
Secretomic Insights into the Pathophysiology of Venturia inaequalis: The Causative Agent of Scab, a Devastating Apple Tree Disease
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Repeat Annotation
2.2. Gene Prediction
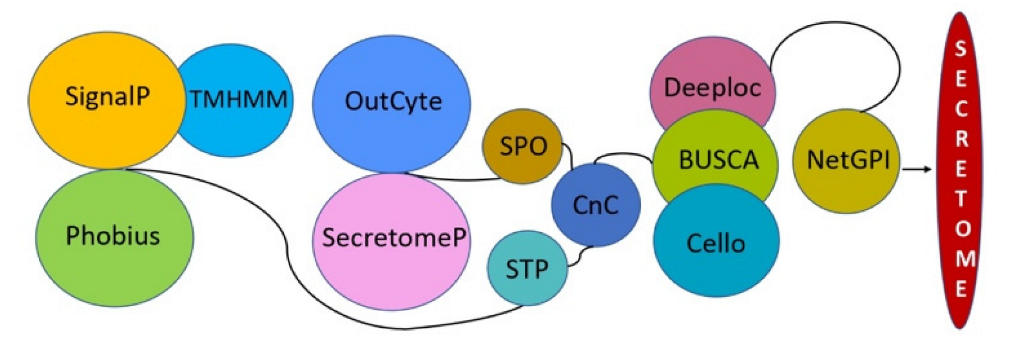
2.3. Secretome Analysis
2.4. Identification of Candidate Effectors
2.5. Functional Annotation
2.6. Preparation of Samples
2.7. RNA Isolation and Library Preparation
2.8. Transcriptome Profiling
3. Results and Discussion
3.1. V. inaequalis Transposable Elements Represent Half of the Overall Genome Size
3.2. Annotated Genome of V. inaequalis (EU-B04) with a BUSCO Completeness Score of 99.1%
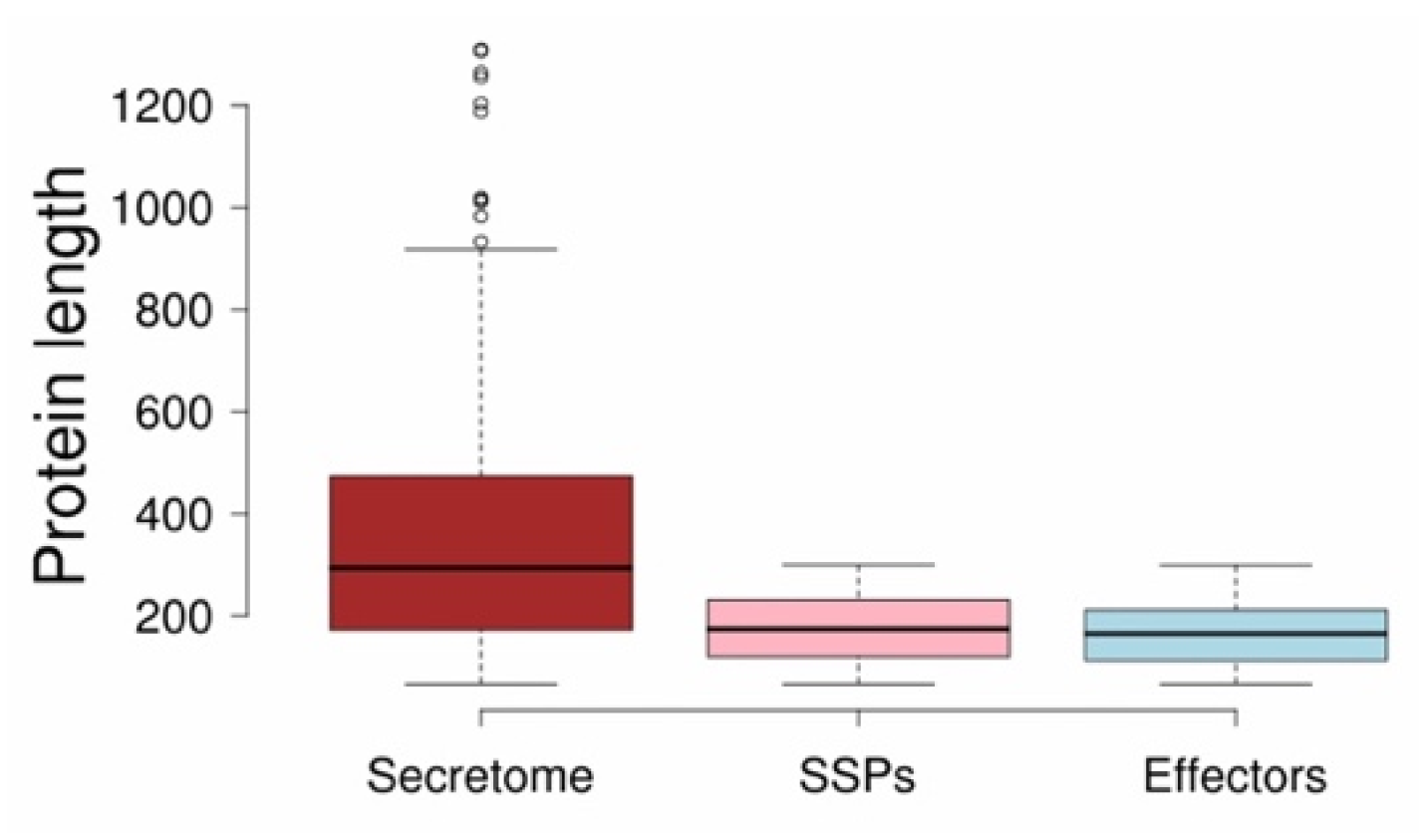
3.3. V. inaequalis Secretome Accounts for Approximately 5% of Its Total Proteome Size
3.4. Three-Quarters of V. inaequalis Small Secreted Proteins (76.52%) Are Effectors That May Contribute to Its Virulence
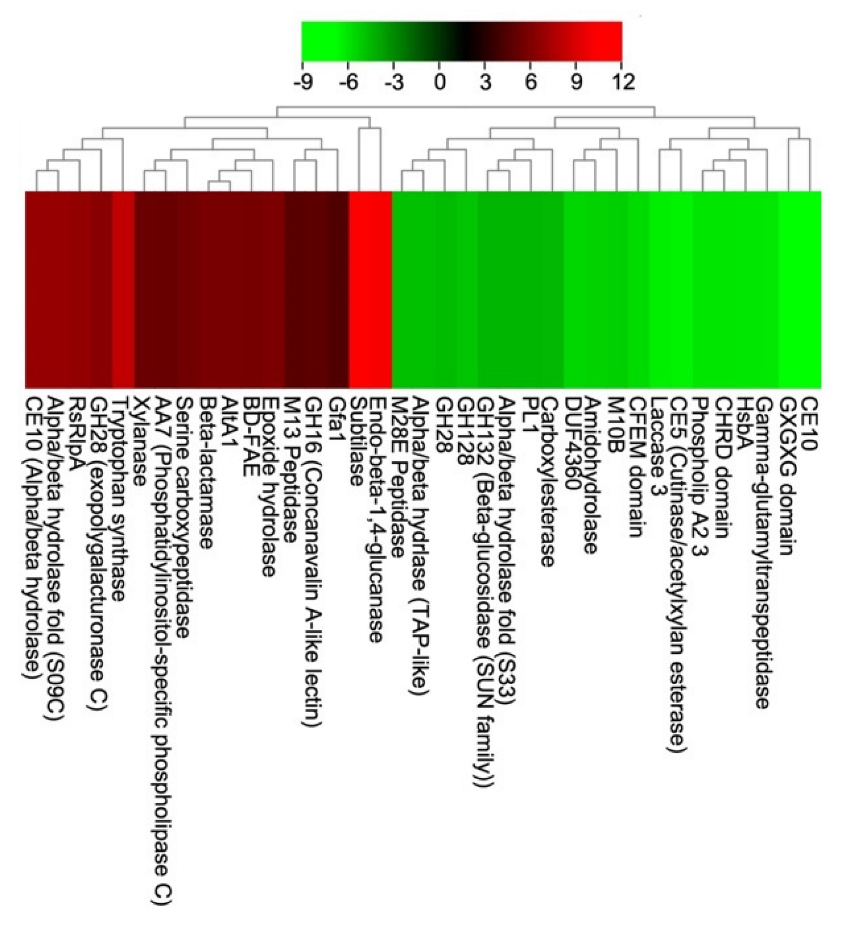
3.5. Functional Annotations of V. inaequalis Secretome and Effectome
3.6. Expression Profiling of V. inaequalis Secretome
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- González-Domínguez, E.; Armengol, J.; Rossi, V. Biology and Epidemiology of Venturia Species Affecting Fruit Crops: A Review. Front. Plant Sci. 2017, 8, 1496. [Google Scholar] [CrossRef]
- MacHardy, W. Inheritance of resistance to Venturia inaequalis. In Apple Scab, Biology, Epidemiology and Management; APS: St. Paul, MN, USA, 1996; pp. 61–103. [Google Scholar]
- Beresford, R.M.; Wright, P.J.; Wood, P.N.; Park, N.M. Sensitivity of Venturia inaequalis to myclobutanil, penconazole and dodine in relation to fungicide use in Hawke’s Bay apple orchards. NZ Plant Prot. 2012, 65, 106–113. [Google Scholar]
- Patocchi, A.; Wehrli, A.; Dubuis, P.-H.; Auwerkerken, A.; Leida, C.; Cipriani, G.; Passey, T.; Staples, M.; Didelot, F.; Philion, V.; et al. Ten Years of VINQUEST: First Insight for Breeding New Apple Cultivars With Durable Apple Scab Resistance. Plant Dis. 2020, 104, 2074–2081. [Google Scholar] [CrossRef] [Green Version]
- Bus, V.G.; Rikkerink, E.H.; Caffier, V.; Durel, C.-E.; Plummer, K.M. Revision of the Nomenclature of the Differential Host-Pathogen Interactions of Venturia inaequalis and Malus. Annu. Rev. Phytopathol. 2011, 49, 391–413. [Google Scholar] [CrossRef] [Green Version]
- Lichtner, F.J.; Jurick, I.W.M.; Ayer, K.M.; Gaskins, V.L.; Villani, S.M.; Cox, K.D. A Genome Resource for Several North American Venturia inaequalis Isolates with Multiple Fungicide Resistance Phenotypes. Phytopathology 2020, 110, 544–546. [Google Scholar] [CrossRef] [Green Version]
- Bowen, J.K.; Mesarich, C.H.; Bus, V.G.M.; Beresford, R.M.; Plummer, K.M.; Templeton, M.D. Venturia inaequalis: The causal agent of apple scab. Mol. Plant Pathol. 2010, 12, 105–122. [Google Scholar] [CrossRef]
- Belete, T.; Boyraz, N. Critical review on apple scab (Venturia inaequalis) biology, epidemiology, economic importance, management and defense mechanisms to the causal agent. J. Plant Physiol. Pathol. 2017, 5, 2. [Google Scholar]
- Ebrahimi, L.; Fotuhifar, K.B.; Javan-Nikkhah, M.; Naghavi, M.R.; Baisakh, N. Population Genetic Structure of Apple Scab (Venturia inaequalis (Cooke) G. Winter) in Iran. PLoS ONE 2016, 11, e0160737. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Tao, F.; Fan, S.; Li, H.; Yang, J.; Gao, L. Genetic diversity of Venturia inaequalis isolates (Apple scab) in China and U.K. determined by SSR markers. PLoS ONE 2021, 16, e0252865. [Google Scholar] [CrossRef]
- Ngou, B.P.M.; Ding, P.; Jones, J.D.G. Thirty years of resistance: Zig-zag through the plant immune system. Plant Cell 2022, 34, 1447–1478. [Google Scholar] [CrossRef]
- Figueroa, M.; Ortiz, D.; Henningsen, E.C. Tactics of host manipulation by intracellular effectors from plant pathogenic fungi. Curr. Opin. Plant Biol. 2021, 62, 102054. [Google Scholar] [CrossRef]
- Zhai, K.; Di Liang, D.; Li, H.; Jiao, F.; Yan, B.; Liu, J.; Lei, Z.; Huang, L.; Gong, X.; Wang, X.; et al. NLRs guard metabolism to coordinate pattern- and effector-triggered immunity. Nature 2021, 601, 245–251. [Google Scholar] [CrossRef]
- Rocafort, M.; Fudal, I.; Mesarich, C.H. Apoplastic effector proteins of plant-associated fungi and oomycetes. Curr. Opin. Plant Biol. 2020, 56, 9–19. [Google Scholar] [CrossRef]
- Tariqjaveed, M.; Mateen, A.; Wang, S.; Qiu, S.; Zheng, X.; Zhang, J.; Bhadauria, V.; Sun, W. Versatile effectors of phytopathogenic fungi target host immunity. J. Integr. Plant Biol. 2021, 63, 1856–1873. [Google Scholar] [CrossRef]
- Khajuria, Y.P.; Kaul, S.; Wani, A.A.; Dhar, M.K. Genetics of resistance in apple against Venturia inaequalis (Wint.) Cke. Tree Genet. Genomes 2018, 14, 1–20. [Google Scholar] [CrossRef]
- Cooper, S.J.; Ashby, A.M. Comparison of cytokinin and cytokinin-O-glucoside cleaving beta-glucosidase production in vitro by Venturia inaequalis and other phytopathogenic fungi with differing modes of nutrition in planta. Physiol. Mol. Plant Pathol. 1998, 53, 61–72. [Google Scholar] [CrossRef]
- Kucheryava, N.; Bowen, J.K.; Sutherland, P.W.; Conolly, J.J.; Mesarich, C.H.; Rikkerink, E.H.; Kemen, E.; Plummer, K.M.; Hahn, M.; Templeton, M.D. Two novel Venturia inaequalis genes induced upon morphogenetic differentiation during infection and in vitro growth on cellophane. Fungal Genet. Biol. 2008, 45, 1329–1339. [Google Scholar] [CrossRef]
- Bowen, J.K.; Mesarich, C.H.; Rees-George, J.; Cui, W.; Win, J.; Fitzgerald, A.; Plummer, K.M.; Templeton, M.D. Candidate effector gene identification in the ascomycete fungal phytopathogen Venturia inaequalis by expressed sequence tag analysis. Mol. Plant Pathol. 2009, 10, 431–448. [Google Scholar] [CrossRef]
- Basenko, E.Y.; Pulman, J.A.; Shanmugasundram, A.; Harb, O.S.; Crouch, K.; Starns, D.; Warrenfeltz, S.; Aurrecoechea, C.; Stoeckert, C.J., Jr.; Kissinger, J.C.; et al. FungiDB: An Integrated Bioinformatic Resource for Fungi and Oomycetes. J. Fungus 2018, 4, 39. [Google Scholar] [CrossRef] [Green Version]
- Thakur, K.; Chawla, V.; Bhatti, S.; Swarnkar, M.K.; Kaur, J.; Shankar, R.; Jha, G. De Novo Transcriptome Sequencing and Analysis for Venturia inaequalis, the Devastating Apple Scab Pathogen. PLoS ONE 2013, 8, e53937. [Google Scholar] [CrossRef]
- Shiller, J.; Van de Wouw, A.P.; Taranto, A.P.; Bowen, J.K.; Dubois, D.; Robinson, A.; Deng, C.H.; Plummer, K.M. A Large Family of AvrLm6-like Genes in the Apple and Pear Scab Pathogens, Venturia inaequalis and Venturia pirina. Front. Plant Sci. 2015, 6, 980. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.H.; Plummer, K.M.; Jones, D.A.B.; Mesarich, C.H.; Shiller, J.; Taranto, A.P.; Robinson, A.J.; Kastner, P.; Hall, N.E.; Templeton, M.D.; et al. Comparative analysis of the predicted secretomes of Rosaceae scab pathogens Venturia inaequalis and V. pirina reveals expanded effector families and putative determinants of host range. BMC Genom. 2017, 18, 1–25. [Google Scholar] [CrossRef]
- Passey, T.A.J.; Armitage, A.D.; Xu, X. Annotated Draft Genome Sequence of the Apple Scab Pathogen Venturia inaequalis. Microbiol. Resour. Announc. 2018, 7, e01062-18. [Google Scholar] [CrossRef] [Green Version]
- Le Cam, B.; Sargent, D.; Gouzy, J.; Amselem, J.; Bellanger, M.N.; Bouchez, O.; Brown, S.; Caffier, V.; De Gracia, M.; Debuchy, R.; et al. Population genome sequencing of the scab fungal species Venturia inaequalis, Venturia pirina, Venturia aucupariae and Venturia asperata. G3 Genes Genom. Genet. 2019, 9, 2405–2414. [Google Scholar]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013-2015. Available online: http://www.repeatmasker.org (accessed on 26 December 2022).
- Bruna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: Automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genome Bioinform. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Bruna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-EP+: Eukaryotic gene prediction with self-training in the space of genes and proteins. NAR Genome Bioinform. 2020, 2, lqaa026. [Google Scholar] [CrossRef]
- Teufel, F.; Armenteros, J.J.A.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Bendtsen, J.D.; Jensen, L.J.; Blom, N.; von Heijne, G.; Brunak, S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng. Des. Sel. 2004, 17, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Poschmann, G.; Waldera-Lupa, D.; Rafie e, N.; Kollmann, M.; Stühler, K. OutCyte: A novel tool for predicting unconventional protein secretion. Sci. Rep. 2019, 9, 19448. [Google Scholar] [CrossRef] [Green Version]
- Savojardo, C.; Martelli, P.L.; Fariselli, P.; Profiti, G.; Casadio, R. BUSCA: An integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 2018, 46, W459–W466. [Google Scholar] [CrossRef]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Sønderby, C.K.; Sønderby, S.K.; Nielsen, H.; Winther, O. DeepLoc: Prediction of protein subcellular localization using deep learning. Bioinformatics 2017, 33, 3387–3395. [Google Scholar] [CrossRef]
- Nielsen, H.; Petsalaki, E.I.; Zhao, L.; Stühler, K. Predicting eukaryotic protein secretion without signals. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2018, 1867, 140174. [Google Scholar] [CrossRef] [Green Version]
- Gíslason, M.H.; Nielsen, H.; Armenteros, J.J.A.; Johansen, A.R. Prediction of GPI-anchored proteins with pointer neural networks. Curr. Res. Biotechnol. 2021, 3, 6–13. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N. EffectorP 3.0: Prediction of Apoplastic and Cytoplasmic Effectors in Fungi and Oomycetes. Mol. Plant-Microbe Interact. 2022, 35, 146–156. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N.; Singh, K.B.; Taylor, J.M. ApoplastP: Prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 2017, 217, 1764–1778. [Google Scholar] [CrossRef] [Green Version]
- Guyon, K.; Balagué, C.; Roby, D.; Raffaele, S. Secretome analysis reveals effector candidates associated with broad host range necrotrophy in the fungal plant pathogen Sclerotinia sclerotiorum. BMC Genom. 2014, 15, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Chomczynski, P.; Mackey, K. Short technical report. Modification of the TRIZOL reagent procedure for isolation of RNA from Polysaccharide- and proteoglycan-rich sources. Biotechnology 1995, 19, 942–945. [Google Scholar]
- Zhong, S.; Joung, J.-G.; Zheng, Y.; Chen, Y.-R.; Liu, B.; Shao, Y.; Xiang, J.Z.; Fei, Z.; Giovannoni, J.J. High-Throughput Illumina Strand-Specific RNA Sequencing Library Preparation. Cold Spring Harb. Protoc. 2011, 2011, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Broggini, G.A.L.; Le Cam, B.; Parisi, L.; Wu, C.; Zhang, H.B.; Gessler, C.; Patocchi, A. Construction of a contig of BAC clones spanning the region of the apple scab avirulence gene AvrVg. Fungal Genet. Biol. 2007, 44, 44–51. [Google Scholar] [CrossRef]
- Borges, A.R.; Link, F.; Engstler, M.; Jones, N.G. The Glycosylphosphatidylinositol Anchor: A Linchpin for Cell Surface Versatility of Trypanosomatids. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Kars, I.; Krooshof, G.H.; Wagemakers, L.; Joosten, R.; Benen, J.A.E.; Van Kan, J.A.L. Necrotizing activity of five Botrytis cinerea endopolygalacturonases produced in Pichia pastoris. Plant J. 2005, 43, 213–225. [Google Scholar] [CrossRef]
- Akhoon, B.A.; Gupta, S.K.; Dhar, M.K. Dissecting the genome, secretome, and effectome repertoires of Monilinia spp.: The causal agent of brown rot disease: A comparative analysis. Postharvest Biol. Technol. 2023, 195, 112120. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, P.; Ma, X.; McDonald, B.A.; Brunner, P.C. Widespread signatures of selection for secreted peptidases in a fungal plant pathogen. BMC Evol. Biol. 2018, 18, 7. [Google Scholar] [CrossRef] [Green Version]
- Dubey, M.; Vélëz, H.; Broberg, M.; Jensen, D.F.; Karlsson, M. LysM Proteins Regulate Fungal Development and Contribute to Hyphal Protection and Biocontrol Traits in Clonostachys rosea. Front. Microbiol. 2020, 11, 679. [Google Scholar] [CrossRef] [Green Version]
- Buck, J.W.; Andrews, J.H. Attachment of the Yeast Rhodosporidium toruloides Is Mediated by Adhesives Localized at Sites of Bud Cell Development. Appl. Environ. Microbiol. 1999, 65, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Elordi, E.S.; Díaz-Peña, E.M.; Vicente Córdoba, C.; Legaz-González, M.E. Effects of concanavalin A on the germination of smut teliospores and on the hyphal growth. Int. J. Pharma Bio Sci. 2018, 9, 50–56. [Google Scholar] [CrossRef]
- Brito, N.; Espino, J.J.; González, C. The endo-β-1, 4-xylanase Xyn11A is required for virulence in Botrytis cinerea. Mol. Plant-Microbe Interact. 2006, 19, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Anasontzis, G.E.; Lebrun, M.H.; Haon, M.; Champion, C.; Kohler, A.; Lenfant, N.; Martin, F.; O’Connell, R.J.; Riley, R.; Grigoriev, I.V.; et al. Broad-specificity GH131 β-glucanases are a hallmark of fungi and oomycetes that colonize plants. Environ. Microbiol. 2019, 21, 2724–2739. [Google Scholar] [CrossRef]
- Arya, G.C.; Srivastava, D.A.; Pandaranayaka, E.P.J.; Manasherova, E.; Prusky, D.B.; Elad, Y.; Frenkel, O.; Dvir, H.; Harel, A. Characterization of the Role of a Non-GPCR Membrane-Bound CFEM Protein in the Pathogenicity and Germination of Botrytis cinerea. Microorganisms 2020, 8, 1043. [Google Scholar] [CrossRef]
- Kou, Y.; Tan, Y.H.; Ramanujam, R.; Naqvi, N.I. Structure–function analyses of the Pth11 receptor reveal an important role for CFEM motif and redox regulation in rice blast. New Phytol. 2016, 214, 330–342. [Google Scholar] [CrossRef]
- Garrido-Arandia, M.; Silva-Navas, J.; Ramírez-Castillejo, C.; Cubells-Baeza, N.; Gómez-Casado, C.; Barber, D.; Pozo, J.C.; Melendi, P.G.; Pacios, L.F.; Díaz-Perales, A. Characterisation of a flavonoid ligand of the fungal protein Alt a 1. Sci. Rep. 2016, 6, 33468. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, X.; Jiao, R.; Xia, Y. The Magas1 gene is involved in pathogenesis by affecting penetration in Metarhizium acridum. J. Microbiol. Biotechnol. 2012, 22, 889–893. [Google Scholar] [CrossRef]
- Venancio, T.; Aravind, L. CYSTM, a novel cysteine-rich transmembrane module with a role in stress tolerance across eukaryotes. Bioinformatics 2009, 26, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yu, Z.; Zhang, D.; Huang, J.; Wu, C.; Yang, G.; Yan, K.; Zhang, S.; Zheng, C. CYSTM, a Novel Non-Secreted Cysteine-Rich Peptide Family, Involved in Environmental Stresses in Arabidopsis thaliana. Plant Cell Physiol. 2017, 59, 423–438. [Google Scholar] [CrossRef] [Green Version]
- Rafiei, V.; Vélëz, H.; Tzelepis, G. The phospholipase VlsPLA2 from the plant pathogen Verticillium longisporum is a virulence factor targeting host nuclei and suppressing PTI-related hypersensitive response. bioRxiv 2022. [Google Scholar] [CrossRef]
- Schumacher, S.; Grosser, K.; Voegele, R.T.; Kassemeyer, H.-H.; Fuchs, R. Identification and Characterization of Nep1-Like Proteins From the Grapevine Downy Mildew Pathogen Plasmopara viticola. Front. Plant Sci. 2020, 11, 65. [Google Scholar] [CrossRef]
- Paulus, J.K.; Kourelis, J.; Ramasubramanian, S.; Homma, F.; Godson, A.; Hörger, A.C.; Hong, T.N.; Krahn, D.; Carballo, L.O.; Wang, S.; et al. Extracellular proteolytic cascade in tomato activates immune protease Rcr3. Proc. Natl. Acad. Sci. USA. 2020, 117, 17409–17417. [Google Scholar] [CrossRef]
- Miao, Y.; Xu, L.; He, X.; Zhang, L.; Shaban, M.; Zhang, X.; Zhu, L. Suppression of tryptophan synthase activates cotton immunity by triggering cell death via promoting SA synthesis. Plant J. 2019, 98, 329–345. [Google Scholar] [CrossRef]
- Charova, S.; Dölfors, F.; Holmquist, L.; Moschou, P.; Dixelius, C.; Tzelepis, G. The RsRlpA Effector Is a Protease Inhibitor Promoting Rhizoctonia solani Virulence through Suppression of the Hypersensitive Response. Int. J. Mol. Sci. 2020, 21, 8070. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khajuria, Y.P.; Akhoon, B.A.; Kaul, S.; Dhar, M.K. Secretomic Insights into the Pathophysiology of Venturia inaequalis: The Causative Agent of Scab, a Devastating Apple Tree Disease. Pathogens 2023, 12, 66. https://doi.org/10.3390/pathogens12010066
Khajuria YP, Akhoon BA, Kaul S, Dhar MK. Secretomic Insights into the Pathophysiology of Venturia inaequalis: The Causative Agent of Scab, a Devastating Apple Tree Disease. Pathogens. 2023; 12(1):66. https://doi.org/10.3390/pathogens12010066
Chicago/Turabian StyleKhajuria, Yash Paul, Bashir Akhlaq Akhoon, Sanjana Kaul, and Manoj Kumar Dhar. 2023. "Secretomic Insights into the Pathophysiology of Venturia inaequalis: The Causative Agent of Scab, a Devastating Apple Tree Disease" Pathogens 12, no. 1: 66. https://doi.org/10.3390/pathogens12010066