
Interferon Epsilon Signaling Confers Attenuated Zika Replication in Human Vaginal Epithelial Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
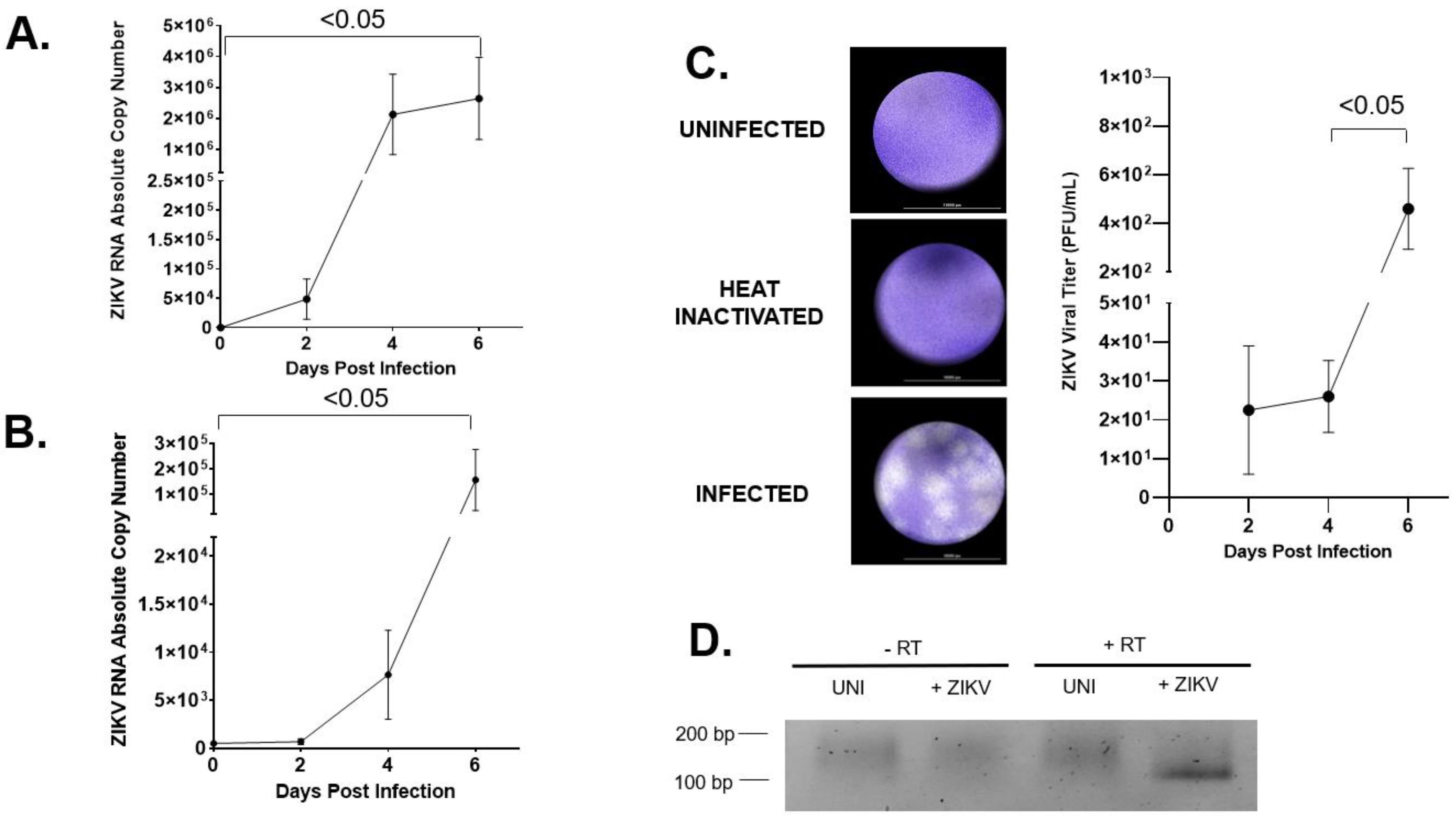
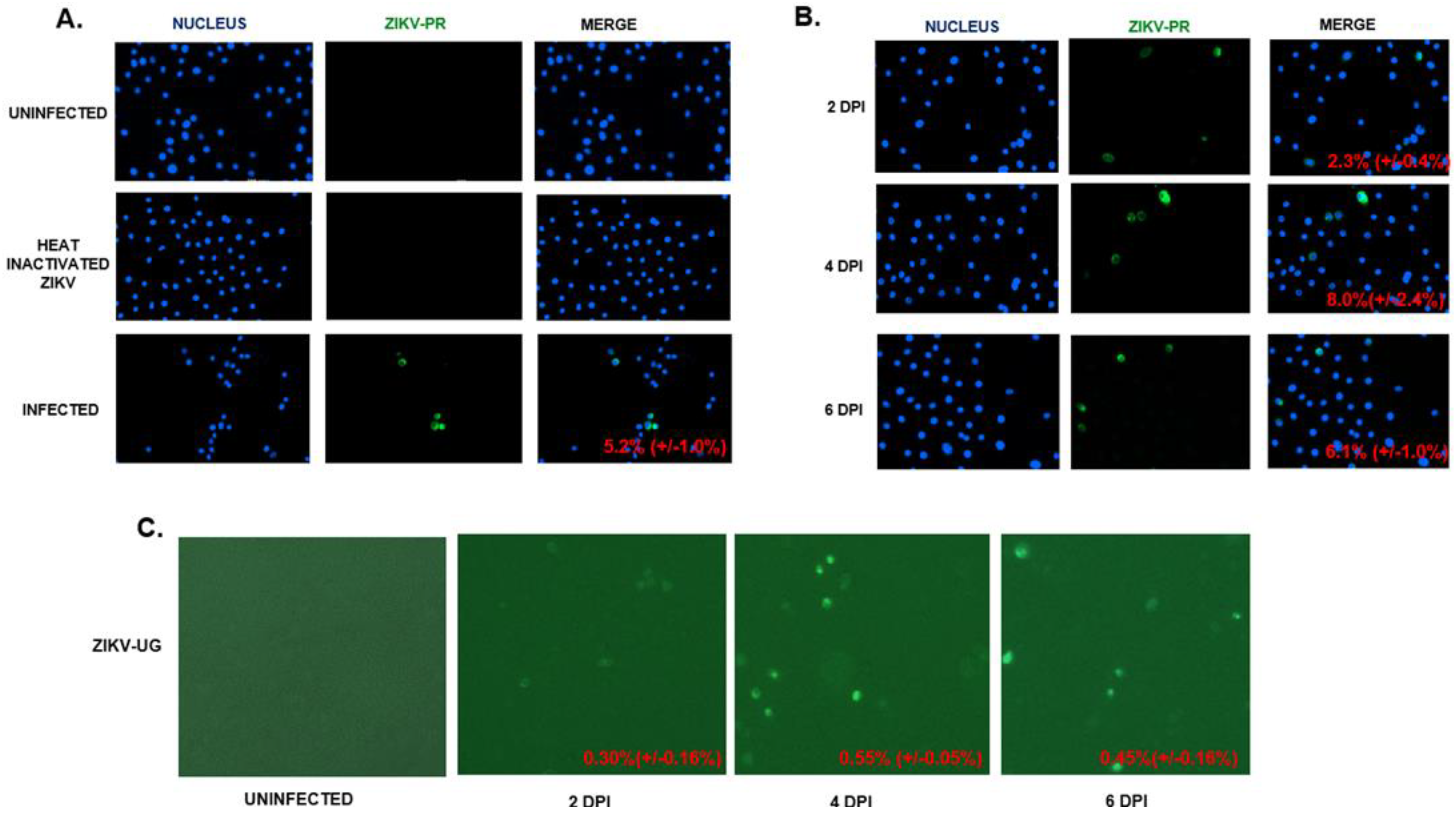
2.1. hVECs Support ZIKV Replication and Viral Production in Vitro
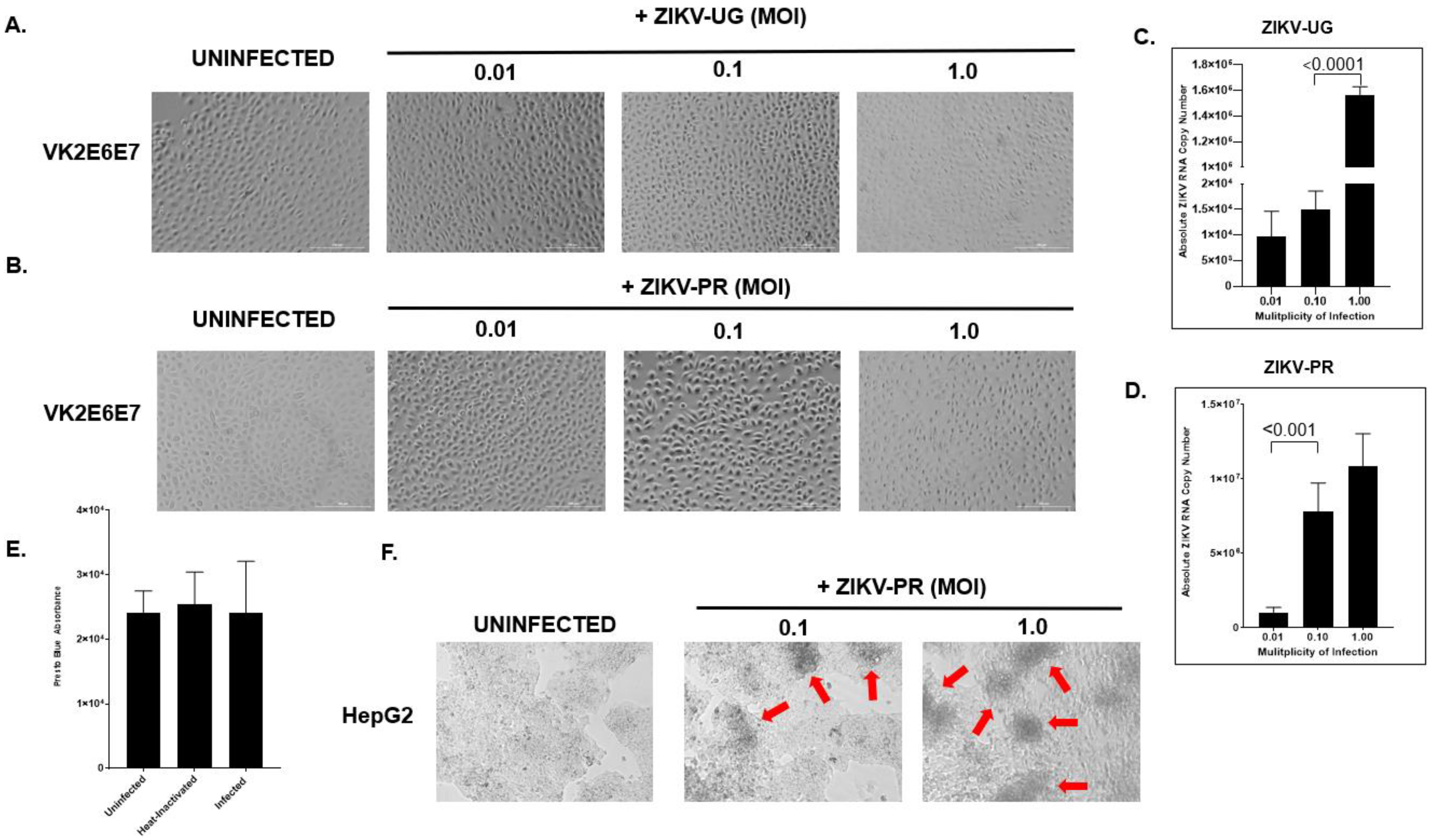
2.2. hVECs Do Not Exhibit Significant Cytopathic Effects following ZIKV Infection
2.3. Interferon Inhibitor Ruxolitinib Enhances ZIKV Replication and Induces Cytopathic Effect in ZIKV-Infected hVECs
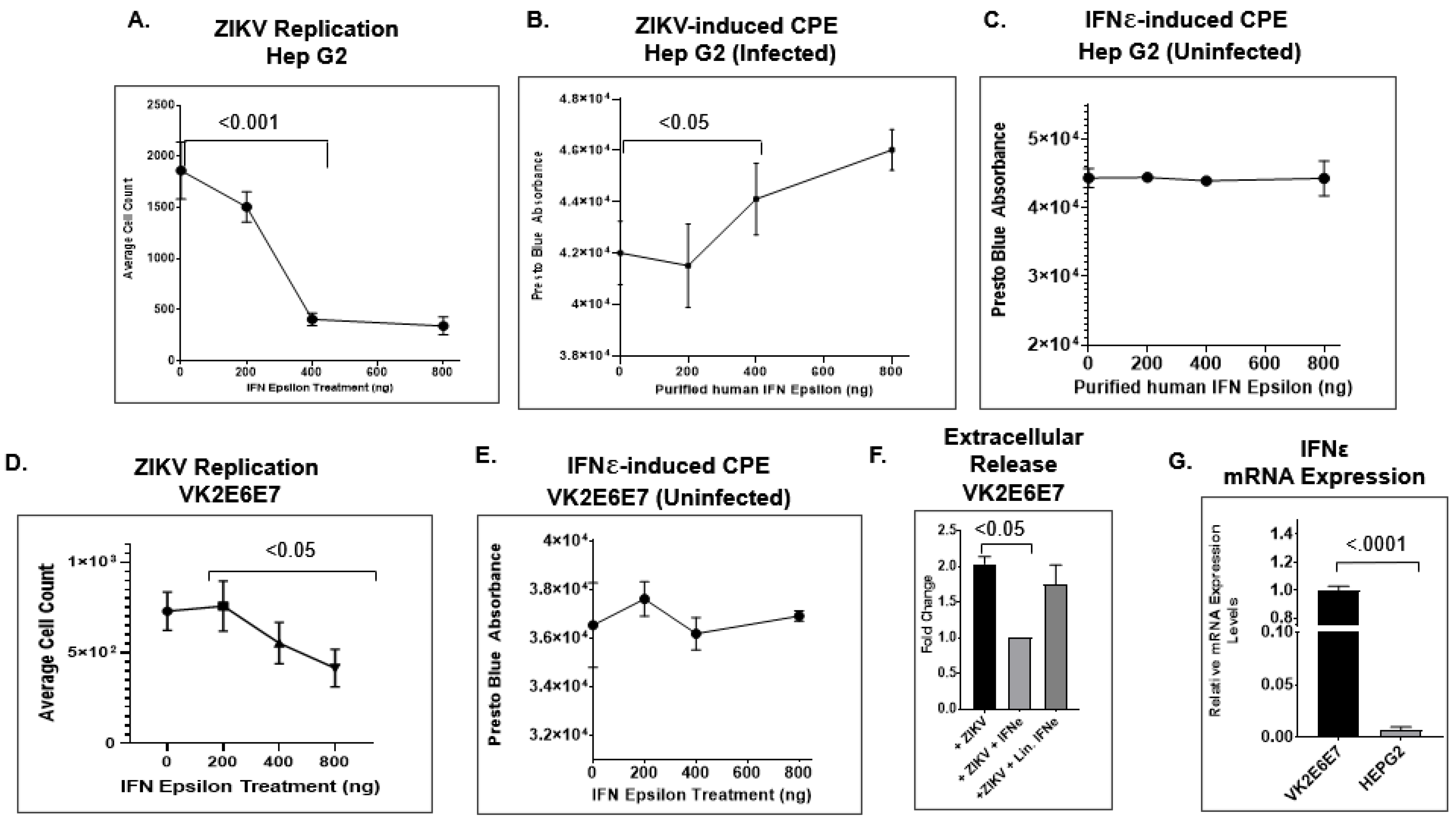
2.4. IFNε Signaling Attenuates ZIKV Replication in Hep G2 Cells
2.5. IFNε Treatment Dampens ZIKV Replication in hVECs via Induction of Type I Interferon-Stimulated Genes
2.6. Primary Human Cervical Cells Are Susceptible to ZIKV Infection
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Song, B.H.; Yun, S.I.; Woolley, M.; Lee, Y.M. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 308, 50–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Sharma, M.; Dhull, D.; Sharma, Y.; Kaushik, S.; Kaushik, S. Zika virus: An emerging challenge to public health worldwide. Can. J. Microbiol. 2020, 66, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runge-Ranzinger, S.; Morrison, A.C.; Manrique-Saide, P.; Horstick, O. Zika transmission patterns: A meta-review. Trop. Med. Int. Health 2019, 24, 523–529. [Google Scholar] [CrossRef]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika Virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Guble, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kublin, J.L.; Whitney, J.B. Zika virus research models. Virus Res. 2018, 254, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Hills, S.L.; Fischer, M.; Petersen, L.R. Epidemiology of Zika Virus Infection. J. Infect. Dis. 2017, 216, S868–S874. [Google Scholar] [CrossRef] [Green Version]
- Dubaut, J.P.; Higuita, N.I.A.; Quaas, A.M. Impact of Zika virus for infertility specialists: Current literature, guidelines, and resources. J. Assist. Reprod. Genet. 2017, 34, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Ibrahim, M.A.; Oluoch, L.; Tekeli, M.; Tekeli, T. Impact of weather seasonality and sexual transmission on the spread of Zika fever. Sci. Rep. 2019, 9, 17055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chibueze, E.C.; Tirado, V.; Lopes, K.d.; Balogun, O.O.; Takemoto, Y.; Swa, T.; Dagvadorj, A.; Nagata, C.; Morisaki, N.; Menendez, C.; et al. Zika virus infection in pregnancy: A systematic review of disease course and complications. Reprod. Health 2017, 14, 28. [Google Scholar] [CrossRef] [Green Version]
- Boyer, S.; Elodie, C.; Thais, C.; Diawo, D.; Anna-Bella, F. An overview of mosquito vectors of Zika virus. Microbes Infect. 2018, 20, 646–660. [Google Scholar] [CrossRef] [PubMed]
- Barrows, N.J.; Campos, R.K.; Liao, K.; Prasanth, K.R.; Soto-Acosta, R.; Yeh, S.; Schott-Lerner, G.; Pompon, J.; Sessions, O.M.; Bradrick, S.S.; et al. Biochemistry and Molecular Biology of Flaviviruses. Chem. Rev. 2018, 118, 4448–4482. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, N.; Hou, W.; Tang, Q. Biological and historical overview of Zika virus. World J. Virol. 2017, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Paixao, E.S.; Barreto, F.; Teixeira, M.D.; Costa, M.D.N.; Rodrigues, L.C. History, Epidemiology, and Clinical Manifestations of Zika: A Systematic Review. Am. J. Public Health 2016, 106, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Haby, M.M.; Pinart, M.; Elias, V.; Reveiz, L. Prevalence of asymptomatic Zika virus infection: A systematic review. Bull. World Health Organ. 2018, 96, 402–413D. [Google Scholar] [CrossRef]
- Britt, W.J. Adverse outcomes of pregnancy-associated Zika virus infection. Semin. Perinatol. 2018, 42, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Zhou, W.; Xiao, Y.; Wu, J. Implication of sexual transmission of Zika on dengue and Zika outbreaks. Math. Biosci. Eng. 2019, 16, 5092–5113. [Google Scholar] [CrossRef] [PubMed]
- Stassen, L.; Armitage, C.W.; van der Heide, D.J.; Beagley, K.W.; Frentiu, F.D. Zika Virus in the Male Reproductive Tract. Viruses 2018, 10, 198. [Google Scholar] [CrossRef] [Green Version]
- Sakkas, H.; Bozidis, P.; Giannakopoulos, X.; Sofikitis, N.; Papadopoulou, C. An Update on Sexual Transmission of Zika Virus. Pathogens 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.O.; Gorchakov, R.; Carlson, A.R.; Berry, R.; Lai, L.; Natrajan, M.; Garcia, M.N.; Correa, A.; Patel, S.M.; Aagaard, K.; et al. Prolonged Detection of Zika Virus in Vaginal Secretions and Whole Blood. Emerg. Infect. Dis. 2017, 23, 99–101. [Google Scholar] [CrossRef]
- Moreira, J.; Peixoto, T.M.; Siqueira, A.M.; Lamas, C.C. Sexually acquired Zika virus: A systematic review. Clin. Microbiol. Infect. 2017, 23, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, E.M.; Duggal, N.K.; Brault, A.C. Pathogenesis and sexual transmission of Spondweni and Zika viruses. PLoS Negl. Trop. Dis. 2017, 11, e0005990. [Google Scholar] [CrossRef] [PubMed]
- Gulland, A. First case of Zika virus spread through sexual contact is detected in UK. BMJ 2016, 355, i6500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, C.J.; Oduyebo, T.; Brault, A.C.; Brooks, J.T.; Chung, K.; Hills, S.; Kuehnert, M.J.; Mead, P.; Meaney-Delman, D.; Rabe, I.; et al. Modes of Transmission of Zika Virus. J. Infect. Dis. 2017, 216, S875–S883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, A.K.; Fikrig, E. Zika Virus and Sexual Transmission: A New Route of Transmission for Mosquito-borne Flaviviruses. Yale J. Biol. Med. 2017, 90, 325–330. [Google Scholar]
- Ferdousi, T.; Cohnstaedt, L.; McVey, D.S.; Scoglio, C. Understanding the survival of Zika virus in a vector interconnected sexual contact network. Sci. Rep. 2019, 9, 7253. [Google Scholar] [CrossRef]
- Counotte, M.J.; Kim, C.R.; Wang, J.; Bernstein, K.; Deal, C.D.; Broutet, N.J.N.; Low, N. Sexual transmission of Zika virus and other flaviviruses: A living systematic review. PLoS Med. 2018, 15, e1002611. [Google Scholar] [CrossRef] [Green Version]
- Blitvich, B.J.; Magalhaes, T.; Laredo-Tiscareño, S.V.; Foy, B.D. Sexual Transmission of Arboviruses: A Systematic Review. Viruses 2020, 12, 933. [Google Scholar] [CrossRef]
- Allard, A.; Althouse, B.M.; Hébert-Dufresne, L.; Scarpino, S.V. The risk of sustained sexual transmission of Zika is underestimated. PLoS Pathog. 2017, 13, e1006633. [Google Scholar] [CrossRef] [Green Version]
- WHO Guidelines for the Prevention of Sexual Transmission of Zika Virus: Executive Summary; World Health Organization (WHO): Geneva, Switzerland, 2019.
- Winkler, C.W.; Woods, T.A.; Rosenke, R.; Scott, D.P.; Best, S.M.; Peterson, K.E. Sexual and Vertical Transmission of Zika Virus in anti-interferon receptor-treated Rag1-deficient mice. Sci. Rep. 2017, 7, 7176. [Google Scholar] [CrossRef]
- Wang, R.; Gornalusse, G.G.; Kim, Y.; Pandey, U.; Hladik1, F.; Vojtech, L. Potent Restriction of Sexual Zika Virus Infection by the Lipid Fraction of Extracellular Vesicles in Semen. Front. Microbiol. 2020, 11, 574054. [Google Scholar] [CrossRef] [PubMed]
- Varese, A.; Dantas, E.; Paletta, A.; Fitzgerald, W.; García, F.D.; Cabrerizo, G.; Diaz, F.E.; Defelipe, L.A.; Pallares, H.; Dodes Traian, M.; et al. Extracellular acidosis enhances Zika virus infection both in human cells and ex-vivo tissue cultures from female re-productive tract. Emerg. Microbes Infect. 2021, 10, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Uraki, R.; Jurado, K.A.; Hwang, J.; Szigeti-Buck, K.; Horvath, T.L.; Iwasaki, A.; Fikrig, E. Fetal Growth Restriction Caused by Sexual Transmission of Zika Virus in Mice. J. Infect. Dis. 2017, 215, 1720–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.W.; Young, M.P.; Mamidi, A.; Regla-Nava, J.A.; Kim, K.; Shresta, S. A Mouse Model of Zika Virus Sexual Transmission and Vaginal Viral Replication. Cell Rep. 2016, 17, 3091–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strange, D.P.; Jiyarom, B.; Zarandi, N.P.; Xie, X.; Baker, C.; Sadri-Ardekani, H.; Shi, P.; Verma, S. Axl Promotes Zika Virus Entry and Modulates the Antiviral State of Human Sertoli Cells. mBio 2019, 10, e01372-19. [Google Scholar] [CrossRef] [Green Version]
- Spencer, J.L.; Lahon, A.; Tran, L.L.; Arya, R.P.; Kneubehl, A.R.; Vogt, M.B.; Xavier, D.; Rowley, D.R.; Kimata, J.T.; Rico-Hesse, R.R. Replication of Zika Virus in Human Prostate Cells: A Potential Source of Sexually Transmitted Virus. J. Infect. Dis. 2018, 217, 538–547. [Google Scholar] [CrossRef] [Green Version]
- Siemann, D.N.; Strange, D.P.; Maharaj, P.N.; Shi, P.; Verma, S. Zika Virus Infects Human Sertoli Cells and Modulates the Integrity of the In Vitro Blood-Testis Barrier Model. J. Virol. 2017, 91, e00623-17. [Google Scholar] [CrossRef] [Green Version]
- Si, L.; Meng, Y.; Tian, F.; Li, W.; Zou, P.; Wang, Q.; Xu, W.; Wang, Y.; Xia, M.; Hu, J.; et al. A Peptide-Based Virus Inactivator Protects Male Mice Against Zika Virus-Induced Damage of Testicular Tissue. Front. Microbiol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.M.; Lebratti, T.J.; Richner, J.M.; Jiang, X.; Fernandez, E.; Zhao, H.; Fremont, D.H.; Diamond, M.S.; Shin, H. Cellular and Humoral Immunity Protect against Vaginal Zika Virus Infection in Mice. J. Virol. 2018, 92, e00038-18. [Google Scholar] [CrossRef] [Green Version]
- Robinson, C.L.; Chong, A.C.N.; Ashbrook, A.W.; Jeng, G.; Jin, J.; Chen, H.; Tang, E.I.; Martin, L.A.; Kim, R.S.; Kenyon, R.M.; et al. Male germ cells support long-term propagation of Zika virus. Nat. Commun. 2018, 9, 2090. [Google Scholar] [CrossRef] [PubMed]
- Peregrine, J.; Gurung, S.; Lindgren, M.C.; Husain, S.; Zavy, M.T.; Myers, D.A.; Papin, J.F. Zika Virus Infection, Reproductive Organ Targeting, and Semen Transmission in the Male Olive Baboon. J. Virol. 2019, 94, e01434-19. [Google Scholar] [CrossRef]
- Pagani, I.; Ghezzi, S.; Ulisse, A.; Rubio, A.; Turrini, F.; Garavaglia, E.; Candiani, M.; Castilletti, C.; Ippolito, G.; Poli, G.; et al. Human Endometrial Stromal Cells Are Highly Permissive To Productive Infection by Zika Virus. Sci. Rep. 2017, 7, 44286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, J.A.; Harms, M.; Krüger, F.; Groß, R.; Joas, S.; Hayn, M.; Dietz, A.N.; Lippold, S.; von Einem, J.; Schubert, A.; et al. Semen inhibits Zika virus infection of cells and tissues from the anogenital region. Nat. Commun. 2018, 9, 2207. [Google Scholar] [CrossRef]
- Mlera, L.; Bloom, M.E. Differential Zika Virus Infection of Testicular Cell Lines. Viruses 2019, 11, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, E.M.; Duggal, N.K.; Delorey, M.J.; Oksanish, J.; Ritter, J.M.; Brault, A.C. Duration of seminal Zika viral RNA shedding in immunocompetent mice inoculated with Asian and African genotype viruses. Virology 2019, 535, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Matusali, G.; Houzet, L.; Satie, A.; Mahé, D.; Aubry, F.; Couderc, T.; Frouard, J.; Bourgeau, S.; Bensalah, K.; Lavoué, S.; et al. Zika virus infects human testicular tissue and germ cells. J. Clin. Investig. 2018, 128, 4697–4710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Jovel, J.; Lopez-Orozco, J.; Limonta, D.; Airo, A.M.; Hou, S.; Stryapunina, I.; Fibke, C.; Moore, R.B.; Hobman, T.C. Human Sertoli cells support high levels of Zika virus replication and persistence. Sci. Rep. 2018, 8, 5477. [Google Scholar] [CrossRef] [PubMed]
- Hubert, M.; Chiche, A.; Legros, V.; Jeannin, P.; Montange, T.; Gessain, A.; Ceccaldi, P.; Vidy, A. Productive Infection of Mouse Mammary Glands and Human Mammary Epithelial Cells by Zika Virus. Viruses 2019, 11, 950. [Google Scholar] [CrossRef] [Green Version]
- Duggal, N.K.; Ritter, J.M.; Pestorius, S.E.; Zaki, S.R.; Davis, B.S.; Chang, G.J.; Bowen, R.A.; Brault, A.C. Frequent Zika Virus Sexual Transmission and Prolonged Viral RNA Shedding in an Immunodeficient Mouse Model. Cell Rep. 2017, 18, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Duggal, N.K.; McDonald, E.M.; Ritter, J.M.; Brault, A.C. Sexual transmission of Zika virus enhances in utero transmission in a mouse model. Sci. Rep. 2018, 8, 4510. [Google Scholar] [CrossRef] [Green Version]
- Caine, E.A.; Scheaffer, S.M.; Arora, N.; Zaitsev, K.; Artyomov, M.N.; Coyne, C.B.; Moley, K.H.; Diamond, M.S. Interferon lambda protects the female reproductive tract against Zika virus infection. Nat. Commun. 2019, 10, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amerson-Brown, M.H.; Miller, A.L.; Maxwell, C.A.; White, M.M.; Vincent, K.L.; Bourne, N.; Pyles, R.B. Cultivated Human Vaginal Microbiome Communities Impact Zika and Herpes Simplex Virus Replication in ex vivo Vaginal Mucosal Cultures. Front. Microbiol. 2018, 9, 3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcendor, D.J. Zika Virus Infection of the Human Glomerular Cells: Implications for Viral Reservoirs and Renal Pathogenesis. J. Infect. Dis. 2017, 216, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gornalusse, G.G.; Zhang, M.; Wang, R.; Rwigamba, E.; Kirby, A.C.; Fialkow, M.; Nance, E.; Hladik, F.; Vojtech, L. HSV-2 Infection Enhances Zika Virus Infection of Primary Genital Epithelial Cells Independently of the Known Zika Virus Receptor AXL. Front. Microbiol. 2021, 12, 825049. [Google Scholar] [CrossRef]
- Carroll, T.; Lo, M.; Lanteri, M.; Dutra, J.; Zarbock, K.; Silveira, P.; Rourke, T.; Ma, Z.; Fritts, L.; O’Connor, S.; et al. Zika virus preferentially replicates in the female reproductive tract after vaginal inoculation of rhesus macaques. PLoS Pathog. 2017, 13, e1006537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, C.A.; Dulson, S.J.; Lazear, H.M. Zika virus replicates in the vagina of mice with 1 intact interferon signaling. bioRxiv 2022, 48, 392. [Google Scholar]
- Nguyen, P.V.; Kafka, J.K.; Ferreira, V.H.; Roth, K.; Kaushic, C. Innate and adaptive immune responses in male and female reproductive tracts in homeostasis and following HIV infection. Cell. Mol. Immunol. 2014, 11, 410–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, A. Antiviral immune responses in the genital tract: Clues for vaccines. Nat. Rev. Immunol. 2010, 10, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Kinlock, B.L.; Wang, Y.; Turner, T.M.; Wang, C.; Liu, B. Transcytosis of HIV-1 through vaginal epithelial cells is dependent on trafficking to the endocytic recycling pathway. PLoS ONE 2014, 9, e96760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, F.C.; Sridhar, P.R.; Baldridge, M.T. Differential roles of interferons in innate responses to mucosal viral infections. Trends Immunol. 2021, 42, 1009–1023. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, A.; MacIntyre, D.A.; Marchesi, J.R.; Lee, Y.S.; Bennett, P.R.; Kyrgiou, M. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: What do we know and where are we going next? Microbiome 2016, 4, 58. [Google Scholar] [CrossRef] [Green Version]
- Groeger, S.; Meyle, J. Oral Mucosal Epithelial Cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Savidis, G.; Perreira, J.M.; Portmann, J.M.; Meraner, P.; Guo, Z.; Green, S.; Brass, A.L. The IFITMs Inhibit Zika Virus Replication. Cell Rep. 2016, 15, 2323–2330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Li, S.F.; Gong, M.; Zhao, F.; Shao, J.; Xie, Y.; Zhang, Y.; Chang, H. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol. Biochem. 2018, 51, 2377–2396. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Miorin, L.; Maestre, A.M.; Fernandez-Sesma, A.; García-Sastre, A. Antagonism of type I interferon by flaviviruses. Biochem. Biophys. Res. Commun. 2017, 492, 587–596. [Google Scholar] [CrossRef]
- Thurmond, S.; Wang, B.; Song, J.; Hai, R. Suppression of Type I Interferon Signaling by Flavivirus NS5. Viruses 2018, 10, 712. [Google Scholar] [CrossRef] [Green Version]
- Xi, Y.; Da, S.L.; Jackson, R.J.; Ranasinghe, C. Role of novel type I interferon epsilon in viral infection and mucosal immunity. Mucosal Immunol. 2012, 5, 610–622. [Google Scholar] [CrossRef] [Green Version]
- Tasker, C.; Subbian, S.; Gao, P.; Couret, J.; Levine, C.; Ghanny, S.; Soteropoulos, P.; Zhao, X.; Landau, N.; Lu, W.; et al. IFN-epsilon protects primary macrophages against HIV infection. JCI Insight 2016, 1, e88255. [Google Scholar] [CrossRef] [Green Version]
- Stifter, S.A.; Matthews, A.Y.; Mangan, N.E.; Fung, K.Y.; Drew, A.; Tate, M.D.; da Costa, T.P.S.; Hampsey, D.; Mayall, J.; Hansbro, P.M.; et al. Defining the distinct, intrinsic properties of the novel type I interferon Epsilon. J. Biol. Chem. 2018, 293, 3168–3179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.W.; Duan, Z.; Zheng, L.; Xie, Z.; Gao, H.; Zhang, H.; Li, W.; Hou, Y. Purification of recombinant human interferon-epsilon and oligonucleotide microarray analysis of interfer-on-epsilon-regulated genes. Protein Expr. Purif. 2007, 53, 356–362. [Google Scholar] [CrossRef]
- Matsumiya, T.; Xing, F.; Ebina, M.; Hayakari, R.; Imaizumi, T.; Yoshida, H.; Kikuchi, H.; Topham, M.K.; Satoh, K.; Stafforini, D.M. Novel role for molecular transporter importin 9 in posttranscriptional regulation of IFN-epsilon expression. J. Immunol. 2013, 191, 1907–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumiya, T.; Prescott, S.M.; Stafforini, D.M. IFN-epsilon mediates TNF-alpha-induced STAT1 phosphorylation and induction of retinoic acid-inducible gene-I in human cervical cancer cells. J. Immunol. 2007, 179, 4542–4549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermant, P.; Francius, C.; Clotman, F.; Michiels, T. IFN-epsilon is constitutively expressed by cells of the reproductive tract and is inefficiently secreted by fibroblasts and cell lines. PLoS ONE 2013, 8, e71320. [Google Scholar] [CrossRef] [Green Version]
- Harris, B.D.; Schreiter, J.; Chevrier, M.; Jordan, J.L.; Walter, M.R. Human interferon- and interferon-kappa exhibit low potency and low affinity for cell-surface IFNAR and the poxvirus antagonist B18R. J. Biol. Chem. 2018, 293, 16057–16068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, A.; Kang, G.; Ma, F.; Lu, W.; Yuan, Z.; Li, Y.; Lewis, M.; Kraiselburd, E.N.; Montaner, L.; Li, Q. The mucosal expression pattern of interferon-epsilon in rhesus macaques. J. Leukoc. Biol. 2014, 96, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Day, S.L.; Ramshaw, I.A.; Ramsay, A.J.; Ranasinghe, C. Differential effects of the type I interferons alpha4, beta, and epsilon on antiviral activity and vaccine efficacy. J. Immunol. 2008, 180, 7158–7166. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Rodríguez, D.; Baeza-Xochihua, V.; Taniguchi-Ponciano, K.; Huerta-Padilla, V.; Ponce-Navarrete, G.; Mantilla, A.; Hernandez, D.; Hernandez, A.; Gomez-Gutierrez, G.; Serna-Reyna, L. Interferon epsilon mRNA expression could represent a potential molecular marker in cervical cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 1979–1988. [Google Scholar]
- Bourke, N.M.; Achilles, S.L.; Huang, S.U.; Cumming, H.E.; Papageorgio, I.; Gearing, L.J.; Thakore, S.; Mangan, N.E.; Mesiano, S.; Hertzog, P.J. Human IFNε: Spaciotemporal expression, hormone regulation and innate immunity in the female reproductive tract. bioRxiv Prepr. 2018. [Google Scholar] [CrossRef]
- Afsar, C.U.; Afsar, S. SARS-CoV-2 (Covid-19): Interferon-epsilon may be responsible of decreased mortality in females. J. Reprod. Immunol. 2020, 141, 103154. [Google Scholar] [CrossRef] [PubMed]
- Abdulhaqq, S.A.; Zorrilla, C.; Kang, G.; Yin, X.; Tamayo, V.; Seaton, K.E.; Joseph, J.; Garced, S.; Tomaras, G.D.; Linn, K.A. Linn, K.A. HIV-1-negative female sex workers sustain high cervical IFN epsilon, low immune activation, and low expression of HIV-1-required host genes. Mucosal Immunol. 2016, 9, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Fung, K.Y.; Mangan, N.E.; Cumming, H.; Horvat, J.C.; Mayall, J.R.; Stifter, S.A.; de Weerd, N.; Roisman, L.C.; Rossjohn, J.; Robertson, S.A.; et al. Interferon-epsilon protects the female reproductive tract from viral and bacterial infection. Science 2013, 339, 1088–1092. [Google Scholar] [CrossRef] [Green Version]
- Raina, N.; Fichorova, J.G.R.; Deborah, J.A. Generation of Papillomavirus-lmmortalized Cell Lines from Normal Human Ectocervical, Endocervical, and Vaginal Epithelium That Maintain Expression of Tissue-Specific Differentiation Proteins. Biol. Reprod. 1997, 57, 847–855. [Google Scholar]
- Berry, A.; Hall, J.V. The complexity of interactions between female sex hormones and Chlamydia trachomatis infections. Curr. Clin. Microbiol. Rep. 2019, 6, 67–75. [Google Scholar] [CrossRef]
- Brabin, L. Hormonal markers of susceptibility to sexually transmitted infections: Are we taking them seriously? Br. Med. J. 2001, 323, 394–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, X.; Shao, Q.; Chaudhry, A.R.; Kinlock, B.L.; Izban, M.G.; Zhang, H.; Villalta, F.; Hildreth, J.E.K.; Liu, B. Medroxyprogesterone Acetate (MPA) Enhances HIV-1 Accumulation and Release in Primary Cervical Epithelial Cells by Inhibiting Lysosomal Activity. Pathogens 2021, 10, 1192. [Google Scholar] [CrossRef]
- Wira, C.R.; Fahey, J.V.; Ghosh, M.; Patel, M.V.; Hickey, D.K.; Ochiel, D.O. Sex hormone regulation of innate immunity in the female reproductive tract: The role of epithelial cells in balancing reproductive potential with protection against sexually transmitted pathogens. Am. J. Reprod. Immunol. 2010, 63, 544–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, K.E.; Rouster, S.D.; Kong, L.X.; Aliota, M.T.; Blackard, J.T.; Dean, G.E. Zika virus replication and cytopathic effects in liver cells. PLoS ONE 2019, 14, e0214016. [Google Scholar] [CrossRef]
- Stefanik, M.; Formanova, P.; Bily, T.; Vancova, M.; Eyer, L.; Palus, M.; Salat, J.; Braconi, C.T.; Zanotto, P.M.; Gould, A.E.; et al. Characterisation of Zika virus infection in primary human astrocytes. BMC Neurosci. 2018, 19, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-San Martin, C.; Li, T.; Bouquet, J.; Streithorst, J.; Yu, G.; Paranjpe, A.; Chiu, C.Y. Differentiation enhances Zika virus infection of neuronal brain cells. Sci. Rep. 2018, 8, 14543. [Google Scholar] [CrossRef] [PubMed]
- Salinas, S.; Erkilic, N.; Damodar, K.; Molès, J.; Fournier-Wirth, C.; van de Perre, P.; Kalatzis, V.; Simonin, Y. Zika Virus Efficiently Replicates in Human Retinal Epithelium and Disturbs Its Permeability. J. Virol. 2017, 91, e02144-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, F.; Josey, B.; Sayitoglu, E.C.; Potens, R.; Sultu, T.; Duru, A.D.; Beljanski, V. Characterization of zika virus infection of human fetal cardiac mesenchymal stromal cells. PLoS ONE 2020, 15, e0239238. [Google Scholar] [CrossRef] [PubMed]
- Roach, T.; Alcendor, D.J. Zika virus infection of cellular components of the blood-retinal barriers: Implications for viral associated congenital ocular disease. J. Neuroinflammation 2017, 14, 43. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Liu, B.; Yves, T.D.; He, Y.; Wang, S.; Tang, H.; Ren, H.; Zhao, P.; Qi, Z.; Qin, Z. Zika Virus Induces Autophagy in Human Umbilical Vein Endothelial Cells. Viruses 2018, 10, 259. [Google Scholar] [CrossRef] [Green Version]
- Monel, B.; Compton, A.A.; Bruel, T.; Amraoui, S.; Burlaud-Gaillard, J.; Roy, N.; Guivel-Benhassine, F.; Porrot, F.; Génin, P.; Meertens, L.; et al. Zika virus induces massive cytoplasmic vacuolization and paraptosis-like death in infected cells. EMBO J. 2017, 36, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.; Meyer, K.; Steele, R.; Ray, R.B.; Ray, R. Zika virus infection dysregulates human neural stem cell growth and inhibits differentiation into neuroprogenitor cells. Cell Death Dis. 2017, 8, e3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.F.-W.; Yip, C.C.-Y.; Tsang, J.O.-L.; Tee, K.-M.; Cai, J.-P.; Chik, K.K.-H.; Zhu, Z.; Chan, C.C.-S.; Choi, G.K.-Y.; Sridhar, S.; et al. Differential cell line susceptibility to the emerging Zika virus: Implications for disease pathogenesis, non-vector-borne human transmission and animal reservoirs. Emerg. Microbes Infect. 2016, 5, e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katze, M.G.; He, Y.; Gale, M.J. Viruses and interferon: A fight for supremacy. Nat. Rev. Immunol. 2002, 2, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.P.; wczarek, C.M.O.; Jermiin, L.S.; Ejdebäck, M.; Hertzog, P.J. Characterization of the type I interferon locus and identification of novel genes. Genomics 2004, 84, 331–345. [Google Scholar] [CrossRef]
- “What We Know about Zika and Pregnancy.” CDC: Zika and Pregnancy, National Center on Birth Defects and Developmental Disabilities, Centers for Disease Control and Prevention. Available online: www.cdc.gov/pregnancy/zika/pregnancy.html (accessed on 1 July 2022).
- Martinez, L.E.; Garcia, G., Jr.; Contreras, D.; Gong, D.; Sun, R.; Arumugaswami, V. Zika Virus Mucosal Infection Provides Protective Immunity. J. Virol. 2020, 94, e00067-20. [Google Scholar] [CrossRef]
- Cherpes, T.L.; Busch, J.L.; Sheridan, B.S.; Harvey, S.A.K.; Hendricks, R.L. Medroxyprogesterone acetate inhibits CD8+ T cell viral specific effector function and induces herpes simplex virus type 1. J. Immunol. 2008, 18, 969–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas Albrecht, M.F.; Boldogh, I.; Alan, S.R. Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; Chapter 44. [Google Scholar]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T.D.A., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Plociennikowska, A.; Frankish, J.; Moraes, T.; del Prete, D.; Kahnt, F.; Acuna, C.; Slezak, M.; Binder, M.; Bartenschlager, R. TLR3 activation by Zika virus stimulates inflammatory cytokine production which dampens the antiviral response induced by RIG-I-like receptors. J. Virol. 2021, 95, 10. [Google Scholar] [CrossRef]
- Arevalo Romero, H.; Pavía, T.A.V.; Cervantes, M.A.V.; Pliego, A.F.; Repetto, A.C.H.; Juárez, M.L. The Dual Role of the Immune Response in Reproductive Organs During Zika Virus Infection. Front. Immunol. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika Virus Prototype Strain with a Cytomegalovirus Promoter-Driven cDNA Clone. mSphere 2016, 1, e00246-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claire, E.; Stewart, R.E.; Catherine, R.; Adamson, S. Inhibitors of the Interferon Response Enhance Virus Replication In Vitro. PLoS ONE 2014, 9, e112014. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mungin, J.W., Jr.; Chen, X.; Liu, B. Interferon Epsilon Signaling Confers Attenuated Zika Replication in Human Vaginal Epithelial Cells. Pathogens 2022, 11, 853. https://doi.org/10.3390/pathogens11080853
Mungin JW Jr., Chen X, Liu B. Interferon Epsilon Signaling Confers Attenuated Zika Replication in Human Vaginal Epithelial Cells. Pathogens. 2022; 11(8):853. https://doi.org/10.3390/pathogens11080853
Chicago/Turabian StyleMungin, James W., Jr., Xin Chen, and Bindong Liu. 2022. "Interferon Epsilon Signaling Confers Attenuated Zika Replication in Human Vaginal Epithelial Cells" Pathogens 11, no. 8: 853. https://doi.org/10.3390/pathogens11080853