
Focal Adhesion Kinase Binds to the HPV E2 Protein to Regulate Initial Replication after Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
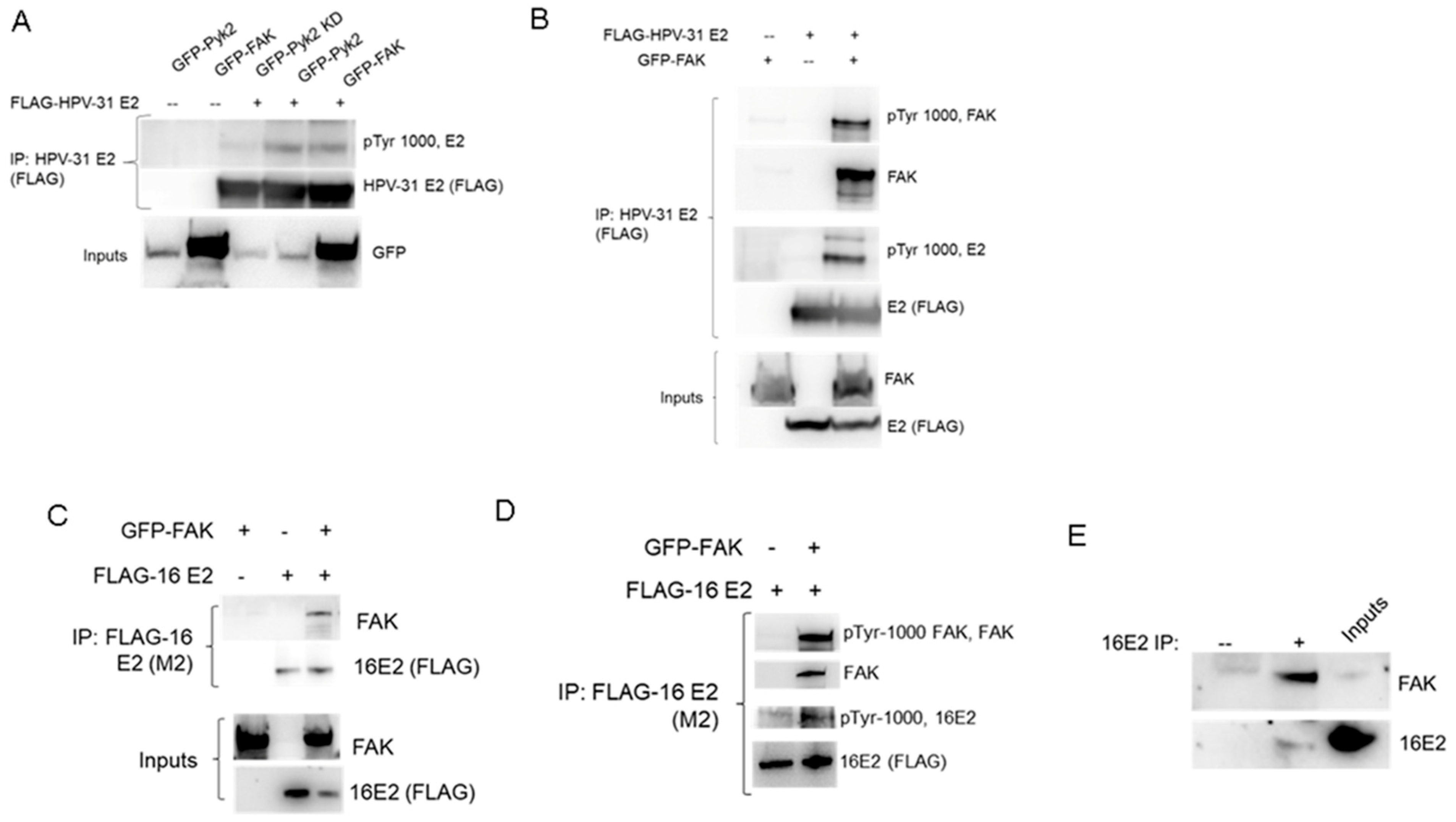
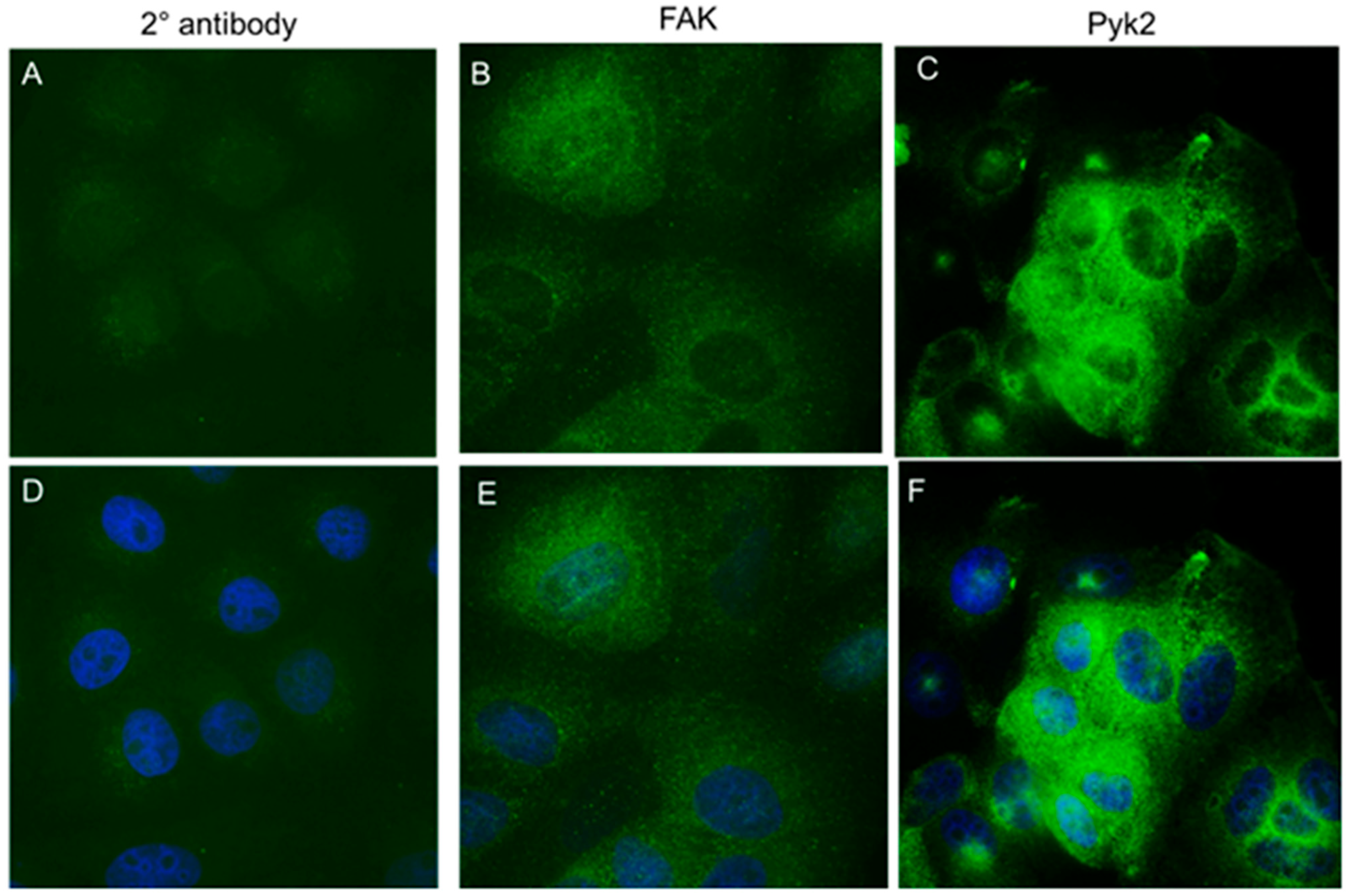
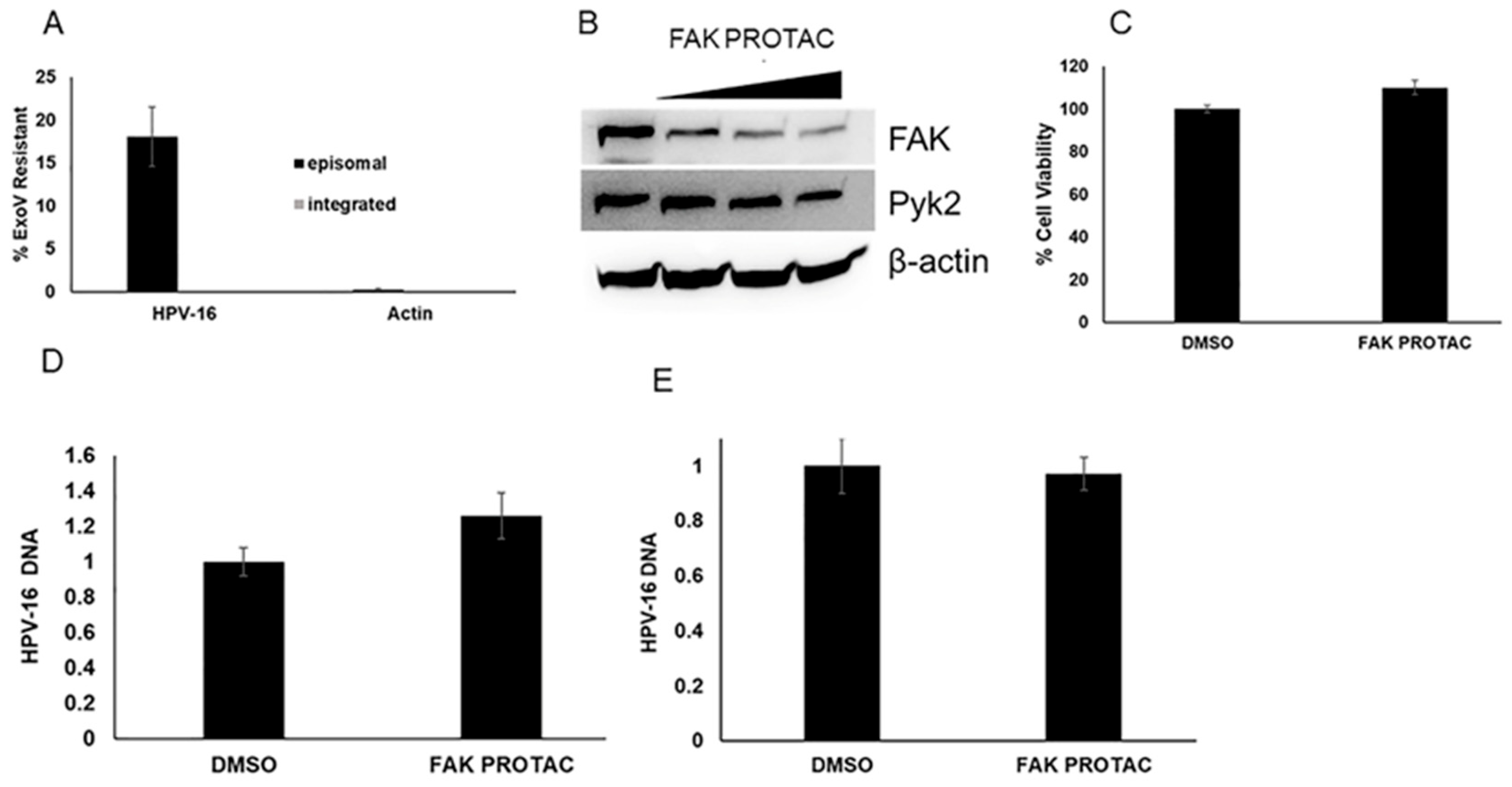
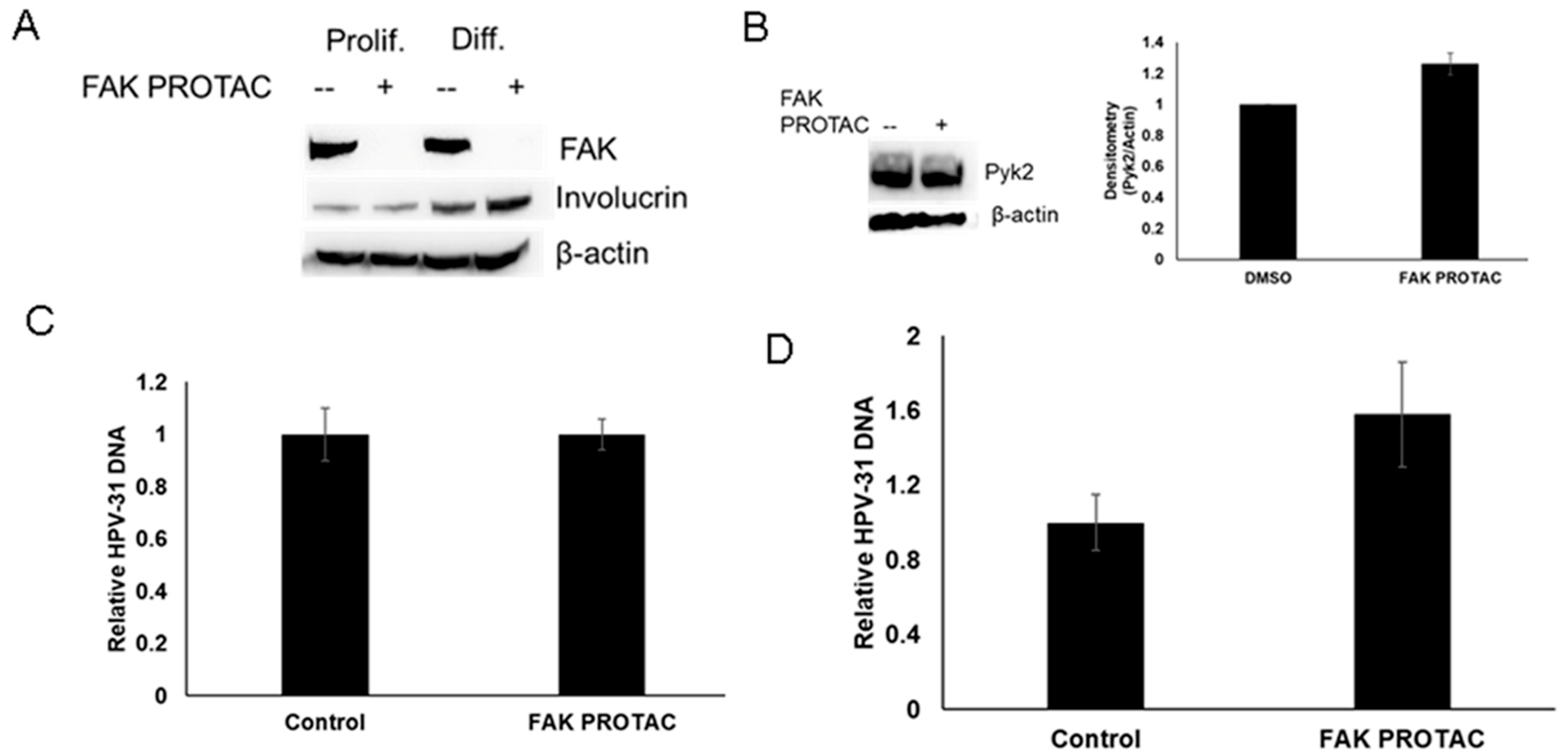
2. Results
3. Discussion
4. Materials and Methods
4.1. Plasmids and Antibodies
4.2. Cell Culture
4.3. Immunofluorescence
4.4. Co-Immunoprecipitations and Immunoblotting
4.5. HPV DNA Quantification
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKinney, C.; Hussmann, K.; McBride, A. The Role of the DNA Damage Response throughout the Papillomavirus Life Cycle. Viruses 2015, 7, 2450. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of Human Papillomavirus-Induced Oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Senapati, S.; Bose, K. Insights into the mechanism of human papillomavirus E2-induced procaspase-8 activation and cell death. Sci. Rep. 2016, 6, 21408. [Google Scholar] [CrossRef]
- Webster, K.; Parish, J.; Pandya, M.; Stern, P.L.; Clarke, A.R.; Gaston, K. The Human Papillomavirus (HPV) 16 E2 Protein Induces Apoptosis in the Absence of Other HPV Proteins and via a p53-dependent Pathway. J. Biol. Chem. 2000, 275, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human Papillomaviruses Activate the ATM DNA Damage Pathway for Viral Genome Amplification upon Differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The Papillomavirus E1 Helicase Activates a Cellular DNA Damage Response in Viral Replication Foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef]
- DeSmet, M.; Kanginakudru, S.; Rietz, A.; Wu, W.H.; Roden, R.; Androphy, E.J. The Replicative Consequences of Papillomavirus E2 Protein Binding to the Origin Replication Factor ORC2. PLoS Pathog. 2016, 12, e1005934. [Google Scholar] [CrossRef]
- Frattini, M.G.; Laimins, L.A. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein. Proc. Natl. Acad. Sci. 1994, 91, 12398. [Google Scholar] [CrossRef]
- Hoffmann, R.; Hirt, B.; Bechtold, V.; Beard, P.; Raj, K. Different modes of human papillomavirus DNA replication during maintenance. J. Virol. 2006, 80, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Henno, L.; Toots, M.; Ustav, M., Jr.; Ustav, M. The Cell Cycle Timing of Human Papillomavirus DNA Replication. PLoS ONE 2015, 10, e0131675. [Google Scholar] [CrossRef] [PubMed]
- Hubert, W.G.; Kanaya, T.; Laimins, L.A. DNA replication of human papillomavirus type 31 is modulated by elements of the upstream regulatory region that lie 5’ of the minimal origin. J. Virol. 1999, 73, 1835–1845. [Google Scholar] [CrossRef] [PubMed]
- Stubenrauch, F.; Lim, H.B.; Laimins, L.A. Differential Requirements for Conserved E2 Binding Sites in the Life Cycle of Oncogenic Human Papillomavirus Type 31. J. Virol. 1998, 72, 1071. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The Papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, E.J.; Culleton, S.P.; Wu, S.Y.; Chiang, C.M.; Androphy, E.J. Acetylation of conserved lysines in bovine papillomavirus E2 by p300. J. Virol. 2013, 87, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Culleton, S.P.; Kanginakudru, S.; DeSmet, M.; Gilson, T.; Xie, F.; Wu, S.Y.; Chiang, C.M.; Qi, G.; Wang, M.; Androphy, E.J. Phosphorylation of the Bovine Papillomavirus E2 Protein on Tyrosine Regulates Its Transcription and Replication Functions. J. Virol. 2017, 91, e01854-16. [Google Scholar] [CrossRef]
- Chang, S.W.; Liu, W.C.; Liao, K.Y.; Tsao, Y.P.; Hsu, P.H.; Chen, S.L. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PloS ONE 2014, 9, e110882. [Google Scholar] [CrossRef]
- Thomas, Y.; Androphy, E.J. Human Papillomavirus Replication Regulation by Acetylation of a Conserved Lysine in the E2 Protein. J. Virol. 2018, 92, 01912–01917. [Google Scholar] [CrossRef]
- Schuck, S.; Ruse, C.; Stenlund, A. CK2 phosphorylation inactivates DNA binding by the papillomavirus E1 and E2 proteins. J. Virol. 2013, 87, 7668–7679. [Google Scholar] [CrossRef]
- Prabhakar, A.T.; James, C.D.; Das, D.; Otoa, R.; Day, M.; Burgner, J.; Fontan, C.T.; Wang, X.; Glass, S.H.; Wieland, A.; et al. CK2 Phosphorylation of Human Papillomavirus 16 E2 on Serine 23 Promotes Interaction with TopBP1 and Is Critical for E2 Interaction with Mitotic Chromatin and the Viral Life Cycle. mBio 2021, 12, e0116321. [Google Scholar] [CrossRef] [PubMed]
- Jose, L.; Androphy, E.J.; DeSmet, M. SETD6 Regulates E2-Dependent Human Papillomavirus Transcription. J. Virol. 2022, 96, e0129522. [Google Scholar] [CrossRef] [PubMed]
- Jose, L.; Gilson, T.; Androphy, E.J.; DeSmet, M. Regulation of the Human Papillomavirus Lifecyle through Post-Translational Modifications of the Viral E2 Protein. Pathogens 2021, 10, 793. [Google Scholar] [CrossRef] [PubMed]
- Jose, L.; DeSmet, M.; Androphy, E.J. Pyk2 Regulates Human Papillomavirus Replication by Tyrosine Phosphorylation of the E2 Protein. J. Virol. 2020, 94, e01110–e01120. [Google Scholar] [CrossRef] [PubMed]
- Naser, R.; Aldehaiman, A.; Díaz-Galicia, E.; Arold, S.T. Endogenous Control Mechanisms of FAK and PYK2 and Their Relevance to Cancer Development. Cancers 2018, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal adhesion kinase signaling in unexpected places. Curr. Opin. Cell Biol. 2017, 45, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.T.; Chen, X.L.; Lim, Y.; Hanson, D.A.; Vo, T.T.; Howerton, K.; Larocque, N.; Fisher, S.J.; Schlaepfer, D.D.; Ilic, D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol. Cell 2008, 29, 9–22. [Google Scholar] [CrossRef]
- Mousson, A.; Sick, E.; Carl, P.; Dujardin, D.; De Mey, J.; Rondé, P. Targeting Focal Adhesion Kinase Using Inhibitors of Protein-Protein Interactions. Cancers 2018, 10, 278. [Google Scholar] [CrossRef]
- Zhou, J.; Yi, Q.; Tang, L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: A focused review. J. Exp. Clin. Cancer Res. 2019, 38, 250. [Google Scholar] [CrossRef]
- Golubovskaya, V.M.; Finch, R.; Cance, W.G. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J. Biol. Chem. 2005, 280, 25008–25021. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.W.; Zhang, C.; Zhang, B.; Kim, C.H.; Qiu, Y.Z.; Du, Q.S.; Mei, L.; Xiong, W.C. Regulation of heterochromatin remodelling and myogenin expression during muscle differentiation by FAK interaction with MBD2. Embo J. 2009, 28, 2568–2582. [Google Scholar] [CrossRef]
- Gnani, D.; Romito, I.; Artuso, S.; Chierici, M.; De Stefanis, C.; Panera, N.; Crudele, A.; Ceccarelli, S.; Carcarino, E.; D’Oria, V.; et al. Focal adhesion kinase depletion reduces human hepatocellular carcinoma growth by repressing enhancer of zeste homolog 2. Cell Death Differ. 2017, 24, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.T.; Wu, J.; Turnage, R.H. FAK induction in keratinocytes in an in vitro model of reepithelialization. J. Surg. Res. 2001, 96, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.E.; Guidry, J.T.; Scott, M.L.; Zwolinska, K.; Raikhy, G.; Prasai, K.; Bienkowska-Haba, M.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Detecting episomal or integrated human papillomavirus 16 DNA using an exonuclease V-qPCR-based assay. Virology 2019, 537, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004, 78, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Pastrana, D.V.; Lowy, D.R.; Schiller, J.T. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 2005, 119, 445–462. [Google Scholar] [CrossRef]
- Buck, C.B.; Thompson, C.D.; Pang, Y.Y.; Lowy, D.R.; Schiller, J.T. Maturation of papillomavirus capsids. J. Virol. 2005, 79, 2839–2846. [Google Scholar] [CrossRef]
- Brendle, S.; Cladel, N.; Balogh, K.; Alam, S.; Christensen, N.; Meyers, C.; Hu, J. A Comparative Study on Delivery of Externally Attached DNA by Papillomavirus VLPs and Pseudoviruses. Vaccines 2021, 9, 1501. [Google Scholar] [CrossRef]
- Malboeuf, C.M.; Simon, D.A.; Lee, Y.E.; Lankes, H.A.; Dewhurst, S.; Frelinger, J.G.; Rose, R.C. Human papillomavirus-like particles mediate functional delivery of plasmid DNA to antigen presenting cells in vivo. Vaccine 2007, 25, 3270–3276. [Google Scholar] [CrossRef]
- Abban, C.Y.; Meneses, P.I. Usage of heparan sulfate, integrins, and FAK in HPV16 infection. Virology 2010, 403, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, E.Y.; Meneses, P.I. A Dual Role for the Nonreceptor Tyrosine Kinase Pyk2 during the Intracellular Trafficking of Human Papillomavirus 16. J. Virol. 2015, 89, 9103–9114. [Google Scholar] [CrossRef] [PubMed]
- Giannone, G.; Rondé, P.; Gaire, M.; Beaudouin, J.; Haiech, J.; Ellenberg, J.; Takeda, K. Calcium rises locally trigger focal adhesion disassembly and enhance residency of focal adhesion kinase at focal adhesions. J. Biol. Chem. 2004, 279, 28715–28723. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, X.; Yang, S. Clinical significance of focal adhesion kinase (FAK) in cervical cancer progression and metastasis. Int. J. Clin. Exp. Pathol. 2020, 13, 2586–2592. [Google Scholar] [PubMed]
- Schneider-Maunoury, S.; Croissant, O.; Orth, G. Integration of human papillomavirus type 16 DNA sequences: A possible early event in the progression of genital tumors. J. Virol. 1987, 61, 3295–3298. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Haugen, T.H.; Turk, J.P.; Tabatabai, F.; Schmid, P.G., 3rd; Dürst, M.; Gissmann, L.; Roman, A.; Turek, L.P. Transcriptional regulation of the human papillomavirus-16 E6-E7 promoter by a keratinocyte-dependent enhancer, and by viral E2 trans-activator and repressor gene products: Implications for cervical carcinogenesis. Embo J. 1987, 6, 3745–3753. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Ono, T.; Ishimoto, A.; Dowhanick, J.J.; Frizzell, M.A.; Howley, P.M.; Sakai, H. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J. Virol. 2000, 74, 3752–3760. [Google Scholar] [CrossRef]
- Ozbun, M.A. Human papillomavirus type 31b infection of human keratinocytes and the onset of early transcription. J. Virol. 2002, 76, 11291–11300. [Google Scholar] [CrossRef]
- McKinney, C.C.; Kim, M.J.; Chen, D.; McBride, A.A. Brd4 Activates Early Viral Transcription upon Human Papillomavirus 18 Infection of Primary Keratinocytes. mBio 2016, 7, e01644-16. [Google Scholar] [CrossRef]
- Riggs, D.; Yang, Z.; Kloss, J.; Loftus, J.C. The Pyk2 FERM regulates Pyk2 complex formation and phosphorylation. Cell. Signal. 2011, 23, 288–296. [Google Scholar] [CrossRef]
- Gu, J.; Tamura, M.; Pankov, R.; Danen, E.H.; Takino, T.; Matsumoto, K.; Yamada, K.M. Shc and FAK differentially regulate cell motility and directionality modulated by PTEN. J. Cell Biol. 1999, 146, 389–403. [Google Scholar] [CrossRef]
- Siddiqa, A.; Léon, K.C.; James, C.D.; Bhatti, M.F.; Roberts, S.; Parish, J.L. The human papillomavirus type 16 L1 protein directly interacts with E2 and enhances E2-dependent replication and transcription activation. J. Gen. Virol. 2015, 96, 2274–2285. [Google Scholar] [CrossRef]
- Jose, L.; Androphy, E.J.; DeSmet, M. Phosphorylation of the Human Papillomavirus E2 Protein at Tyrosine 138 Regulates Episomal Replication. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Van Doorslaer, K.; Porter, S.; McKinney, C.; Stepp, W.H.; McBride, A.A. Novel recombinant papillomavirus genomes expressing selectable genes. Sci. Rep. 2016, 6, 37782. [Google Scholar] [CrossRef]
- Gilson, T.D.; Gibson, R.T.; Androphy, E.J. Optimization of human papillomavirus-based pseudovirus techniques for efficient gene transfer. Sci. Rep. 2020, 10, 15517. [Google Scholar] [CrossRef]
- Xie, F.; DeSmet, M.; Kanginakudru, S.; Jose, L.; Culleton, S.P.; Gilson, T.; Li, C.; Androphy, E.J. Kinase Activity of Fibroblast Growth Factor Receptor 3 Regulates Activity of the Papillomavirus E2 Protein. J. Virol. 2017, 91, e01066-17. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jose, L.; Gonzalez, J.; Kessinger, E.; Androphy, E.J.; DeSmet, M. Focal Adhesion Kinase Binds to the HPV E2 Protein to Regulate Initial Replication after Infection. Pathogens 2023, 12, 1203. https://doi.org/10.3390/pathogens12101203
Jose L, Gonzalez J, Kessinger E, Androphy EJ, DeSmet M. Focal Adhesion Kinase Binds to the HPV E2 Protein to Regulate Initial Replication after Infection. Pathogens. 2023; 12(10):1203. https://doi.org/10.3390/pathogens12101203
Chicago/Turabian StyleJose, Leny, Jessica Gonzalez, Emma Kessinger, Elliot J. Androphy, and Marsha DeSmet. 2023. "Focal Adhesion Kinase Binds to the HPV E2 Protein to Regulate Initial Replication after Infection" Pathogens 12, no. 10: 1203. https://doi.org/10.3390/pathogens12101203
APA StyleJose, L., Gonzalez, J., Kessinger, E., Androphy, E. J., & DeSmet, M. (2023). Focal Adhesion Kinase Binds to the HPV E2 Protein to Regulate Initial Replication after Infection. Pathogens, 12(10), 1203. https://doi.org/10.3390/pathogens12101203