
Genomics of Tenacibaculum Species in British Columbia, Canada

 , , , ,
, , , ,
Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Level | Sequencing Platform | Isolate | Contigs | N50 (Mb) | L50 | Genbank Accession Number | Citation |
|---|---|---|---|---|---|---|---|
| Complete | PacBio Sequel | Tenacibaculum sp. AHE14PA | 1 | 2.8 | 1 | GCA_019278465.1 | [20] |
| Tenacibaculum sp. AHE15PA | 1 | 2.8 | 1 | GCA_019278445.1 | |||
| T. mesophilum bac2 | 1 | 3.4 | 1 | GCA_024181065.1 | NA | ||
| PacBio single-molecule real-time | T. dicentrarchi AY7486TD | 1 | 2.9 | 1 | GCA_001483385.1 | [21] | |
| PacBio RSII | T. todarodis strain LPB0136T | 1 | 3.0 | 1 | GCA_CP018155.1 | [22] | |
| PacBio RSII and Illumina HiSeq | T. maritimum NCIMB 2154T | 1 | 3.4 | 1 | GCA_900119795.1 | [7] | |
| PacBio RSII and Illumina MiSeq | T. mesophilum DSM 13764T | 1 | 3.5 | 1 | GCA_009362255.1 | [23] | |
| PacBio | T. maritimum TM-KORJJ | 1 | 3.3 | 1 | GCA_004803875.1 | NA | |
| NA | T. jejuense strain KCTC 22618T | 1 | 4.6 | 1 | GCA_900198195.1 | NA | |
| Incomplete | Various | Tenacibaculum spp.* | 1 – 1196 | 3.9 × −03–4.5 | 1-191 | Various | NA |
2. Materials and Methods
2.1. Isolate Selection
2.2. DNA Extractions
2.3. MinION Sequencing
2.4. MinION Post-Processing and Quality Control
2.5. Genome Assembly and Polishing
- Subsample each barcode independently into 12 maximally independent read sets.
- Cluster the contigs produced by each assembly per barcode (resulting in a hierarchical cluster dendrogram, from which subjective decisions were made).
- Reconcile (circularize and align to a consistent start position) all of the contigs included in the cluster from the previous step per barcode.
- Compute the multiple sequence alignments between all of the reconciled contigs per cluster.
- Partition the initial read files (complete sets per barcode) into their appropriate clusters (i.e., chromosome reads to chromosome cluster, plasmid reads to plasmid cluster, etc.).
- Compute the consensus for each cluster, for each barcode. Use the reconciled contigs (step 4), alignments (step 5), and raw reads for the given cluster (step 6).
- Medaka polishing (v.1.4.3) per barcode.
2.6. Genomic Annotation
2.7. Phylogenomic Investigations
2.7.1. 16S rDNA
2.7.2. atpA and fusA
2.7.3. Multilocus Sequence Analysis (MLSA)
2.7.4. Average Nucleotide Identity (ANI)
2.8. Virulence, Antimicrobial Resistance, and Genomic Island Investigations
2.9. Gene Content Investigation
3. Results
3.1. Isolate Selection
3.2. MinION Post-Processing and Quality Control
3.3. Genome Assembly and Annotation
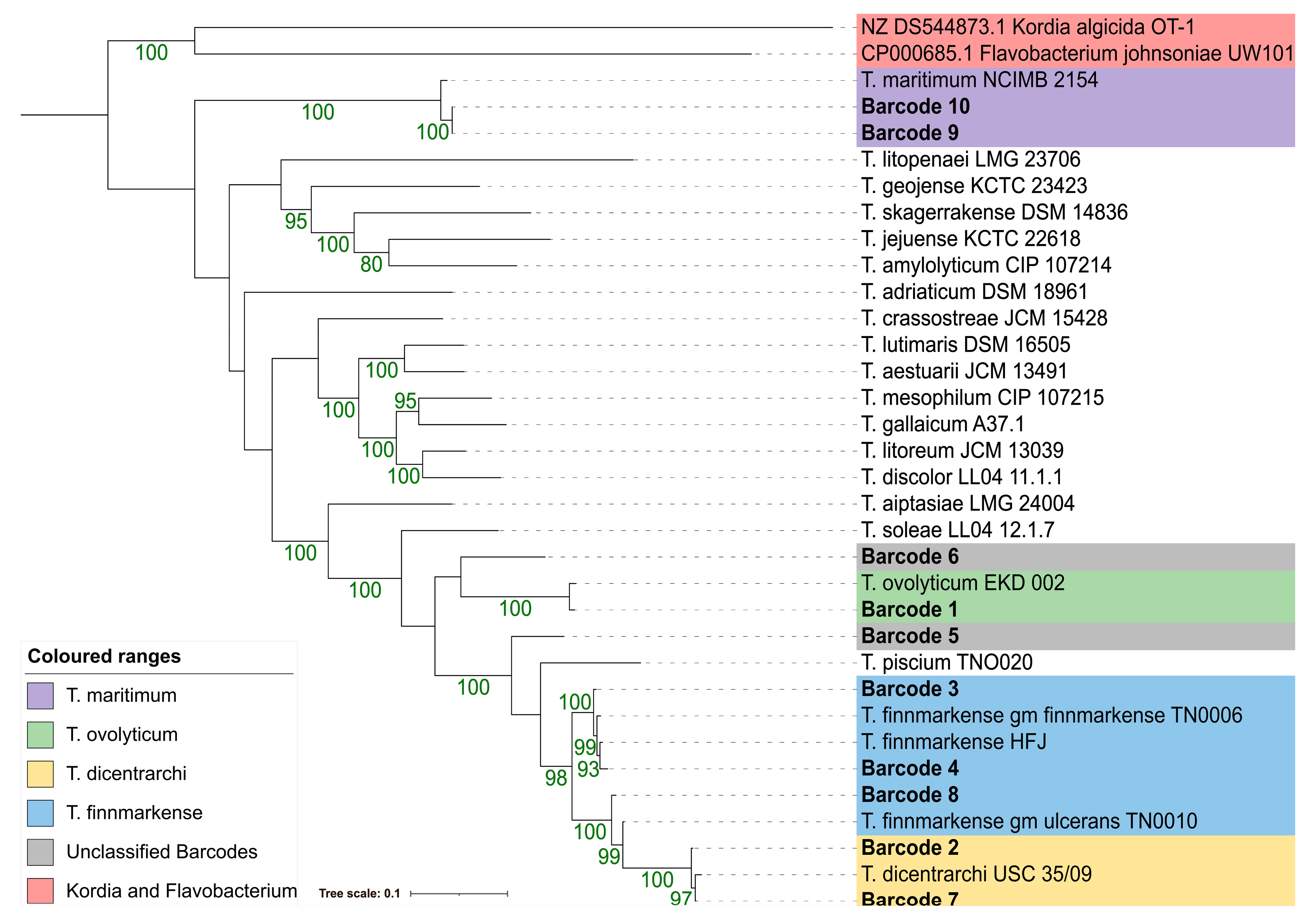
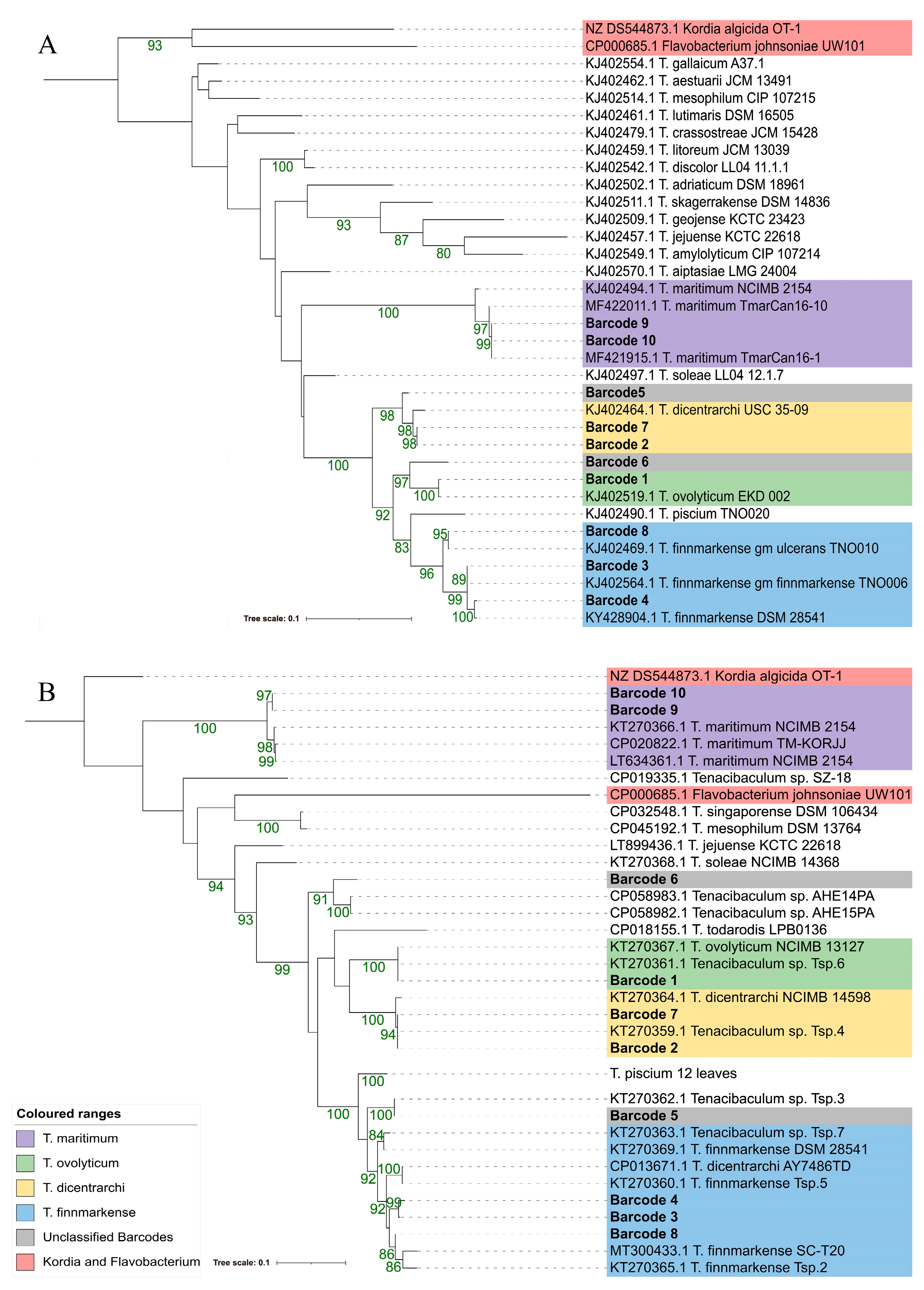
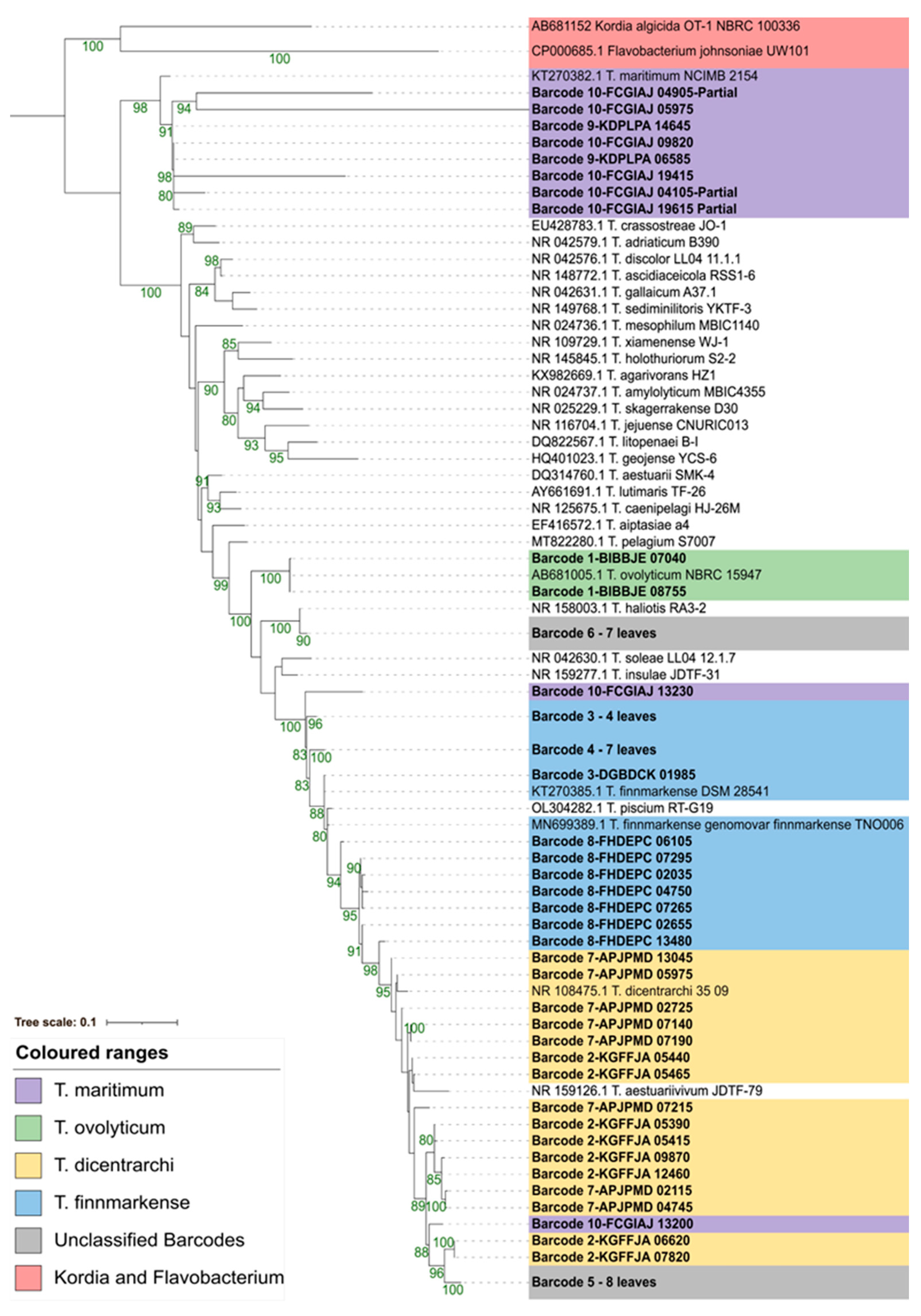
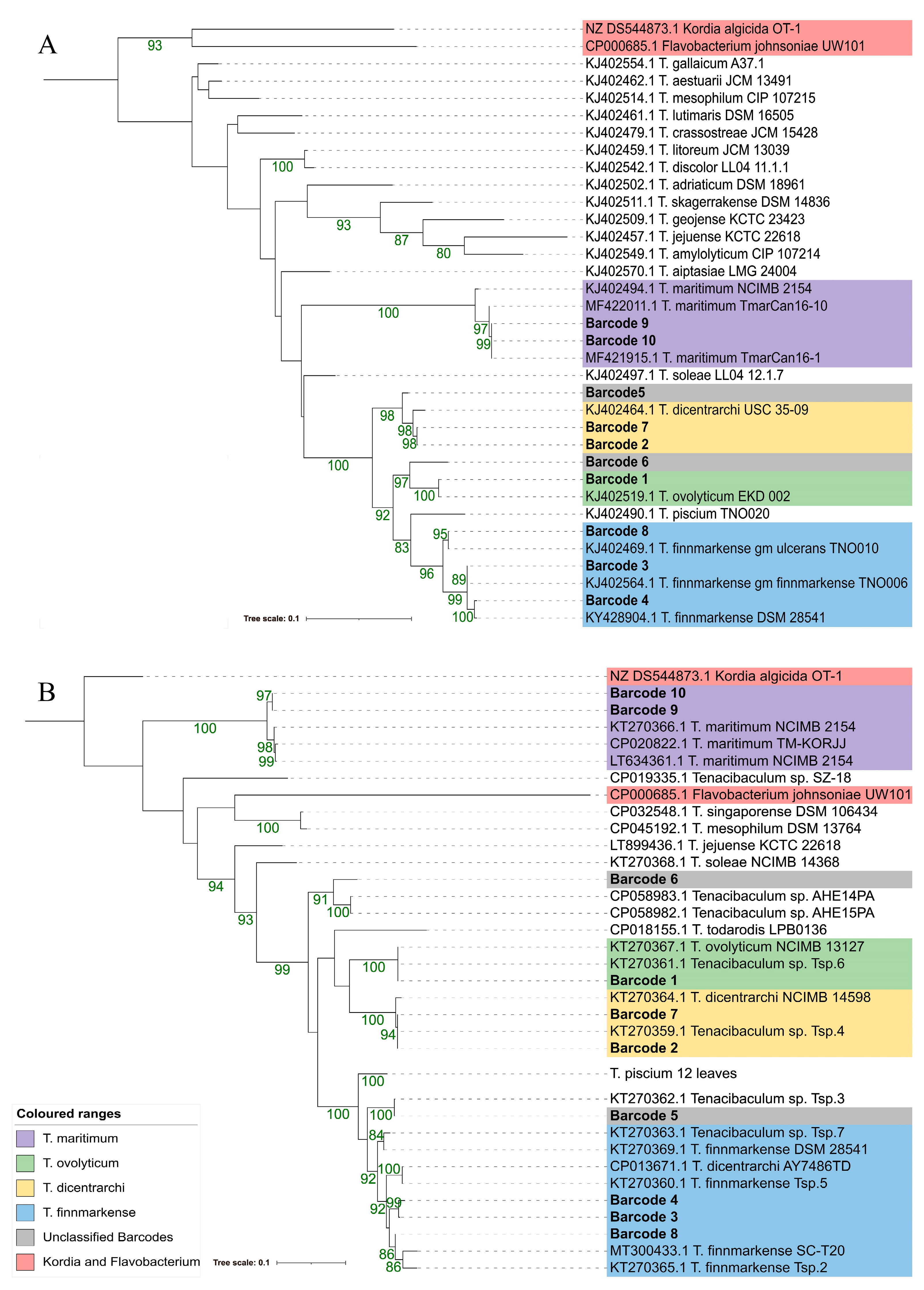
3.4. Phylogenetic Resolution Varies with Tenacibaculum Identification Methods
3.4.1. Average Nucleotide Identity (ANI)
3.4.2. Multilocus Sequence Analysis (MLSA)
3.4.3. atpA and fusA
3.4.4. 16S rDNA
3.5. Virulence, Antimicrobial Resistance, and Genomic Islands
3.5.1. Virulence Related Genes
3.5.2. Antimicrobial Resistance Genes
3.5.3. Genomic Islands
3.6. Gene Content Investigation
4. Discussion
4.1. Genomic Assembly Provides Novel Circular Genomes
4.2. Variable Genomic Resolution and Novel Species
4.3. Putative Virulence Factors of BC Tenacibaculum Species
4.3.1. Iron-Related Genes
4.3.2. Transport and Secretion Systems
4.3.3. Toxins
4.4. Putative Antimicrobial Resistance Determinants of BC Tenacibaculum Species
4.5. Numerous Genomic Islands among Tenacibaculum Species
4.6. Gene Content Analysis Indicating Diversity among Tenacibaculum Species
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nowlan, J.P.; Lumsden, J.S.; Russell, S. Advancements in characterizing Tenacibaculum infections in Canada. Pathogens 2020, 9, 1029. [Google Scholar] [CrossRef]
- Frisch, K.; Småge, S.B.; Vallestad, C.; Duesund, H.; Brevik, J.; Klevan, A.; Olsen, R.H.; Sjaatil, S.T.; Gauthier, D.; Brudeseth, B.; et al. Experimental induction of mouthrot in Atlantic salmon smolts using Tenacibaculum maritimum from Western Canada. J. Fish Dis. 2018, 41, 1247–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Småge, S.B.; Frisch, K.; Vold, V.; Duesund, H.; Brevik, J.; Olsen, R.H.; Sjaatil, S.T.; Klevan, A.; Brudeseth, B.; Watanabe, K.; et al. Induction of tenacibaculosis in Atlantic salmon smolts using Tenacibaculum finnmarkense and the evaluation of a whole cell inactivated vaccine. Aquaculture 2018, 495, 858–864. [Google Scholar] [CrossRef]
- Nowlan, J.P.; Britney, S.R.; Lumsden, J.S.; Russell, S. Experimental Induction of Tenacibaculosis in Atlantic Salmon (Salmo salar L.) using Tenacibaculum maritimum, T. dicentrarchi, and T. finnmarkense. Pathogens 2021, 10, 1439. [Google Scholar] [CrossRef]
- Piñeiro-Vidal, M.; Gijón, D.; Zarza, C.; Santos, Y. Tenacibaculum dicentrarchi sp. nov., a marine bacterium of the family Flavobacteriaceae isolated from European sea bass. Int. J. Syst. Evol. Microbial. 2012, 62, 425–429. [Google Scholar] [CrossRef] [Green Version]
- Faílde, L.D.; Losada, A.P.; Bermúdez, R.; Santos, Y.; Quiroga, M.I. Tenacibaculum maritimum infection: Pathology and immunohistochemistry in experimentally challenged turbot (Psetta maxima L.). Microb. Pathog. 2013, 65, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pascual, D.; Lunazzi, A.; Magdelenat, G.; Rouy, Z.; Roulet, A.; Lopez-Roques, C.; Larocque, R.; Barbeyron, T.; Gobet, A.; Michel, G.; et al. The complete genome sequence of the fish pathogen Tenacibaculum maritimum provides insights into virulence mechanisms. Front. Microbial. 2017, 8, 1542. [Google Scholar] [CrossRef] [Green Version]
- Teramoto, M.; Zhai, Z.; Komatsu, A.; Shibayama, K.; Suzuki, M. Genome sequence of the psychrophilic bacterium Tenacibaculum ovolyticum strain da5A-8 isolated from deep seawater. Genome Announc. 2016, 4, e00644-e16. [Google Scholar] [CrossRef] [Green Version]
- Avendaño-Herrera, R.; Saldarriaga-Córdoba, M.; Irgang, R. Draft genome sequence of Tenacibaculum ovolyticum To-7Br, Recovered from a farmed Atlantic salmon (Salmo salar). Microbiol. Resour. Announc. 2022, 11, e0025422. [Google Scholar] [CrossRef]
- Bridel, S.; Olsen, A.-B.; Nilsen, H.; Bernardet, J.-F.; Achaz, G.; Avendaño-Herrera, R.; Duchaud, E. Comparative genomics of Tenacibaculum dicentrarchi and “Tenacibaculum finnmarkense” highlights intricate evolution of fish-pathogenic species. Genome Biol. Evol. 2018, 10, 452–457. [Google Scholar] [CrossRef]
- Olsen, A.B.; Spilsberg, B.; Nilsen, H.K.; Lagesen, K.; Gulla, S.; Avendaño-Herrera, R.; Irgang, R.; Duchaud, E.; Colquhoun, D.J. Tenacibaculum piscium sp. nov., isolated from skin ulcers of sea-farmed fish, and description of Tenacibaculum finnmarkense sp. nov. with subdivision into genomovars finnmarkense and ulcerans. Int. J. Syst. Evol. 2020, 70, 6079–6090. [Google Scholar] [CrossRef]
- Avendaño-Herrera, R.; Olsen, A.B.; Saldarriaga-Cordoba, M.; Colquhoun, D.J.; Reyes, V.; Rivera-Bohle, J.; Duchaud, E.; Irgang, R. Isolation, identification, virulence potential and genomic features of Tenacibaculum piscium isolates recovered from Chilean salmonids. Transbound. Emerg. Dis. 2022, 69, e3305–e3315. [Google Scholar] [CrossRef]
- Saha, R.; Saha, N.; Donofrio, R.S.; Bestervelt, L.L. Microbial siderophores: A mini review. J. Basic Microbiol. 2013, 53, 303–317. [Google Scholar] [CrossRef]
- Green, E.R.; Mecsas, J. Bacterial secretion systems: An overview. Microbiol. Spectr. 2016, 4, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Albertsen, M.; Anslan, S.; Callahan, B. Perspectives and benefits of high-throughput long-read sequencing in microbial ecology. Appl. Environ. Microbiol. 2021, 87, e0062621. [Google Scholar] [CrossRef]
- Oxford Nanopore Technologies (ONT), Nanopore Sequencing. The Advantages of Long Reads for Genome Assembly. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwi9r5f8i4j5AhXCAzQIHdfkBMUQFnoECAkQAw&url=https%3A%2F%2Fnanoporetech.com%2Fsites%2Fdefault%2Ffiles%2Fs3%2Fwhite-papers%2FWGS_Assembly_white_paper.pdf%3FsubmissionGuid%3D40a7546b-9e51-42e7-bde9-b5ddef3c3512&usg=AOvVaw1S_sBMns-u-WlA4qyAZoRS (accessed on 1 September 2022).
- Ashton, P.M.; Nair, S.; Dallman, T.; Rubino, S.; Rabsch, W.; Mwaigwisya, S.; Wain, J.; O’Grady, J. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat. Biotechnol. 2015, 33, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Rahube, T.O.; Cameron, A.D.S.; Lerminiaux, N.A.; Bhat, S.V.; Alexander, K.A. Globally Disseminated Multidrug Resistance Plasmids Revealed by Complete Assembly of Multidrug Resistant Escherichia coli and Klebsiella pneumoniae Genomes from Diarrheal Disease in Botswana. Appl. Microbiol. 2022, 2, 934–949. [Google Scholar] [CrossRef]
- Tan, S.; Dvorak, C.M.; Estrada, A.A.; Gebhart, C.; Marthaler, D.G.; Murtaugh, M.P. MinION sequencing of Streptococcus suis allows for functional characterization of bacteria by multilocus sequence typing and antimicrobial resistance profiling. J. Microbiol. Methods 2020, 169, 105817. [Google Scholar] [CrossRef]
- Bartlau, N.; Wichels, A.; Krohne, G.; Adriaenssens, E.M.; Heins, A.; Fuchs, B.M.; Amann, R.; Moraru, C. Highly diverse flavobacterial phages isolated from North Sea spring blooms. ISME J. 2022, 16, 555–568. [Google Scholar] [CrossRef]
- Grothusen, H.; Castillo, A.; Henríquez, P.; Navas, E.; Bohle, H.; Araya, C.; Bustamante, F.; Bustos, P.; Mancilla, M. First complete genome sequence of Tenacibaculum dicentrarchi, an emerging bacterial pathogen of salmonids. Genome Announc. 2016, 4, e01756-e15. [Google Scholar] [CrossRef]
- Shin, S.-K.; Kim, E.; Yi, H. Tenacibaculum todarodis sp. nov., isolated from a squid. Int. J. Syst. Evol. Microbiol. 2018, 68, 1479–1483. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Soh, M.; Azman, M.N.; Ngoh, S.Y.; Orbán, L.; Seedorf, H. Insights into the microbiome of farmed Asian sea bass (Lates calcarifer) with symptoms of tenacibaculosis and description of Tenacibaculum singaporense sp. nov. Antonie Leeuwenhoek 2020, 113, 737–752. [Google Scholar] [CrossRef]
- Nowlan, J.P.; Lumsden, J.S.; Russell, S. Quantitative PCR for Tenacibaculum dicentrarchi and T. finnmarkense. J. Fish Dis. 2021, 44, 655–659. [Google Scholar] [CrossRef]
- Fringuelli, E.; Savage, P.D.; Gordon, A.; Baxter, E.J.; Rodger, H.D.; A Graham, D. Development of a quantitative real-time PCR for the detection of Tenacibaculum maritimum and its application to field samples. J. Fish Dis. 2012, 35, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Nowlan, J.P.; Heese, B.M.; Wilson, M.J.; Britney, S.R.; Lumsden, J.S.; Russell, S. Tenacibaculum ovolyticum 16S rDNA Quantitative-PCR Assay Development and Field Testing. Fishes 2022, 7, 303. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Cerdeira, L.T.; Hawkey, J.; Méric, G.; Ben Vezina, B.; Wyres, K.L.; Holt, K.E. Trycycler: Consensus long-read assemblies for bacterial genomes. Genome Biol. 2021, 22, 266. [Google Scholar] [CrossRef] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Vaser, R.; Šikić, M. Raven: A de novo genome assembler for long reads. BioRxiv 2021. [Google Scholar] [CrossRef]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.-J.; Luo, H.; Gao, F. Ori-Finder 2022: A comprehensive web server for prediction and analysis of bacterial replication origins. Genom. Proteom. Bioinform. 2022. [Google Scholar] [CrossRef]
- Schwengers, O.; Jelonek, L.; Dieckmann, M.A.; Beyvers, S.; Blom, J.; Goesmann, A. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genom. 2021, 7, 000685. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stothard, P.; Wishart, D.S. Circular genome visualization and exploration using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Habib, C.; Houel, A.; Lunazzi, A.; Bernardet, J.-F.; Olsen, A.B.; Nilsen, H.; Toranzo, A.E.; Castro, N.; Nicolas, P.; Duchaud, E. Multilocus sequence analysis of the marine bacterial genus Tenacibaculum suggests parallel evolution of fish pathogenicity and endemic colonization of aquaculture systems. Appl. Environ. Microbiol. 2014, 80, 5503–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Småge, S.B.; Brevik, J.; Duesund, H.; Ottem, K.F.; Watanabe, K.; Nylund, A. Tenacibaculum finnmarkense sp. nov., a fish pathogenic bacterium of the family Flavobacteriaceae isolated from Atlantic salmon. Antonie Leeuwenhoek 2016, 109, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbial. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Sies, A.N.; Nowlan, J.P.; Britney, S.R.; Cameron, A.D.S.; Siah, A.; Russell1, S. Long-read sequencing reveals complete plasmids in three Tenacibaculum spp. manuscript in preparation.
- Garber, A.I.; Nealson, K.H.; Okamoto, A.; McAllister, S.M.; Chan, C.S.; Barco, R.A.; Merino, N. FeGenie: A comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front. Microbiol. 2020, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbial. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Tetzschner, A.M.M.; Johnson, J.R.; Johnston, B.D.; Lund, O.; Scheutz, F. In silico genotyping of Escherichia coli isolates for extraintestinal virulence genes by use of whole-genome sequencing data. J. Clin. Microbial. 2020, 58, e01269-e20. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.-M. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L.; Simon Fraser University Research Computing Group. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Soares, S.C.; Geyik, H.; Ramos, R.T.; de Sá, P.H.; Barbosa, E.G.; Baumbach, J.; Figueiredo, H.C.; Miyoshi, A.; Tauch, A.; Silva, A.; et al. GIPSy: Genomic island prediction software. J. Biotechnol. 2016, 232, 2–11. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 2020, 21, 1–21. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- De Maio, N.; Shaw, L.P.; Hubbard, A.; George, S.; Sanderson, N.D.; Swann, J.; Wick, R.; AbuOun, M.; Stubberfield, E.; Hoosdally, S.J.; et al. Comparison of long-read sequencing technologies in the hybrid assembly of complex bacterial genomes. Microb. Genom. 2019, 5, e000294. [Google Scholar] [CrossRef]
- Chen, Z.; Erickson, D.L.; Meng, J. Benchmarking hybrid assembly approaches for genomic analyses of bacterial pathogens using Illumina and Oxford Nanopore sequencing. BMC Genom. 2020, 21, 631. [Google Scholar] [CrossRef]
- Bridel, S.; Bourgeon, F.; Marie, A.; Saulnier, D.; Pasek, S.; Nicolas, P.; Bernardet, J.-F.; Duchaud, E. Genetic diversity and population structure of Tenacibaculum maritimum, a serious bacterial pathogen of marine fish: From genome comparisons to high throughput MALDI-TOF typing. Vet. Res. 2020, 51, 60. [Google Scholar] [CrossRef]
- Lopez, P.; Bridel, S.; Saulnier, D.; David, R.; Magariños, B.; Torres, B.S.; Bernardet, J.F.; Duchaud, E. Genomic characterization of Tenacibaculum maritimum O-antigen gene cluster and development of a multiplex PCR-based serotyping scheme. Transbound. Emerg. Dis. 2022, 69, e2876–e2888. [Google Scholar] [CrossRef] [PubMed]
- Avendaño-Herrera, R.; Collarte, C.; Saldarriaga-Córdoba, M.; Irgang, R. New salmonid hosts for Tenacibaculum species: Expansion of tenacibaculosis in Chilean aquaculture. J. Fish Dis. 2020, 43, 1077–1085. [Google Scholar] [CrossRef]
- Barriel, V.; Tassy, P. Rooting with multiple outgroups: Consensus versus parsimony. Cladistics 1998, 14, 193–200. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Shaffer, H.B. Troubleshooting molecular phylogenetic analyses. Annu. Rev. Ecol. Evol. Syst 2002, 33, 49–72. [Google Scholar] [CrossRef]
- Saldarriaga-Córdoba, M.; Irgang, R.; Avendaño-Herrera, R. Comparison between genome sequences of Chilean Tenacibaculum dicentrarchi isolated from red conger eel (Genypterus chilensis) and Atlantic salmon (Salmo salar) focusing on bacterial virulence determinants. J. Fish Dis. 2021, 44, 1843–1860. [Google Scholar] [CrossRef]
- Smith, J.L. The physiological role of ferritin-like compounds in bacteria. Crit. Rev. Microbiol. 2004, 30, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Halsey, T.A.; Vazquez-Torres, A.; Gravdahl, D.J.; Fang, F.C.; Libby, S.J. The ferritin-like Dps protein is required for Salmonella enterica serovar Typhimurium oxidative stress resistance and virulence. Infect. Immun. 2004, 72, 1155–1158. [Google Scholar] [CrossRef] [Green Version]
- Velayudhan, J.; Castor, M.; Richardson, A.; Main-Hester, K.L.; Fang, F.C. The role of ferritins in the physiology of Salmonella enterica sv. Typhimurium: A unique role for ferritin B in iron-sulphur cluster repair and virulence. Mol. Microbiol. 2007, 63, 1495–1507. [Google Scholar] [CrossRef]
- Mey, A.R.; Wyckoff, E.E.; Kanukurthy, V.; Fisher, C.R.; Payne, S.M. Iron and fur regulation in Vibrio cholerae and the role of fur in virulence. Infect. Immun. 2005, 73, 8167–8178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olczak, T.; Siudeja, K.; Olczak, M. Purification and initial characterization of a novel Porphyromonas gingivalis HmuY protein expressed in Escherichia coli and insect cells. Protein Expr. Purif. 2006, 49, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Olczak, T.; Sroka, A.; Potempa, J.; Olczak, M. Porphyromonas gingivalis HmuY and HmuR: Further characterization of a novel mechanism of heme utilization. Arch. Microbiol. 2008, 189, 197–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójtowicz, H.; Wojaczyński, J.; Olczak, M.; Króliczewski, J.; Latos-Grażyński, L.; Olczak, T. Heme environment in HmuY, the heme-binding protein of Porphyromonas gingivalis. Biochem. Biophys. Res. Commun. 2009, 383, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lechardeur, D.; Bernardet, J.-F.; Kerouault, B.; Guérin, C.; Rigaudeau, D.; Nicolas, P.; Duchaud, E.; Rochat, T. Two functionally distinct heme/iron transport systems are virulence determinants of the fish pathogen Flavobacterium psychrophilum. Virulence 2022, 13, 1221–1241. [Google Scholar] [CrossRef]
- D’Onofrio, A.; Crawford, J.M.; Stewart, E.J.; Witt, K.; Gavrish, E.; Epstein, S.; Clardy, J.; Lewis, K. Siderophores from neighboring organisms promote the growth of uncultured bacteria. Chem. Biol. 2010, 17, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Avendaño-Herrera, R.; Toranzo, A.E.; Romalde, J.L.; Lemos, M.L.; Magariños, B. Iron uptake mechanisms in the fish pathogen Tenacibaculum maritimum. Appl. Environ. Microbiol. 2005, 71, 6947–6953. [Google Scholar] [CrossRef] [Green Version]
- Baxa, D.V.; Kawai, K.; Kusuda, R. In vitro and in vivo activities of Flexibacter maritimus toxins. Bull. Mar. Sci. 1988, 10, 1–8. [Google Scholar]
- Vences, A.; Rivas, A.J.; Lemos, M.L.; Husmann, M.; Osorio, C.R. Chromosome-encoded hemolysin, phospholipase, and collagenase in plasmidless isolates of Photobacterium damselae subsp. damselae contribute to virulence for fish. Appl. Environ. Microbiol. 2017, 83, e00401–e00417. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Okino, N.; Tani, M. New insight into the structure, reaction mechanism, and biological functions of neutral ceramidase. Biochim. Biophys. Acta Biomembr. 2014, 1841, 682–691. [Google Scholar] [CrossRef]
- Oda, M.; Takahashi, M.; Matsuno, T.; Uoo, K.; Nagahama, M.; Sakurai, J. Hemolysis induced by Bacillus cereus sphingomyelinase. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, L.R.; Tiirola, M.; Valtonen, E.T. Chondroitin AC lyase activity is related to virulence of fish pathogenic Flavobacterium columnare. J. Fish Dis. 2006, 29, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.C.; Garbe, J.; Collin, M. Cysteine proteinase SpeB from Streptococcus pyogenes—A potent modifier of immunologically important host and bacterial proteins. Biol. Chem. 2011, 392, 1077–1088. [Google Scholar] [CrossRef]
- Mally, M.; Shin, H.; Paroz, C.; Landmann, R.; Cornelis, G.R. Capnocytophaga canimorsus: A human pathogen feeding at the surface of epithelial cells and phagocytes. PLoS Pathog. 2008, 4, e1000164. [Google Scholar] [CrossRef]
- Gilbert, R.J.C. Cholesterol-Dependent Cytolysins. In Advances in Experimental Medicine and Biology; Proteins Membrane Binding and Pore Formation: Cholesterol-Dependent Cytolysins; Anderluh, G., Lakey, J., Eds.; Springer: New York, NY, USA, 2010; Volume 677, pp. 56–66. [Google Scholar] [CrossRef]
- Veith, P.D.; Glew, M.D.; Gorasia, D.G.; Reynolds, E.C. Type IX secretion: The generation of bacterial cell surface coatings involved in virulence, gliding motility and the degradation of complex biopolymers. Mol. Microbiol. 2017, 106, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Koronakis, V.; Koronakis, E.; Hughes, C. Isolation and analysis of the C-terminal signal directing export of Escherichia coli hemolysin protein across both bacterial membranes. EMBO J. 1989, 8, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Champion, P.A.D.; Stanley, S.A.; Champion, M.M.; Brown, E.J.; Cox, J.S. C-terminal signal sequence promotes virulence factor secretion in Mycobacterium tuberculosis. Science 2006, 313, 1632–1636. [Google Scholar] [CrossRef]
- Veith, P.D.; Muhammad, N.A.N.; Dashper, S.G.; Likić, V.A.; Gorasia, D.G.; Chen, D.; Byrne, S.J.; Catmull, D.V.; Reynolds, E.C. Protein substrates of a novel secretion system are numerous in the Bacteroidetes phylum and have in common a cleavable C-terminal secretion signal, extensive post-translational modification, and cell-surface attachment. J. Proteome Res. 2013, 12, 4449–4461. [Google Scholar] [CrossRef]
- Wu, A.K.; Kropinski, A.M.; Lumsden, J.S.; Dixon, B.; MacInnes, J.I. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum ATCC 49418T. Stand. Genom. Sci. 2015, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Miyoshi, S.I.; Shinoda, S. Microbial metalloproteases and pathogenesis. Microbes Infect. 2000, 2, 91–98. [Google Scholar] [CrossRef]
- Pidde-Queiroz, G.; Magnoli, F.C.; Portaro, F.C.V.; Serrano, S.M.T.; Lopes, A.S.; Leme, A.F.P.; Berg, C.W.V.D.; Tambourgi, D.V. PI snake venom metalloproteinase is able to activate the complement system by direct cleavage of central components of the cascade. PLOS Negl. Trop. Dis. 2013, 7, e2519. [Google Scholar] [CrossRef] [PubMed]
- Duregotti, E.; Zanetti, G.; Scorzeto, M.; Megighian, A.; Montecucco, C.; Pirazzini, M.; Rigoni, M. Snake and spider toxins induce a rapid recovery of function of botulinum neurotoxin paralysed neuromuscular junction. Toxins 2015, 7, 5322–5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustillo, S.; Van de Velde, A.C.; Perfumo, V.M.; Gay, C.C.; Leiva, L.C. Apoptosis induced by a snake venom metalloproteinase from Bothrops alternatus venom in C2C12 muscle cells. Apoptosis 2017, 22, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Chaves-Moreira, D.; Senff-Ribeiro, A.; Wille, A.C.M.; Gremski, L.H.; Chaim, O.M.; Veiga, S.S. Highlights in the knowledge of brown spider toxins. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 6. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Cheng, Y.; Wang, R.-J.; Du, J.; Volovych, O.; Li, J.-C.; Hu, Y.; Lu, Z.-Y.; Lu, Z.; Zou, Z. A metalloprotease homolog venom protein from a parasitoid wasp suppresses the toll pathway in host hemocytes. Front. Immunol. 2018, 9, 2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britney, S.R.; Nowlan, J.P.; Lumsden, J.S.; Russell, S. Minimum Inhibitory Concentration of florfenicol for Canadian Tenacibaculum dicentrarchi field isolates. Dis. Aquat. Org. 2022. submitted. [Google Scholar]
- Irgang, R.; Mancilla, M.; Avendaño-Herrera, R. Florfenicol and oxytetracycline susceptibility patterns in Chilean isolates of Tenacibaculum dicentrarchi: An emerging pathogen for farmed salmonids. J. Fish Dis. 2021, 44, 1043–1046. [Google Scholar] [CrossRef] [PubMed]
- Kehrenberg, C.; Schwarz, S. Distribution of florfenicol resistance genes fexA and cfr among chloramphenicol-resistant Staphylococcus isolates. Antimicrob. Agents Chemother. 2006, 50, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.C. Tetracycline resistance determinants: Mechanisms of action, regulation of expression, genetic mobility, and distribution. FEMS Microbiol. Rev. 1996, 19, 1–24. [Google Scholar] [CrossRef]
- Ramos, J.L.; Martínez-Bueno, M.; Molina-Henares, A.J.; Terán, W.; Watanabe, K.; Zhang, X.; Gallegos, M.T.; Brennan, R.; Tobes, R. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 2005, 69, 326–356. [Google Scholar] [CrossRef] [Green Version]
- Cuthbertson, L.; Nodwell, J.R. The TetR family of regulators. Microbiol. Mol. Biol. Rev. 2013, 77, 440–475. [Google Scholar] [CrossRef] [PubMed]
- Nadella, R.K.; Panda, S.K.; Badireddy, M.R.; Kurcheti, P.P.; Raman, R.P.; Mothadaka, M.P. Multi-drug resistance, integron and transposon-mediated gene transfer in heterotrophic bacteria from Penaeus vannamei and its culture environment. Environ. Sci. Pollut. Res. 2022, 29, 37527–37542. [Google Scholar] [CrossRef] [PubMed]
- da Silva Filho, A.C.; Raittz, R.T.; Guizelini, D.; De Pierri, C.R.; Augusto, D.W.; dos Santos-Weiss, I.C.R.; Marchaukoski, J.N. Comparative analysis of genomic island prediction tools. Front. Genet. 2018, 9, 619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawato, Y.; Istiqomah, I.; Gaafar, A.Y.; Hanaoka, M.; Ishimaru, K.; Yasuike, M.; Nishiki, I.; Nakamura, Y.; Fujiwara, A.; Nakai, T. A novel jumbo Tenacibaculum maritimum lytic phage with head-fiber-like appendages. Arch. Virol. 2020, 165, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Prévots, F.; Daloyau, M.; Bonin, O.; Dumont, X.; Tolou, S. Cloning and sequencing of the novel abortive infection gene abiH of Lactococcus lactis ssp. lactis biovar. diacetylactis S94. FEMS Microbiol. Lett. 1996, 142, 295–299. [Google Scholar] [CrossRef]
- Inglin, R.C.; Meile, L.; Stevens, M.J. Clustering of pan-and core-genome of Lactobacillus provides novel evolutionary insights for differentiation. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef]
- Kumar, R.; Bröms, J.E.; Sjöstedt, A. Exploring the diversity within the genus Francisella—An integrated pan-genome and genome-mining approach. Front. Microbiol. 2020, 11, 1928. [Google Scholar] [CrossRef]



| Bacterial Sequence | Genbank Accession Number | NCBI Name |
|---|---|---|
| Tenacibaculum adriaticum DSM 18961T | GCF_008124875.1 | ASM812487v1 |
| Tenacibaculum agarivorans HZ1T | GCF_001936575.1 | ASM193657v1 |
| Tenacibaculum aiptasiae a4T | GCF_008806755.1 | ASM880675v1 |
| Tenacibaculum caenipelagi CECT 8283T | GCF_004363005.1 | ASM436300v1 |
| Tenacibaculum dicentrarchi TD3509T | GCF_900239455.1 | TD3509TV1 |
| Tenacibaculum dicentrarchi TdCh04 | GCF_018616285.1 | ASM1861628v1 |
| Tenacibaculum dicentrarchi TNO021 | GCF_900239305.1 | TNO021V1 |
| Tenacibaculum dicentrarchi QCR29 | GCF_018616555.1 | ASM1861655v1 |
| Tenacibaculum discolor DSM 18842T | GCF_003664185.1 | ASM366418v1 |
| Tenacibaculum finnmarkense gm. finnmarkense TNO006T | GCF_900239185.1 | TNO006V1 |
| Tenacibaculum finnmarkense gm. ulcerans TNO010T | GCF_900239495.1 | TNO010V1 |
| Tenacibaculum finnmarkense AY7486TD | GCF_001483385.1 | ASM148338v1 |
| Tenacibaculum finnmarkense TFHFJ T | GCF_900239485.1 | TFHFJTV1 |
| Tenacibaculum gallaicum DSM 18841T | GCF_003387615.1 | ASM338761v1 |
| Tenacibaculum holothuriorum S2-2T | GCF_002120225.1 | ASM212022v1 |
| Tenacibaculum jejuense KCTC 22618T | GCF_900198195.1 | TjejuenseV1 |
| Tenacibaculum lutimaris DSM 16505T | GCF_003610735.1 | ASM361073v1 |
| Tenacibaculum maritimum NCIMB 2154T | GCF_900119795.1 | MARITPRJEB17743 |
| Tenacibaculum maritimum TM-KORJJ | GCF_004803875.1 | ASM480387v1 |
| Tenacibaculum mesophilum DSM 13764T | GCF_003867075.1 | ASM386707v1 |
| Tenacibaculum ovolyticum da5A-8 | GCF_001641405.1 | ASM164140v1 |
| Tenacibaculum ovolyticum DSM 18103T | GCF_000430545.1 | ASM43054v1 |
| Tenacibaculum ovolyticum To-7Br | GCF_021852385.1 | ASM2185238v1 |
| Tenacibaculum pelagium S7007T | GCF_014062345.1 | ASM1406234v1 |
| Tenacibaculum piscium RT-G24 | GCF_021390715.1 | ASM2139071v1 |
| Tenacibaculum piscium SC-I4 | GCF_021390755.1 | ASM2139075v1 |
| Tenacibaculum piscium TNO020T | GCF_900239505.1 | TNO020V1 |
| Tenacibaculum piscium TNO070 | GCF_015143395.1 | ASM1514339v1 |
| Tenacibaculum singaporense DSM 106434T | GCF_003867015.1 | ASM386701v1 |
| Tenacibaculum skagerrakense DSM 14836T | GCF_004345825.1 | ASM434582v1 |
| Tenacibaculum soleae UCD-KL19 | GCF_001693415.1 | ASM169341v1 |
| Tenacibaculum todarodis LPB0136T | GCF_001889045.1 | ASM188904v1 |
| Tenacibaculum sp. SZ-18 | GCF_002813915.1 | ASM281391v1 |
| Tenacibaculum sp. AHE14PA | GCF_019278465.1 | ASM1927846v1 |
| Tenacibaculum sp. AHE15PA | GCF_019278445.1 | ASM1927844v1 |
| Isolate Name | Genomic Identification | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| qPCR (+/−) 2 | 16S (27F, 1492R) NCBI BLAST 3 | Presumed Identity | |||||||
| MAR | DICEN | FIN | OVO | Closest Match | Query Cover %, Similarity %, E-value | ||||
| 20-4135-2 | − | − | − | + | Tenacibaculum ovolyticum da5A-8 | 100 | 99.9 | 0 | T. ovolyticum |
| 20-4116-9 | − | + | − | − | Tenacibaculum dicentrarchi TdChD04 | 99 | 98.8 | 0 | T. dicentrarchi |
| 20-4106-2 | − | − | + | − | Tenacibaculum finnmarkense Tsp.2 | 100 | 99 | 0 | T. finnmarkense |
| 17-2576-1 | − | + | + | − | Tenacibaculum sp. RTG-16 | 98 | 99.1 | 0 | T. finnmarkense |
| 18-2881-A | − | − | − | − | Tenacibaculum sp. Tsp.4 | 100 | 98.3 | 0 | Tenacibaculum sp. |
| 18-3228-7B | − | − | − | − | NA | NA | NA | NA | Tenacibaculum sp. |
| 18-3141 | − | + | − | − | NA | NA | NA | NA | T. dicentrarchi |
| LI C6 FM3-F | − | + | + | − | Tenacibaculum finnmarkense AY7486TD | 100 | 99.9 | 0 | T. finnmarkense |
| T.mar 2.1C | + | − | − | − | Tenacibaculum maritimum NLF-15 | 99 | 100 | 0 | T. maritimum |
| T.mar ATR 174 1B | + | − | − | − | Tenacibaculum maritimum TFA4 | 99 | 98.9 | 0 | T. maritimum |
| Barcode # | Isolate Name | Number of Reads | Total Basepairs | Estimated Chromosome Size | Estimated Chromosome Coverage | Read Length N50 | Mean Read Length | Max Read Length | Mean Q Score |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 20-4135-2 | 46,497 | 345,317,009 | 4,100,000 | 84.22 | 13,519 | 7426.7 | 101,783 | 15.6 |
| 2 | 20-4116-9 | 112,128 | 732,515,487 | 2,700,000 | 271.3 | 9074 | 6532.9 | 81,239 | 15.2 |
| 3 | 20-4106-2 | 144,584 | 823,300,941 | 2,700,000 | 304.9 | 7801 | 5694.3 | 65,495 | 15.2 |
| 4 | 17-2576-1 | 216,705 | 994,359,562 | 2,700,000 | 368.3 | 6496 | 4588.5 | 63,050 | 15 |
| 5 | 18-2881-A | 39,459 | 348,772,821 | 3,500,000 | 99.6 | 18,051 | 8838.9 | 117,965 | 15.3 |
| 6 | 18-3228-7B | 57,597 | 460,320,495 | 3,500,000 | 131.5 | 15,516 | 7992.1 | 122,145 | 15.3 |
| 7 | 18-3141 | 108,120 | 763,100,153 | 2,700,000 | 282.6 | 9862 | 7057.9 | 64,381 | 15.3 |
| 8 | LI C6 FM3-F | 335,237 | 1233,306,653 | 2,700,000 | 456.8 | 4938 | 3678.9 | 66,955 | 15.2 |
| 9 | T.mar 2.1C | 154,709 | 663,990,855 | 3,300,000 | 201.2 | 6913 | 4291.9 | 119,184 | 15.3 |
| 10 | T.mar ATR 174 1B | 439,613 | 508,915,182 | 3,300,000 | 154.2 | 1255 | 1157.6 | 84,467 | 15.5 |
| Barcode # | Assembler 1 and Polishing Tool 2 | Number of Identified Contigs | Complete Chromosomal Contig Size | Putative Plasmid Sizes * |
|---|---|---|---|---|
| 1 | Trycycler + Medaka | 1 | 4.20 Mb | - |
| 2 | Raven + Medaka | 5 | 1.6 Mb, 984 Kb | 28 Kb, 14 Kb, 2 Kb |
| 3 | Trycycler + Medaka | 2 | 2.86 Mb | 22 Kb |
| 4 | Trycycler + Medaka | 4 | 2.83 Mb | 131 Kb; 14 Kb, 3 Kb |
| 5 | Trycycler + Medaka | 1 | 2.79 Mb | - |
| 6 | Trycycler + Medaka | 1 | 2.89 Mb | - |
| 7 | Trycycler + Medaka | 1 | 2.78 Mb | - |
| 8 | Trycycler + Medaka | 2 | 2.93 Mb | 154 Kb |
| 9 | Raven + Medaka | 5 | 1.9 Mb, 1.6 Mb, 700 Kb | 5 Kb, 4 Kb |
| 10 | Raven + Medaka | 45 | Unclear | Unclear |
| ANI Comparison | Reference | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| barcode1 | barcode2 | barcode3 | barcode4 | barcode5 | barcode6 | barcode7 | barcode8 | barcode9 | barcode10 | T. maritimum 2154T | T. ovolyticum DSM 18103T | T. dicentrarchi TD3509T | T. finnmarkense gm. finnmarkense TNO006T | T. finnmarkense gm. ulcerans TNO010T | T. piscium TNO020T | T. soleae UCD-KL19T | T. mesophilum DSM 13764T | T. discolor DSM 18842T | T. gallaicum DSM 18841T | ||
| Query | barcode1 | 100.0 | 81.4 | 81.1 | 81.2 | 82.5 | 83.4 | 81.3 | 81.2 | 78.4 | 78.3 | 78.3 | 97.6 | 81.5 | 81.1 | 81.3 | 80.6 | 83.5 | 78.3 | 79.6 | 79.3 |
| barcode2 | 81.6 | 100.0 | 94.1 | 94.1 | 88.3 | 82.0 | 99.2 | 94.1 | 79.2 | 79.7 | 78.1 | 81.5 | 98.2 | 94.1 | 94.1 | 87.3 | 81.7 | 79.0 | 79.1 | 79.0 | |
| barcode3 | 81.2 | 94.1 | 100.0 | 98.6 | 87.5 | 81.9 | 93.9 | 96.9 | 78.4 | 78.0 | 78.0 | 81.0 | 93.4 | 98.2 | 96.9 | 87.7 | 81.4 | 78.5 | 78.8 | 78.6 | |
| barcode4 | 81.2 | 93.9 | 98.6 | 100.0 | 87.2 | 81.9 | 93.9 | 96.9 | 78.1 | 78.6 | 77.8 | 80.9 | 93.3 | 98.2 | 96.8 | 87.4 | 81.3 | 78.5 | 78.7 | 78.7 | |
| barcode5 | 82.6 | 88.3 | 87.4 | 87.2 | 100.0 | 83.1 | 88.1 | 87.3 | 78.1 | 78.3 | 78.0 | 82.4 | 87.9 | 87.2 | 87.3 | 85.6 | 82.2 | 79.4 | 79.5 | 79.3 | |
| barcode6 | 83.6 | 82.1 | 81.9 | 81.8 | 82.8 | 100.0 | 82.0 | 81.7 | 78.1 | 78.0 | 78.1 | 83.5 | 81.8 | 81.8 | 81.6 | 81.3 | 85.3 | 79.8 | 80.0 | 79.7 | |
| barcode7 | 81.5 | 99.2 | 94.1 | 94.0 | 88.0 | 82.0 | 100.0 | 94.1 | 78.4 | 78.1 | 78.2 | 81.5 | 98.1 | 94.1 | 94.2 | 87.1 | 81.6 | 79.0 | 79.0 | 79.1 | |
| barcode8 | 81.3 | 94.0 | 96.9 | 96.9 | 87.2 | 82.0 | 93.8 | 100.0 | 78.0 | 78.6 | 77.9 | 81.1 | 93.5 | 96.8 | 98.6 | 87.9 | 81.2 | 78.7 | 78.8 | 78.9 | |
| barcode9 | 78.2 | 79.0 | 78.1 | 78.3 | 78.0 | 78.0 | 77.9 | 78.1 | 100.0 | 99.9 | 97.4 | 78.0 | 78.2 | 78.0 | 78.2 | 78.4 | 78.1 | 78.0 | 78.0 | 77.8 | |
| barcode10 | 78.3 | 80.0 | 78.7 | 79.3 | 78.4 | 78.2 | 78.8 | 79.0 | 99.9 | 100.0 | 97.4 | 78.1 | 79.0 | 78.7 | 79.0 | 78.9 | 78.2 | 78.1 | 77.9 | 77.8 | |
| Barcode # | # Of Copies | # Of SNP | # Of INDEL | Length (bp) | Locus Tags | NCBI BLAST | |||
|---|---|---|---|---|---|---|---|---|---|
| Top BLAST Match | Query Cover | E-value | % Identity | ||||||
| 1 | 7 | 1 | 0 | 1518 | BIBBJE_07040 BIBBJE_14780 | T. ovolyticum da5A-8 (LC144619.1) | 100 | 0 | 99.93 |
| BIBBJE_08755 BIBBJE_14755 BIBBJE_15960 BIBBJE_15985 BIBBJE_17670 | T. ovolyticum da5A-8 (LC144619.1) | 100 | 0 | 100 | |||||
| 2 | 9 | 28 | 1 | 1518 | KGFFJA_05390 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.88 |
| KGFFJA_05415 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.75 | |||||
| KGFFJA_05440 KGFFJA_10540 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 98.94 | |||||
| KGFFJA_05465 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 98.91 | |||||
| KGFFJA_07820 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 98.28 | |||||
| KGFFJA_09870 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.75 | |||||
| KGFFJA_12460 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.88 | |||||
| 1517 | KGFFJA_06620 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 98.21 | ||||
| 3 | 7 | 22 | 0 | 1518 | DGBDCK_01985 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 99.74 |
| DGBDCK_02660 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.81 | |||||
| DGBDCK_04865 DGBDCK_06085 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.88 | |||||
| DGBDCK_07235 DGBDCK_13085 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.75 | |||||
| DGBDCK_07260 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.81 | |||||
| 4 | 8 | 16 | 14 | 1520 | CAJKLB_03075 CAJKLB_03965 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.29 |
| 1520 | CAJKLB_07030 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.35 | ||||
| 1522 | CAJKLB_08670 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.16 | ||||
| 1525 | CAJKLB_10260 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.36 | ||||
| 1525 | CAJKLB_10285 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.16 | ||||
| 1520 | CAJKLB_10310 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.22 | ||||
| 1522 | CAJKLB_18445 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 97.5 | ||||
| 5 | 9 | 13 | 2 | 1518 | JAJPGM_00905 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.62 |
| 1516 | JAJPGM_01550 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.28 | ||||
| 1518 | JAJPGM_03165 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.28 | ||||
| 1518 | JAJPGM_04825 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.62 | ||||
| 1518 | JAJPGM_06265 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.62 | ||||
| 1518 | JAJPGM_06295 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.68 | ||||
| 1518 | JAJPGM_07025 JAJPGM_11610 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.68 | ||||
| 1518 | JAJPGM_07055 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 97.68 | ||||
| 6 | 7 | 14 | 2 | 1516 | KPHGJK_03565 | Tenacibaculum sp. AHE14PA (CP058983.1) | 100 | 0 | 98.48 |
| 1518 | KPHGJK_07470 | Tenacibaculum sp. AHE15PA (CP058982.1) | 100 | 0 | 98.15 | ||||
| 1518 | KPHGJK_07495 | Tenacibaculum sp. AHE15PA (CP058982.1) | 100 | 0 | 98.02 | ||||
| 1518 | KPHGJK_07520 | Tenacibaculum sp. AHE15PA (CP058982.1) | 100 | 0 | 98.22 | ||||
| 1516 | KPHGJK_07545 | Tenacibaculum sp. AHE15PA (CP058982.1) | 100 | 0 | 98.08 | ||||
| 1516 | KPHGJK_13405 | Tenacibaculum sp. AHE15PA (CP058982.1) | 100 | 0 | 97.95 | ||||
| 1518 | KPHGJK_13430 | Tenacibaculum sp. AHE15PA (CP058982.1) | 100 | 0 | 98.15 | ||||
| 7 | 9 | 34 | 4 | 1516 | APJPMD_02115 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.81 |
| 1518 | APJPMD_02725 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 99.07 | ||||
| 1518 | APJPMD_04745 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.88 | ||||
| 1518 | APJPMD_05975 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 99.27 | ||||
| 1518 | APJPMD_07140 APJPMD_07165 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 98.81 | ||||
| 1518 | APJPMD_07190 | T. dicentrarchi 35/09T (NR_108475.1) | 99 | 0 | 98.94 | ||||
| 1516 | APJPMD_07215 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.15 | ||||
| 1516 | APJPMD_13045 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.88 | ||||
| 8 | 7 | 33 | 2 | 1518 | FHDEPC_02035 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 99.6 |
| 1517 | FHDEPC_02655 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 99.67 | ||||
| 1518 | FHDEPC_04750 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 99.41 | ||||
| 1517 | FHDEPC_06105 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 99.34 | ||||
| 1517 | FHDEPC_07265 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 99.6 | ||||
| 1518 | FHDEPC_07295 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 99.67 | ||||
| 1517 | FHDEPC_13480 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.68 | ||||
| 9 | 6 | 1 | 0 | 1520 | KDPLPA_06585 KDPLPA_14230 KDPLPA_15870 | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 99.08 |
| 1520 | KDPLPA_14645 KDPLPA_17070 KDPLPA_18190 | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 99.14 | ||||
| 10 | 8 | NA | NA | 793 | FCGIAJ_04105 Partial | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 96.2 |
| 977 | FCGIAJ_04905 Partial | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 92.28 | ||||
| 1422 | FCGIAJ_05975 | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 98.43 | ||||
| 1520 | FCGIAJ_09820 | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 99.08 | ||||
| 1522 | FCGIAJ_13200 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.35 | ||||
| 1524 | FCGIAJ_13230 | T. finnmarkense AY7486TD (CP013671.1) | 100 | 0 | 98.35 | ||||
| 1354 | FCGIAJ_19415 | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 86.87 | ||||
| 1104 | FCGIAJ_19615 Partial | T. maritimum TM-KORJJ (CP020822.1) | 100 | 0 | 98.82 | ||||
| Group | Gene-Cluster Comparison | |||
|---|---|---|---|---|
| 95% | 80% | |||
| Total | Shared | Total | Shared | |
| Genus (barcodes 1-10) | 11,978 | 191 | 9960 | 973 |
| T. maritimum (barcodes 9 & 10) | 4215 | 3821 | 4196 | 3818 |
| T. dicentrarchi (barcodes 2 & 7) | 2591 | 2140 | 2626 | 2140 |
| T. finnmarkense (barcodes 3, 4, & 8) | 3197 | 2043 | 3265 | 2052 |
| T. dicentrarchi/T. finnmarkense (barcodes 2, 3, 4, 7, & 8) | 3655 | 1921 | 3724 | 1928 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nowlan, J.P.; Sies, A.N.; Britney, S.R.; Cameron, A.D.S.; Siah, A.; Lumsden, J.S.; Russell, S. Genomics of Tenacibaculum Species in British Columbia, Canada. Pathogens 2023, 12, 101. https://doi.org/10.3390/pathogens12010101
Nowlan JP, Sies AN, Britney SR, Cameron ADS, Siah A, Lumsden JS, Russell S. Genomics of Tenacibaculum Species in British Columbia, Canada. Pathogens. 2023; 12(1):101. https://doi.org/10.3390/pathogens12010101
Chicago/Turabian StyleNowlan, Joseph P., Ashton N. Sies, Scott R. Britney, Andrew D. S. Cameron, Ahmed Siah, John S. Lumsden, and Spencer Russell. 2023. "Genomics of Tenacibaculum Species in British Columbia, Canada" Pathogens 12, no. 1: 101. https://doi.org/10.3390/pathogens12010101
APA StyleNowlan, J. P., Sies, A. N., Britney, S. R., Cameron, A. D. S., Siah, A., Lumsden, J. S., & Russell, S. (2023). Genomics of Tenacibaculum Species in British Columbia, Canada. Pathogens, 12(1), 101. https://doi.org/10.3390/pathogens12010101