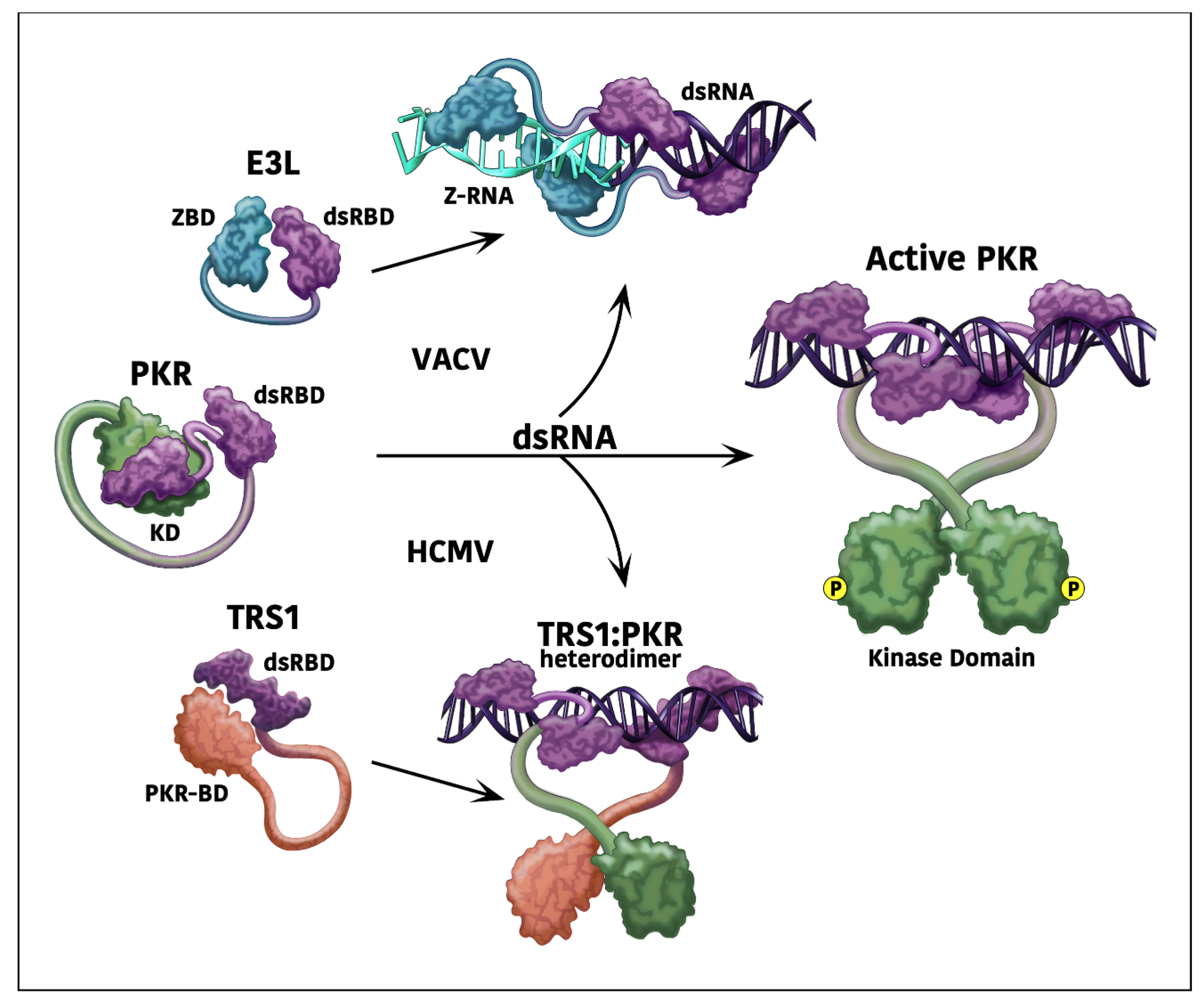
3. Complementation of VACV∆E3L by HCMV
4. Identification of the HCMV PKR and OAS/RNaseL Antagonists Using VACV∆E3L
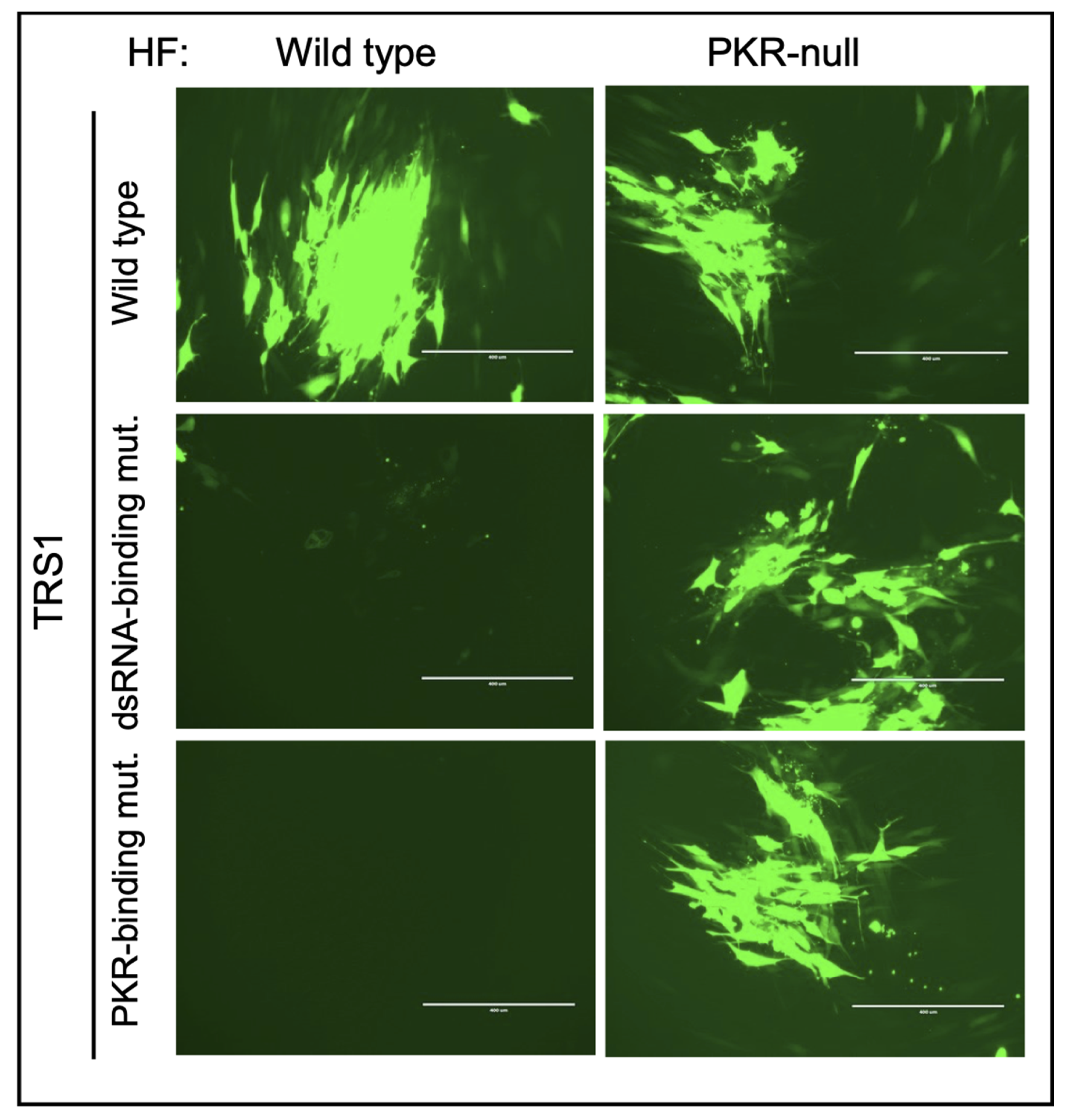
5. Mechanisms of HCMV Antagonism of dsRNA-Activated Host Defenses
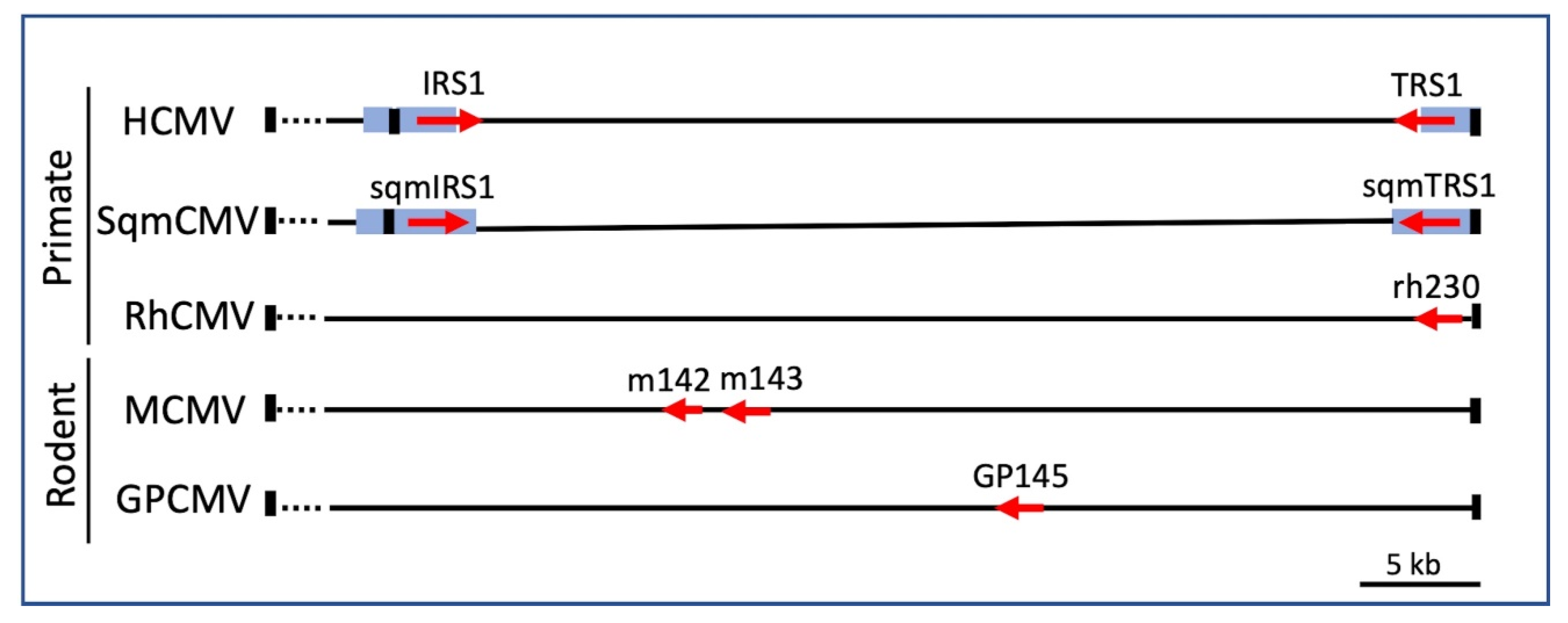
6. Interrogation of the CMV-PKR Evolutionary “Arms Race” Using VACV Recombinants

{kind=link}
{kind=link}
{kind=link}
| Viruses | Human Cell Lines | ||||||
|---|---|---|---|---|---|---|---|
| HeLa | HeLa/ PKRko | HF | HF/ PKRko | HF/ rTRS1 | References | ||
| VACV | + | + | + | + | + | [25,54,80] | |
| VACV∆E3L (VACV∆E3L∆K3L) | − | + | − | + | − | [25,54,82] | |
| VACV∆E3L/HCMV−TRS1 | + | + | + | + | [25,55,82] | ||
| VACV∆E3L/HCMV−IRS1 | + | + | [55] | ||||
| VACV∆E3L/RhCMV−TRS1 | − | + | − | + | + | [25,80,82] | |
| VACV∆E3L/AgmCMV−TRS1 | − | + | − | + | [25] | ||
| VACV∆E3L/SqmCMV−TRS1 | + | + | [25] | ||||
| HCMV | + | + | [23,78] | ||||
| HCMV∆I/∆T | − | + | [23] | ||||
| HCMV∆I/∆T/VACVE3L | + | [13] | |||||
| + | Permissive to virus replication | ||||||
| − | Restricted virus replication | ||||||
| Not Tested | |||||||
7. Role of Gene Dosage in Antagonism of PKR
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moss, B. Poxviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Kaynarcalidan, O.; Moreno Mascaraque, S.; Drexler, I. Vaccinia Virus: From Crude Smallpox Vaccines to Elaborate Viral Vector Vaccine Design. Biomedicines 2021, 9, 1780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Dong, L.; Zhao, C.; Zheng, P.; Zhang, X.; Xu, J. Vaccinia virus-based vector against infectious diseases and tumors. Hum. Vaccin. Immunother. 2021, 17, 1578–1585. [Google Scholar] [CrossRef] [PubMed]
- Torres-Dominguez, L.E.; McFadden, G. Poxvirus oncolytic virotherapy. Expert Opin. Biol. Ther. 2019, 19, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.S.; Lu, B.; Guo, Z.; Giehl, E.; Feist, M.; Dai, E.; Liu, W.; Storkus, W.J.; He, Y.; Liu, Z.; et al. Vaccinia virus-mediated cancer immunotherapy: Cancer vaccines and oncolytics. J. Immunother. Cancer. 2019, 7, 6. [Google Scholar] [CrossRef]
- Smith, E.S.; Shi, S.; Zauderer, M. Construction of cDNA libraries in vaccinia virus. Methods Mol. Biol. 2004, 269, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Belyakov, I.M.; Earl, P.L.; Berzofsky, J.A.; Moss, B. Enhanced cell surface expression, immunogenicity and genetic stability resulting from a spontaneous truncation of HIV Env expressed by a recombinant MVA. Virology 2008, 372, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.S.; Zauderer, M. Antibody library display on a mammalian virus vector: Combining the advantages of both phage and yeast display into one technology. Curr. Drug Discov. Technol. 2014, 11, 48–55. [Google Scholar] [CrossRef]
- Thiel, V.; Siddell, S.G. Reverse genetics of coronaviruses using vaccinia virus vectors. Curr. Top Microbiol. Immunol. 2005, 287, 199–227. [Google Scholar] [CrossRef] [Green Version]
- Mocarski, E.S.; Shenk, T.; Pass, R.F. Cytomegaloviruses. In Fields Virology. Philadelphia; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1961–2014. [Google Scholar]
- Semmes, E.C.; Hurst, J.H.; Walsh, K.M.; Permar, S.R. Cytomegalovirus as an immunomodulator across the lifespan. Curr. Opin. Virol. 2020, 44, 112–120. [Google Scholar] [CrossRef]
- Dever, T.E.; Dar, A.C.; Sicheri, F. The eIF2alpha kinases. In Translational Control in Biology and Medicine; Matthews, M.B., Sonenberg, N., Hershey, J.W.B., Eds.; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 2007; pp. 319–344. [Google Scholar]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [Green Version]
- Son, K.N.; Liang, Z.; Lipton, H.L. Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol. 2015, 89, 9383–9392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, I.J.; Pe’ery, T.; Mathews, M.B. Protein Synthesis and Translational Control during Viral Infection. In Translational Control in Biology and Medicine; Matthews, M.B., Sonenberg, N., Hershey, J.W.B., Eds.; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 2007; pp. 545–599. [Google Scholar]
- Thacore, H.R.; Youngner, J.S. Rescue of vesicular stomatitis virus from interferon-induced resistance by superinfection with vaccinia virus. I. Rescue in cell cultures from different species. Virology 1973, 56, 505–511. [Google Scholar] [CrossRef]
- Whitaker-Dowling, P.; Youngner, J.S. Vaccinia-mediated rescue of encephalomyocarditis virus from the inhibitory effects of interferon. Virology 1986, 152, 50–57. [Google Scholar] [CrossRef]
- Youngner, J.S.; Thacore, H.R.; Kelly, M.E. Sensitivity of ribonucleic acid and deoxyribonucleic acid viruses to different species of interferon in cell cultures. J. Virol. 1972, 10, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.W.; Watson, J.C.; Jacobs, B.L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 4825–4829. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.C.; Chang, H.W.; Jacobs, B.L. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology 1991, 185, 206–216. [Google Scholar] [CrossRef]
- Beattie, E.; Kauffman, E.B.; Martinez, H.; Perkus, M.E.; Jacobs, B.L.; Paoletti, E.; Tartaglia, J. Host-range restriction of vaccinia virus E3L-specific deletion mutants. Virus Genes 1996, 12, 89–94. [Google Scholar] [CrossRef]
- Braggin, J.E.; Child, S.J.; Geballe, A.P. Essential role of protein kinase R antagonism by TRS1 in human cytomegalovirus replication. Virology 2016, 489, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Varga, J.; Deschambault, Y. Poxvirus encoded eIF2alpha homolog, K3 family proteins, is a key determinant of poxvirus host species specificity. Virology 2020, 541, 101–112. [Google Scholar] [CrossRef]
- Carpentier, K.S.; Esparo, N.M.; Child, S.J.; Geballe, A.P. A Single Amino Acid Dictates Protein Kinase R Susceptibility to Unrelated Viral Antagonists. PLoS Pathog. 2016, 12, e1005966. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Jacobs, B.L.; Samuel, C.E. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J. Virol. 2008, 82, 840–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beattie, E.; Denzler, K.L.; Tartaglia, J.; Perkus, M.E.; Paoletti, E.; Jacobs, B.L. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J. Virol. 1995, 69, 499–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, A.D.; Turner, P.C.; Embury, J.E.; Moldawer, L.L.; Baker, H.V.; Moyer, R.W. Roles of vaccinia virus genes E3L and K3L and host genes PKR and RNase L during intratracheal infection of C57BL/6 mice. J. Virol. 2011, 85, 550–567. [Google Scholar] [CrossRef] [Green Version]
- Szczerba, M.; Subramanian, S.; Trainor, K.; McCaughan, M.; Kibler, K.V.; Jacobs, B.L. Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response. Biomedicines 2022, 10, 235. [Google Scholar] [CrossRef]
- Fierro-Monti, I.; Mathews, M.B. Proteins binding to duplexed RNA: One motif, multiple functions. Trends Biochem. Sci. 2000, 25, 241–246. [Google Scholar] [CrossRef]
- Ho, C.K.; Shuman, S. Mutational analysis of the vaccinia virus E3 protein defines amino acid residues involved in E3 binding to double-stranded RNA. J. Virol. 1996, 70, 2611–2614. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.W.; Uribe, L.H.; Jacobs, B.L. Rescue of vaccinia virus lacking the E3L gene by mutants of E3L. J. Virol. 1995, 69, 6605–6608. [Google Scholar] [CrossRef] [Green Version]
- Langland, J.O.; Jacobs, B.L. Inhibition of PKR by vaccinia virus: Role of the N- and C-terminal domains of E3L. Virology 2004, 324, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Brandt, T.A.; Jacobs, B.L. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 2001, 75, 850–856. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Muralinath, M.; Brandt, T.; Pearcy, M.; Hauns, K.; Lowenhaupt, K.; Jacobs, B.L.; Rich, A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 6974–6979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Yin, C.; Fedorov, A.; Qiao, L.; Bao, H.; Beknazarov, N.; Wang, S.; Gautam, A.; Williams, R.M.; Crawford, J.C.; et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 2022, 606, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Koehler, H.; Cotsmire, S.; Zhang, T.; Balachandran, S.; Upton, J.W.; Langland, J.; Kalman, D.; Jacobs, B.L.; Mocarski, E.S. Vaccinia virus E3 prevents sensing of Z-RNA to block ZBP1-dependent necroptosis. Cell Host Microbe 2021, 29, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Thakur, M.; Seo, E.J.; Dever, T.E. Variola virus E3L Zalpha domain, but not its Z-DNA binding activity, is required for PKR inhibition. RNA 2014, 20, 214–227. [Google Scholar] [CrossRef] [Green Version]
- White, S.D.; Jacobs, B.L. The amino terminus of the vaccinia virus E3 protein is necessary to inhibit the interferon response. J. Virol. 2012, 86, 5895–5904. [Google Scholar] [CrossRef] [Green Version]
- Mayo, C.B.; Erlandsen, H.; Mouser, D.J.; Feinstein, A.G.; Robinson, V.L.; May, E.R.; Cole, J.L. Structural Basis of Protein Kinase R Autophosphorylation. Biochemistry 2019, 58, 2967–2977. [Google Scholar] [CrossRef]
- Bierle, C.J.; Semmens, K.M.; Geballe, A.P. Double-stranded RNA binding by the human cytomegalovirus PKR antagonist TRS1. Virology 2013, 442, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Shors, S.T.; Beattie, E.; Paoletti, E.; Tartaglia, J.; Jacobs, B.L. Role of the vaccinia virus E3L and K3L gene products in rescue of VSV and EMCV from the effects of IFN-alpha. J. Interferon Cytokine Res. 1998, 18, 721–729. [Google Scholar] [CrossRef]
- Beattie, E.; Tartaglia, J.; Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology 1991, 183, 419–422. [Google Scholar] [CrossRef]
- Elde, N.C.; Child, S.J.; Geballe, A.P.; Malik, H.S. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 2009, 457, 485–489. [Google Scholar] [CrossRef]
- Park, C.; Peng, C.; Brennan, G.; Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Ann. N. Y. Acad. Sci. 2019, 1438, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Rothenburg, S.; Seo, E.J.; Gibbs, J.S.; Dever, T.E.; Dittmar, K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 2009, 16, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elde, N.C.; Child, S.J.; Eickbush, M.T.; Kitzman, J.O.; Rogers, K.S.; Shendure, J.; Geballe, A.P.; Malik, H.S. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 2012, 150, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Kawagishi-Kobayashi, M.; Silverman, J.B.; Ung, T.L.; Dever, T.E. Regulation of the protein kinase PKR by the vaccinia virus pseudosubstrate inhibitor K3L is dependent on residues conserved between the K3L protein and the PKR substrate eIF2alpha. Mol. Cell. Biol. 1997, 17, 4146–4158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra, S.; Abaitua, F.; Martinez-Sobrido, L.; Esteban, M.; Garcia-Sastre, A.; Rodriguez, D. Host-range restriction of vaccinia virus E3L deletion mutant can be overcome in vitro, but not in vivo, by expression of the influenza virus NS1 protein. PLoS ONE 2011, 6, e28677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langland, J.O.; Pettiford, S.; Jiang, B.; Jacobs, B.L. Products of the porcine group C rotavirus NSP3 gene bind specifically to double-stranded RNA and inhibit activation of the interferon-induced protein kinase PKR. J. Virol. 1994, 68, 3821–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shors, T.; Jacobs, B.L. Complementation of deletion of the vaccinia virus E3L gene by the Escherichia coli RNase III gene. Virology 1997, 227, 77–87. [Google Scholar] [CrossRef]
- Vijaysri, S.; Talasela, L.; Mercer, A.A.; McInnes, C.J.; Jacobs, B.L.; Langland, J.O. The Orf virus E3L homologue is able to complement deletion of the vaccinia virus E3L gene in vitro but not in vivo. Virology 2003, 314, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Hauns, K.; Langland, J.O.; Jacobs, B.L.; Hogue, B.G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 2007, 81, 2554–2563. [Google Scholar] [CrossRef] [Green Version]
- Child, S.J.; Jarrahian, S.; Harper, V.M.; Geballe, A.P. Complementation of vaccinia virus lacking the double-stranded RNA-binding protein gene E3L by human cytomegalovirus. J. Virol. 2002, 76, 4912–4918. [Google Scholar] [CrossRef] [Green Version]
- Child, S.J.; Hakki, M.; De Niro, K.L.; Geballe, A.P. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J. Virol. 2004, 78, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menard, C.; Wagner, M.; Ruzsics, Z.; Holak, K.; Brune, W.; Campbell, A.E.; Koszinowski, U.H. Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J. Virol. 2003, 77, 5557–5570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Child, S.J.; Hanson, L.K.; Brown, C.E.; Janzen, D.M.; Geballe, A.P. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J. Virol. 2006, 80, 10173–10180. [Google Scholar] [CrossRef] [Green Version]
- Ostermann, E.; Warnecke, G.; Waibler, Z.; Brune, W. Knockout of the Host Resistance Gene Pkr Fully Restores Replication of Murine Cytomegalovirus m142 and m143 Mutants In Vivo. J. Virol. 2016, 90, 1144–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valchanova, R.S.; Picard-Maureau, M.; Budt, M.; Brune, W. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J. Virol. 2006, 80, 10181–10190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierle, C.J.; Schleiss, M.R.; Geballe, A.P. Antagonism of the protein kinase R pathway by the guinea pig cytomegalovirus US22-family gene gp145. Virology 2012, 433, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Schleiss, M.R.; Bierle, C.J.; Swanson, E.C.; McVoy, M.A.; Ben Wang, J.; Al-Mahdi, Z.; Geballe, A.P. Vaccination with a Live Attenuated Cytomegalovirus Devoid of a Protein Kinase R Inhibitory Gene Results in Reduced Maternal Viremia and Improved Pregnancy Outcome in a Guinea Pig Congenital Infection Model. J. Virol. 2015, 89, 9727–9738. [Google Scholar] [CrossRef] [Green Version]
- Blankenship, C.A.; Shenk, T. Mutant human cytomegalovirus lacking the immediate-early TRS1 coding region exhibits a late defect. J. Virol. 2002, 76, 12290–12299. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.R.; Muzithras, V.P. A cluster of dispensable genes within the human cytomegalovirus genome short component: IRS1, US1 through US5, and the US6 family. J. Virol. 1992, 66, 2541–2546. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Silva, M.C.; Shenk, T. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. USA 2003, 100, 12396–12401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziehr, B.; Vincent, H.A.; Moorman, N.J. Human Cytomegalovirus pTRS1 and pIRS1 Antagonize Protein Kinase R To Facilitate Virus Replication. J. Virol. 2016, 90, 3839–3848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassady, K.A. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J. Virol. 2005, 79, 8707–8715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budt, M.; Niederstadt, L.; Valchanova, R.S.; Jonjic, S.; Brune, W. Specific inhibition of the PKR-mediated antiviral response by the murine cytomegalovirus proteins m142 and m143. J. Virol. 2009, 83, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaumorcel, M.; Lussignol, M.; Mouna, L.; Cavignac, Y.; Fahie, K.; Cotte-Laffitte, J.; Geballe, A.; Brune, W.; Beau, I.; Codogno, P.; et al. The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 2012, 86, 2571–2584. [Google Scholar] [CrossRef] [Green Version]
- Romanowski, M.J.; Shenk, T. Characterization of the human cytomegalovirus irs1 and trs1 genes: A second immediate-early transcription unit within irs1 whose product antagonizes transcriptional activation. J. Virol. 1997, 71, 1485–1496. [Google Scholar] [CrossRef] [Green Version]
- Stasiak, P.C.; Mocarski, E.S. Transactivation of the cytomegalovirus ICP36 gene promoter requires the alpha gene product TRS1 in addition to IE1 and IE2. J. Virol. 1992, 66, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Vincent, H.A.; Ziehr, B.; Lenarcic, E.M.; Moorman, N.J. Human cytomegalovirus pTRS1 stimulates cap-independent translation. Virology 2019, 537, 246–253. [Google Scholar] [CrossRef]
- Ziehr, B.; Lenarcic, E.; Vincent, H.A.; Cecil, C.; Garcia, B.; Shenk, T.; Moorman, N.J. Human cytomegalovirus TRS1 protein associates with the 7-methylguanosine mRNA cap and facilitates translation. Proteomics 2015, 15, 1983–1994. [Google Scholar] [CrossRef] [Green Version]
- Shors, T.; Kibler, K.V.; Perkins, K.B.; Seidler-Wulff, R.; Banaszak, M.P.; Jacobs, B.L. Complementation of vaccinia virus deleted of the E3L gene by mutants of E3L. Virology 1997, 239, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Hakki, M.; Geballe, A.P. Double-stranded RNA binding by human cytomegalovirus pTRS1. J. Virol. 2005, 79, 7311–7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakki, M.; Marshall, E.E.; De Niro, K.L.; Geballe, A.P. Binding and nuclear relocalization of protein kinase R by human cytomegalovirus TRS1. J. Virol. 2006, 80, 11817–11826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, M.D.; Malik, H.S. Rules of engagement: Molecular insights from host-virus arms races. Annu. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Child, S.J.; Hickson, S.E.; Bayer, A.; Malouli, D.; Fruh, K.; Geballe, A.P. Antagonism of the Protein Kinase R Pathway in Human Cells by Rhesus Cytomegalovirus. J. Virol. 2018, 92, e01793-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Child, S.J.; Greninger, A.L.; Geballe, A.P. Rapid adaptation to human protein kinase R by a unique genomic rearrangement in rhesus cytomegalovirus. PLoS Pathog. 2021, 17, e1009088. [Google Scholar] [CrossRef] [PubMed]
- Brennan, G.; Kitzman, J.O.; Rothenburg, S.; Shendure, J.; Geballe, A.P. Adaptive gene amplification as an intermediate step in the expansion of virus host range. PLoS Pathog. 2014, 10, e1004002. [Google Scholar] [CrossRef]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Child, S.J.; Brennan, G.; Braggin, J.E.; Geballe, A.P. Species specificity of protein kinase r antagonism by cytomegalovirus TRS1 genes. J. Virol. 2012, 86, 3880–3889. [Google Scholar] [CrossRef] [Green Version]
- Sidrauski, C.; Acosta-Alvear, D.; Khoutorsky, A.; Vedantham, P.; Hearn, B.; Li, H.; Gamache, K.; Gallagher, C.M.; Ang, K.K.-H.; Wilson, C.; et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife 2013, 2, e00498. [Google Scholar] [CrossRef]
- Erlandson, K.J.; Cotter, C.A.; Charity, J.C.; Martens, C.; Fischer, E.R.; Ricklefs, S.M.; Porcella, S.F.; Moss, B. Duplication of the A17L locus of vaccinia virus provides an alternate route to rifampin resistance. J. Virol. 2014, 88, 11576–11585. [Google Scholar] [CrossRef] [Green Version]
- Slabaugh, M.; Roseman, N.; Davis, R.; Mathews, C. Vaccinia virus-encoded ribonucleotide reductase: Sequence conservation of the gene for the small subunit and its amplification in hydroxyurea-resistant mutants. J. Virol. 1988, 62, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, G.; Kitzman, J.O.; Shendure, J.; Geballe, A.P. Experimental Evolution Identifies Vaccinia Virus Mutations in A24R and A35R That Antagonize the Protein Kinase R Pathway and Accompany Collapse of an Extragenic Gene Amplification. J. Virol. 2015, 89, 9986–9997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olson, A.T.; Child, S.J.; Geballe, A.P. Antagonism of Protein Kinase R by Large DNA Viruses. Pathogens 2022, 11, 790. https://doi.org/10.3390/pathogens11070790
Olson AT, Child SJ, Geballe AP. Antagonism of Protein Kinase R by Large DNA Viruses. Pathogens. 2022; 11(7):790. https://doi.org/10.3390/pathogens11070790
Chicago/Turabian StyleOlson, Annabel T., Stephanie J. Child, and Adam P. Geballe. 2022. "Antagonism of Protein Kinase R by Large DNA Viruses" Pathogens 11, no. 7: 790. https://doi.org/10.3390/pathogens11070790
APA StyleOlson, A. T., Child, S. J., & Geballe, A. P. (2022). Antagonism of Protein Kinase R by Large DNA Viruses. Pathogens, 11(7), 790. https://doi.org/10.3390/pathogens11070790