1. Introduction
The spread of infections resistant to available antimicrobial drugs is a major threat to human health [
1]. According to the WHO, in less than 30 years, microbial resistance could become the leading cause of death. To avoid microbial resistance, the identification of new targets and innovative drugs with new modes of action that are able to evade resistance mechanisms may represent a valid solution to counteract the continuous emergence and spread of resistant infections [
2] Mapping the spread of resistance, the development of rapid diagnostics to ensure early selection of appropriate therapies, incentives for discovery of new agents, and studies of therapeutic pathways are alternatives to minimize cases of resistance [
3].
Triterpenoids are one of the largest subclasses of terpenoids, with over 14,000 known structures [
4]. These structures are cyclized from squalene precursors oxidized by oxidosqualene cyclases, with wide structural diversity
4. Most triterpenes, predominantly pentacyclics, are widely distributed in plants—in seeds, stem bark, roots, leaves, or the wax-like coating of various fruits and herbs such as thyme, mistletoe, or lavender [
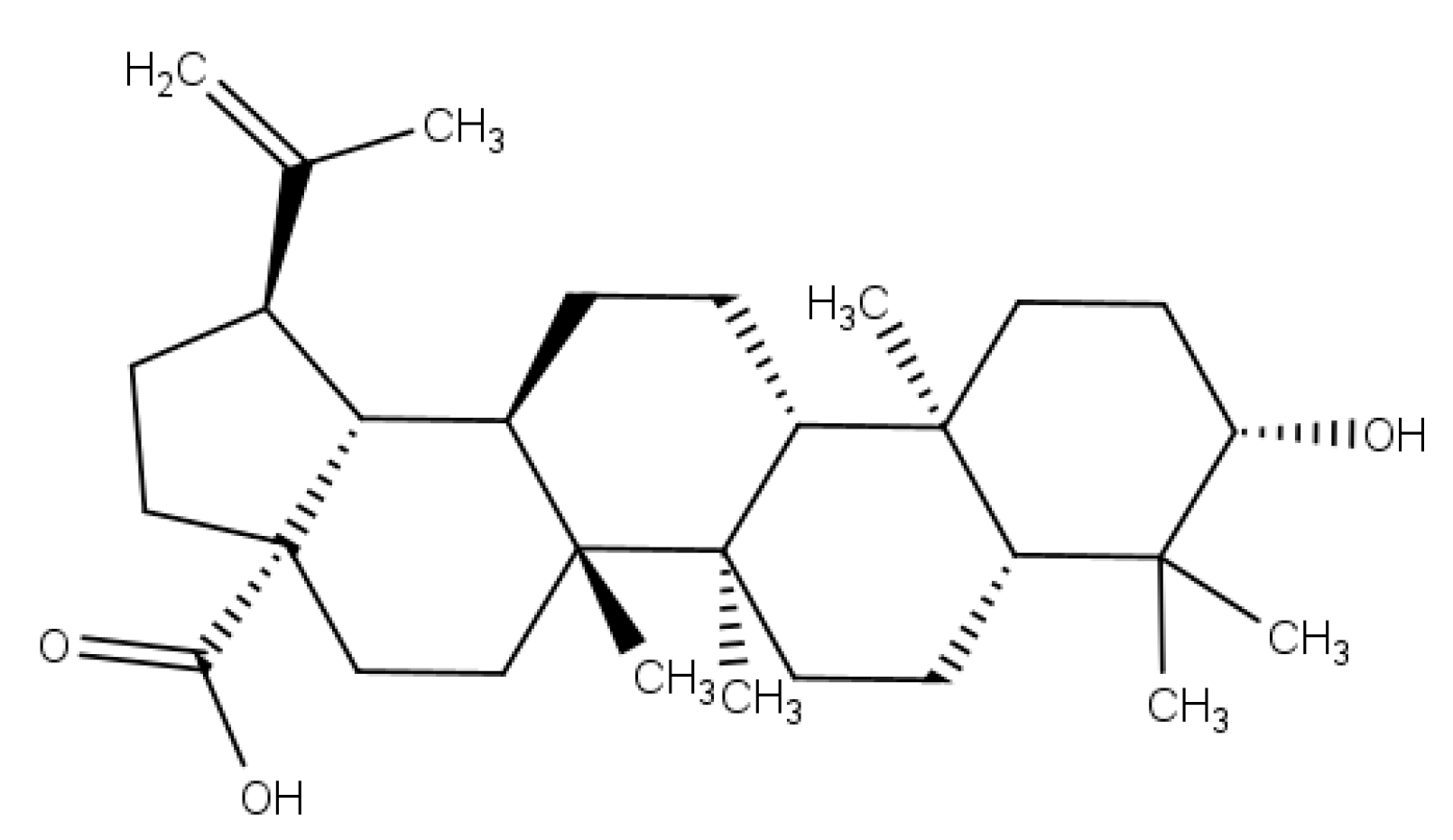
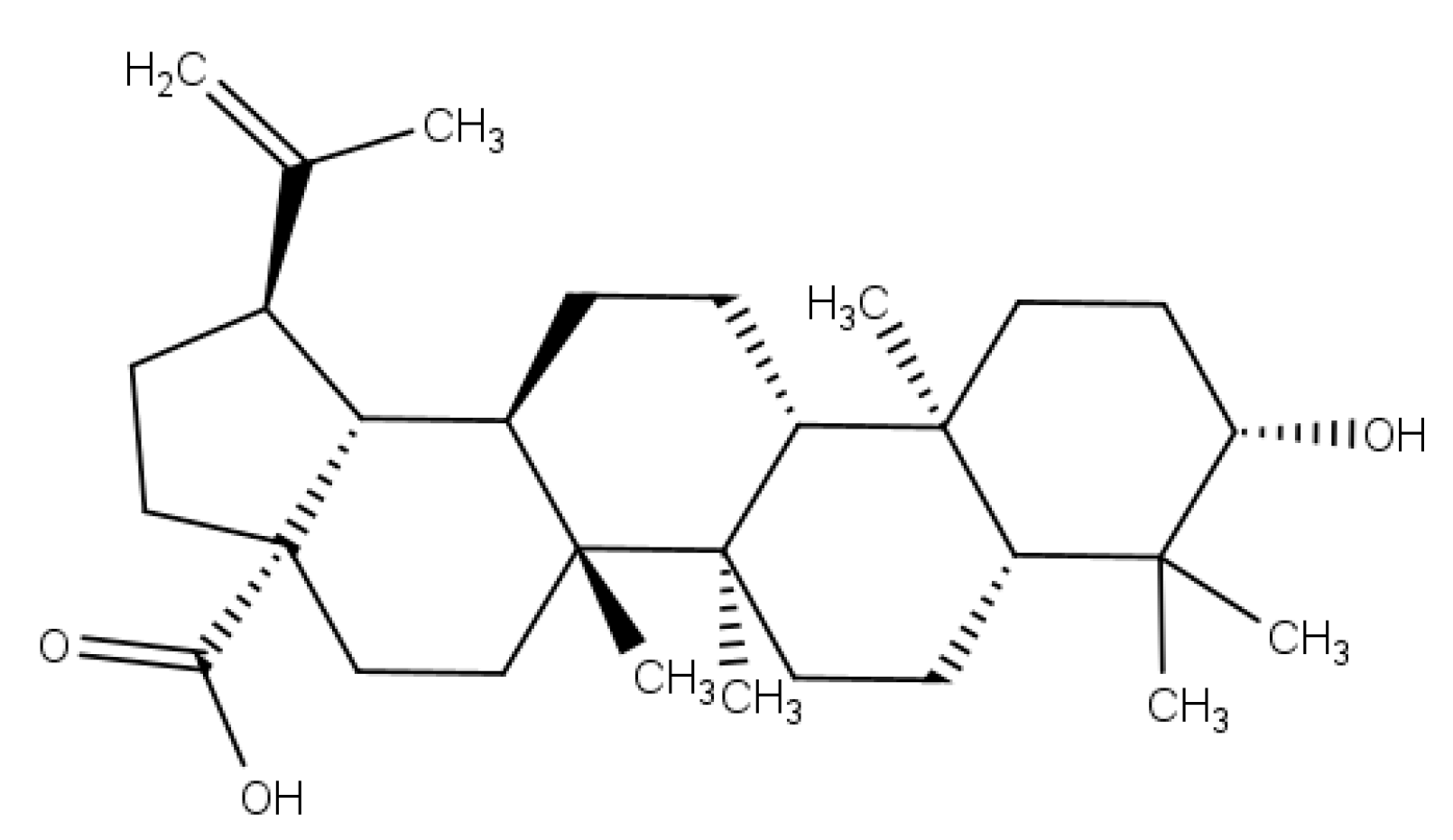
5]. Betulinic acid (BA) (
Figure 1) is a lupane-type pentacyclic triterpenoid that has a variety of medicinal properties [
6]. BA has gained prominence and considerable research interest because of its potent physiological and pharmacological activities [
7]. It has been reported to have a variety of effects, including antitumor [
8], anti-inflammatory [
9], anti-HIV [
10], antidiabetic [
11], antibacterial [
12], antiviral [
13], and antimalarial [
14] activities, among others. BA can be isolated from several plants, such as
Quisqualis fructus,
Coussarea paniculata,
Caesalpinia paraguariensis,
Vitex negundo,
Ilex macropoda,
Anemone raddeana, Doliocarpus schottianus,
Tovomita krukovii,
Chaenomeles lagenaria,
Berlinia grandiflora Vietnamese,
Orthosiphon stamineus, and
Eucalyptus, among others [
15].
Phytochemicals, due to their various pharmacological properties, can act against bacterial and fungal resistance, as reported in several studies [
16,
17]. These agents can act alone or in combination with antibiotics to increase antibacterial activity against a wide range of bacteria [
18]. As for antifungal agents, although new mechanisms of action are at various stages of clinical evolution, their number is relatively small compared to other diseases [
19]. Consideration of the common structural features distinguishing fungi should facilitate the development of antifungal drugs with broad-spectrum activity, and the outcome of systemic fungal infections strongly depends on how quickly treatment is initiated [
20].
Most compounds used in infectious disease treatments are either natural or semi-synthetically modified to improve efficacy by targeting the engineering of natural products into potent antibiotics [
21]. Therefore, it is necessary to screen natural products that may be new therapeutic alternatives for the treatment of infectious diseases [
17]. Among all available options, plant-derived compounds have shown the most potential applications in fighting bacterial and antifungal infections [
18].
According to Matamoros-Recio et al. [
22], it is very difficult to study microorganism structures at the molecular level by experimental techniques, despite the significant development of fast and efficient experimental protocols. Due to the various cases of resistance, it is important to study new routes and new targets and develop new compounds capable of overcoming the factors that contribute to antimicrobial resistance. Therefore, the authors emphasize that computational methods allow the resolution of atomic structures and reveal molecular aspects (interatomic and intermolecular interactions) not accessible by any experimental microscopy. This information significantly contributes to reducing costs in the production of antimicrobial drugs that have high potential. Computational methods contribute to the discovery of more selective, potent, and efficient drugs. Thus, our study sought to investigate the potential of BA against various bacterial and fungal strains through in vitro activity, as well as molecular-level analysis of the likely mechanisms of action through computational studies.
3. Results
3.1. QSAR Modeling
The predictive model against
M. tuberculosis was validated as reported by Cavalcanti et al. [
25]. The predictive model against
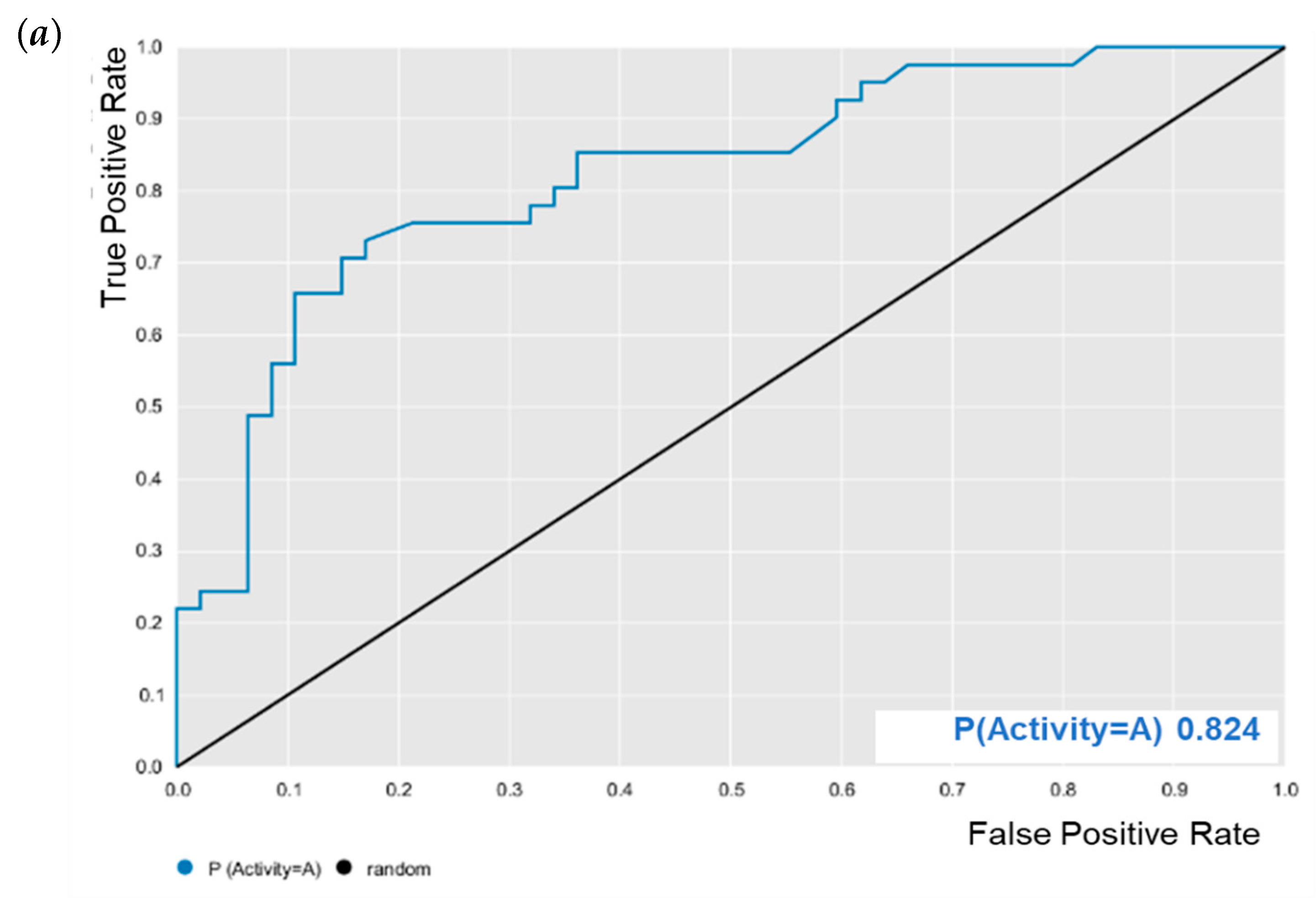
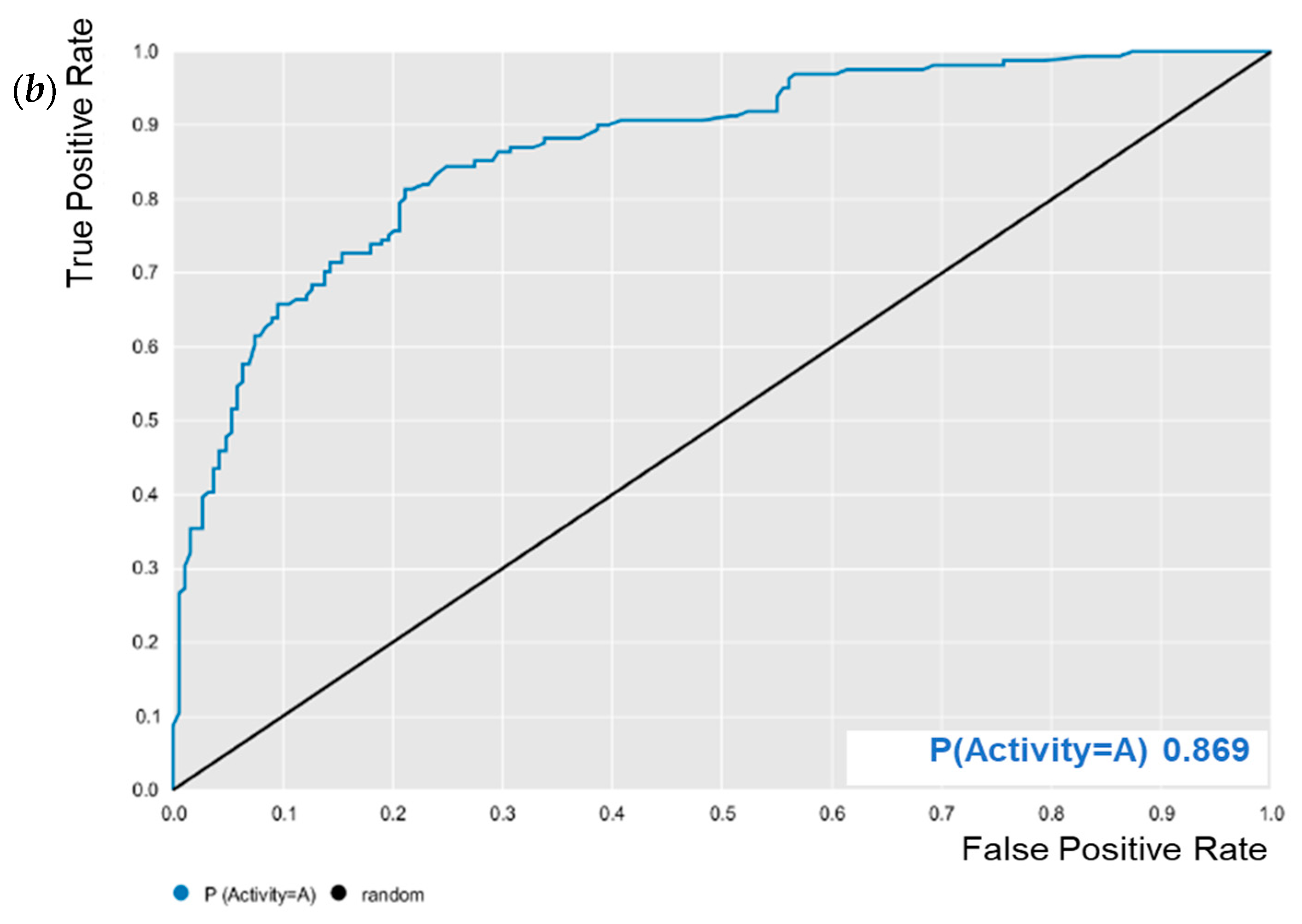
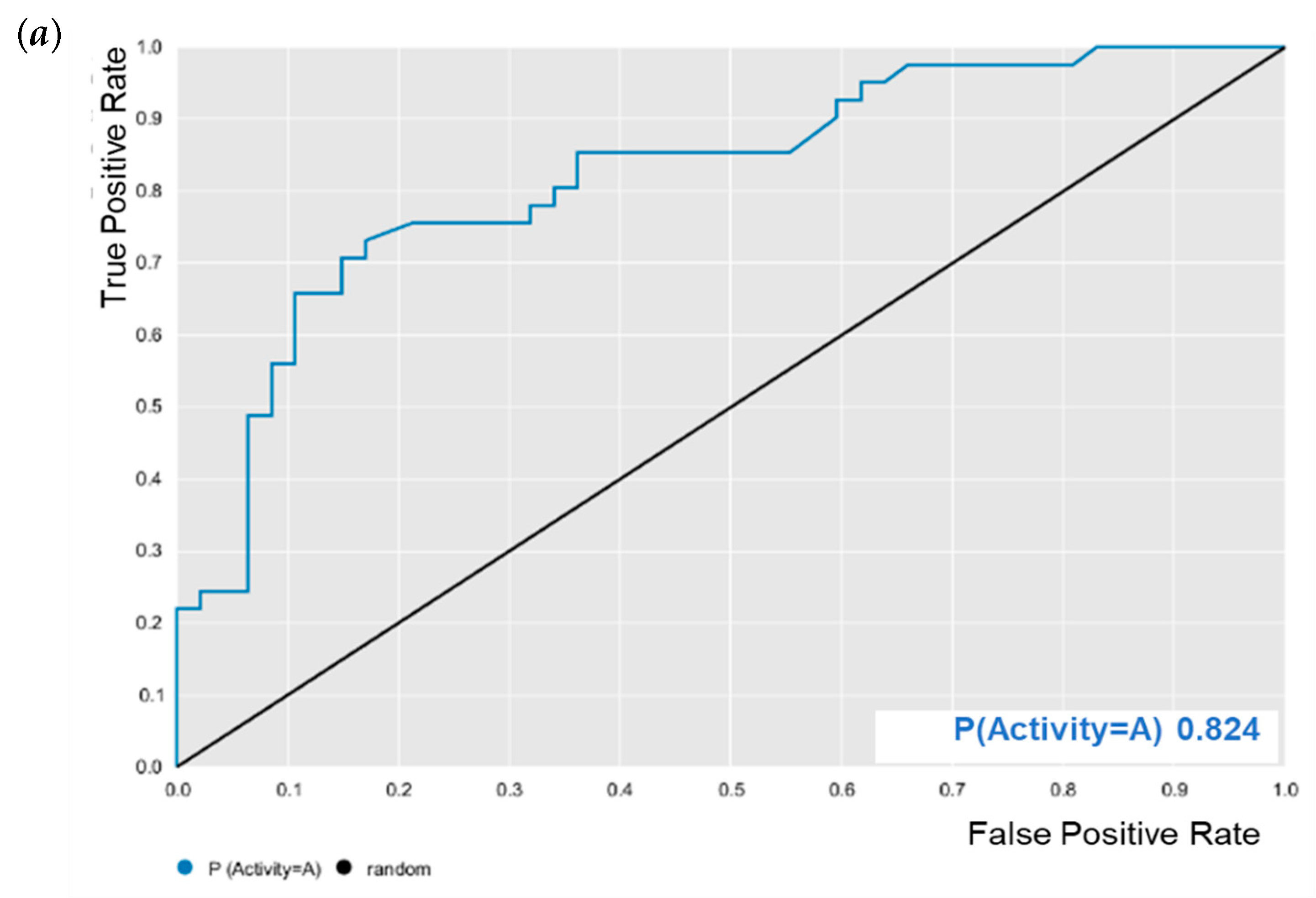
E. coli was evaluated for its predictive powers. The results showed excellent model performance, as represented by the cross-validation values: accuracy = 0.78; specificity = 0.82; sensitivity = 0.72; PPV = 0.78; NPV = 0.78; ROC = 0.86 (
Figure 2); and MCC = 0.55. After validation, BA was subjected to predictive analysis in the built models. The results showed that BA was within the chemical domain and presented biological activity of 63% against
M. tuberculosis, that is, the potential to inhibit the growth of
M. tuberculosis. While BA showed 65% biological activity against
E. coli.
3.2. Antimicrobial Activity Tests
BA at a concentration of 561 µM caused the growth inhibition of 9/12 (75%) microbial species including bacteria and fungi, and the respective MIC was then considered. In parallel, the controls also presented MIC at a concentration of 561 µM in the proportion of 50%/bacteria and 75%/fungi. In general, the MBC and MFC of the products were between 561 and 1122 µM, except for M. tuberculosis. The MIC value of BA against M. tuberculosis was 100 µM. The MIC values for the controls were: MIC = 0.19 µM for moxifloxacin and MIC = 0.15 µM for rifampin. Therefore, the investigated compound had inhibitory activity against M. tuberculosis but was not potent when compared to controls.
The mode of action of the test substance was also evaluated through the MBC/MIC ratio, a methodology used by Hafidh [
39] to specify the nature of the antimicrobial effect, considered as bactericidal and fungicidal when the MBC/MIC and MFC/MIC ratio is between 1:1 and 2:1. On the other hand, if the ratio is greater than 2:1, the mode of action is more likely to be bacteriostatic and fungistatic. The results showed that BA had a bacteriostatic effect against
E. coli and no fungistatic effect for the investigated strains.
3.3. Alignment of Protein Sequences
The alignment of protein sequences contributed to identifying the conserved regions and common residues of the active site in enzymes from selected species. Initially, DNA gyrase protein sequences were evaluated among all bacterial species investigated in this study. It was observed through the alignment that bacterial DNA gyrases share 45.39 to 88.26% identity with each other (
Figure S1). DNA gyrases from
S. aureus and
S. epidermidis share the highest degree of identity at 88.26%. C-terminal regions with low identity that were not part of the active site were excluded from the alignment. According to Broeck et al. [
48], the amino acids Val70, Ile74, Asp88, and Met120 are part of the active site of
E. coli DNA gyrase. We can see from
Figure S2 that 50% of the site’s amino acids are conserved in all aligned sequences. For the beta-lactamase enzyme, the alignment showed identity of 14.66 to 95.38% (
Figure S2). Only bacteria belonging to the same genus of Staphylococcus showed a higher degree of identity between the aligned sequences (95.38%), while most showed a low degree of identity, evidencing the need to design selective inhibitors for each type of bacterial strain. C- and N-terminal regions with low identity were excluded. The alignment of PBP protein sequences showed a degree of identity ranging from 19.31% to 82.63%, with the highest degree of identity remaining for the
Staphylococcus genus (
Figure S3). These results suggest that the DNA gyrase enzyme is the most conserved among the analyzed proteins and that it is necessary to develop selective inhibitors based on the knowledge of the target structure. In addition, the alignment allowed the identification of conserved regions of
S. epidermidis proteins.
Among the enzymes selected for the fungal species investigated in this study, we noticed, through the protein sequence alignment analyses, that the three enzymes present a greater degree of identity between the species. This is due to the fact that most of the analyzed species belong to the same genus,
Candida. The aligned sequences of the CYP51 enzyme showed a degree of identity ranging from 47.14 to 82.95%. Among species of the same genus, the results showed that the CYP51 enzyme from
C. glabrata shares 64.30% identity with
C. albicans and 63.92% with
C. tropicalis. These values are considered low for species of the same genus, showing that there is a need to develop drugs for the target of each species. Moreover,
C. albicans shares 82.95% of its amino acids with
C. tropicalis (
Figure S4). The results also showed that most active site amino acids are conserved across species. Among
Candida species, we observed two active site amino acid substitutions. The residues Phe58 and Ala62 in
C. albicans and
C. tropicalis are replaced by Val and Ile in
C. glabrata, respectively. According to Hargrove et al. [
20],
C. glabrata is more resistant to the drug fluconazole, and small substitutions and/or mutations in the active site can confer drug resistance.
Due to the lack of genomic sequencing data for the SAP-2 enzyme for most of the fungal species in this study, it was possible to align only the sequences of
C. albicans and
C. tropicalis. The results showed that both share only 47.21% identity (
Figure S5). These data indicate that the SAP-2 enzyme of the two species are sequentially quite different. Therefore, the development of inhibitors for these enzymes may have different effects for both species. According to Cultffiel et al. [
49], the A70450 inhibitor binds to SAP2 20 times stronger than pepstatin, and the amino acids Ile82, Tyr84, Gly85, Asp86 and Ser88 make many contacts along the inhibitor.
The alignment results for the DHFR enzyme showed that the sequences share between 27.84 and 81.25% identity.
Trichophyton rubrum and
A. flavus are the species with the enzyme sequences with the lowest identity when aligned with the other species. Another interesting result is that the C. glabrata DHFR shares only 47.37% of residues with
C. albicans and 45.03% of residues with
C. tropicalis (
Figure S6). However,
C. albicans and
C. tropicalis share 81.25% of amino acids.
3.4. Modeling by Homology
In this study, seven homology models were constructed for the species: S. epidermidis (DNA gyrase, beta-lactamase, and PBP); C. tropicalis (SAP-2 and DHFR); A. flavus (CYP51); and T. rubrum (DHFR).
The Ramachandran diagram is one of the most used tools to evaluate the stereochemical quality of three-dimensional protein structures. The diagram represents all possible combinations of dihedral angles Ψ (psi) versus φ (phi) for each amino acid in a protein, with the exception of glycine, which has no side chains. Models that obtain more than 90% of the amino acids present in the allowed and/or favored regions (colored regions of the graph) are considered reliable (
Figure S7). White regions represent outliers, which have bad contacts. The results showed that all the models generated had more than 97% of amino acids in the allowed and favored regions (
Table S3). We also used the Verify 3D software to analyze the compatibility of the three-dimensional (3D) structure with its one-dimensional (1D) amino acid sequence. The results showed that all models had a 3D-1D score of more than 80% (
Table S3), which is considered ideal for an optimal structure.
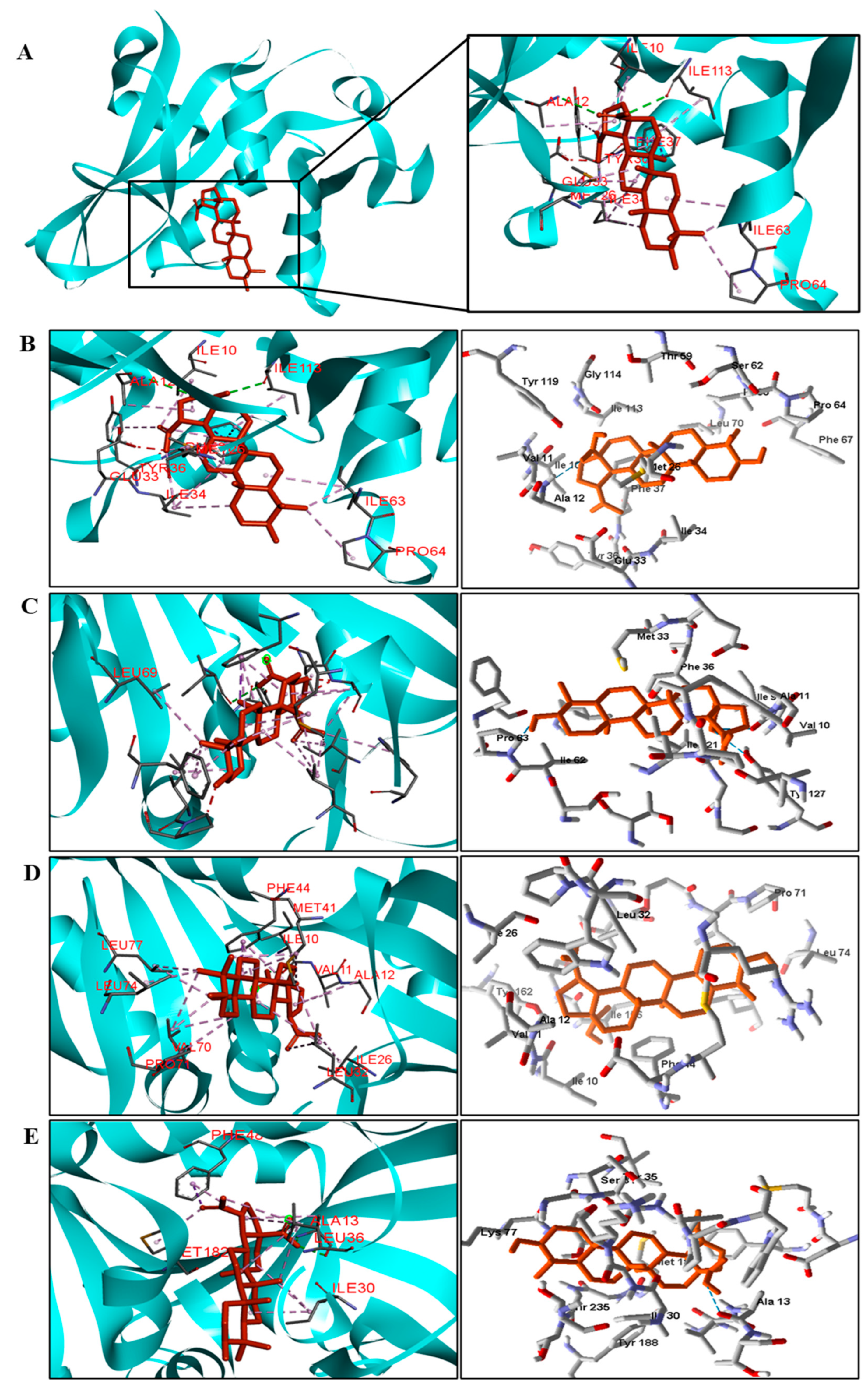
3.5. Molecular Docking
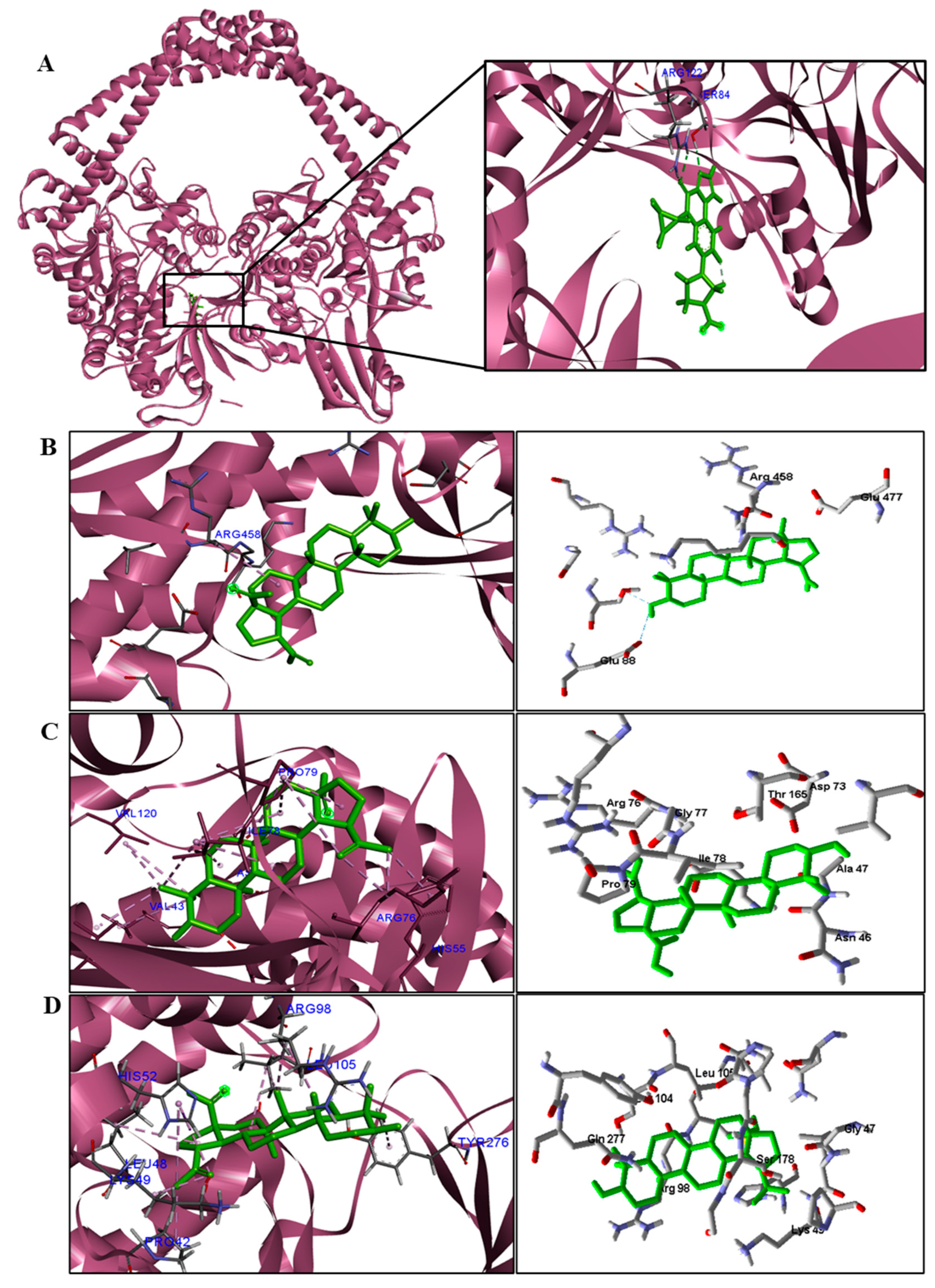
BA was subjected to molecular docking in six types of enzymes to investigate antimicrobial activity against bacteria and fungi. The docking results were generated using the MolDock score function. More negative values indicated better predictions. The protein in which the compound obtained binding energy values higher or close to the standard drug was considered promising for antimicrobial activity.
The generated docking results were validated by redocking the crystallographic ligand with all investigated proteins. The root mean square deviations (RMSDs) of the obtained fit poses were calculated in comparison with the crystal structure. RMSD values less than 2 Å indicate an optimal degree of screening reliability. Information about the starting structures and the results of the redocking validation is shown in
Table S4. The redocking analysis showed that all RMSD values were below 2.0 Å, that is, the generated poses positioned the ligand correctly in the active location. Therefore, the values were considered satisfactory for docking validation. Missing RMSD values are due to the lack of complexed ligands.
The docking results can be seen in
Table 5 and
Table 6. According to the results, of all proteins analyzed for the species of bacteria investigated, BA presented higher binding energy values than the standard drug for the three types of enzymes (
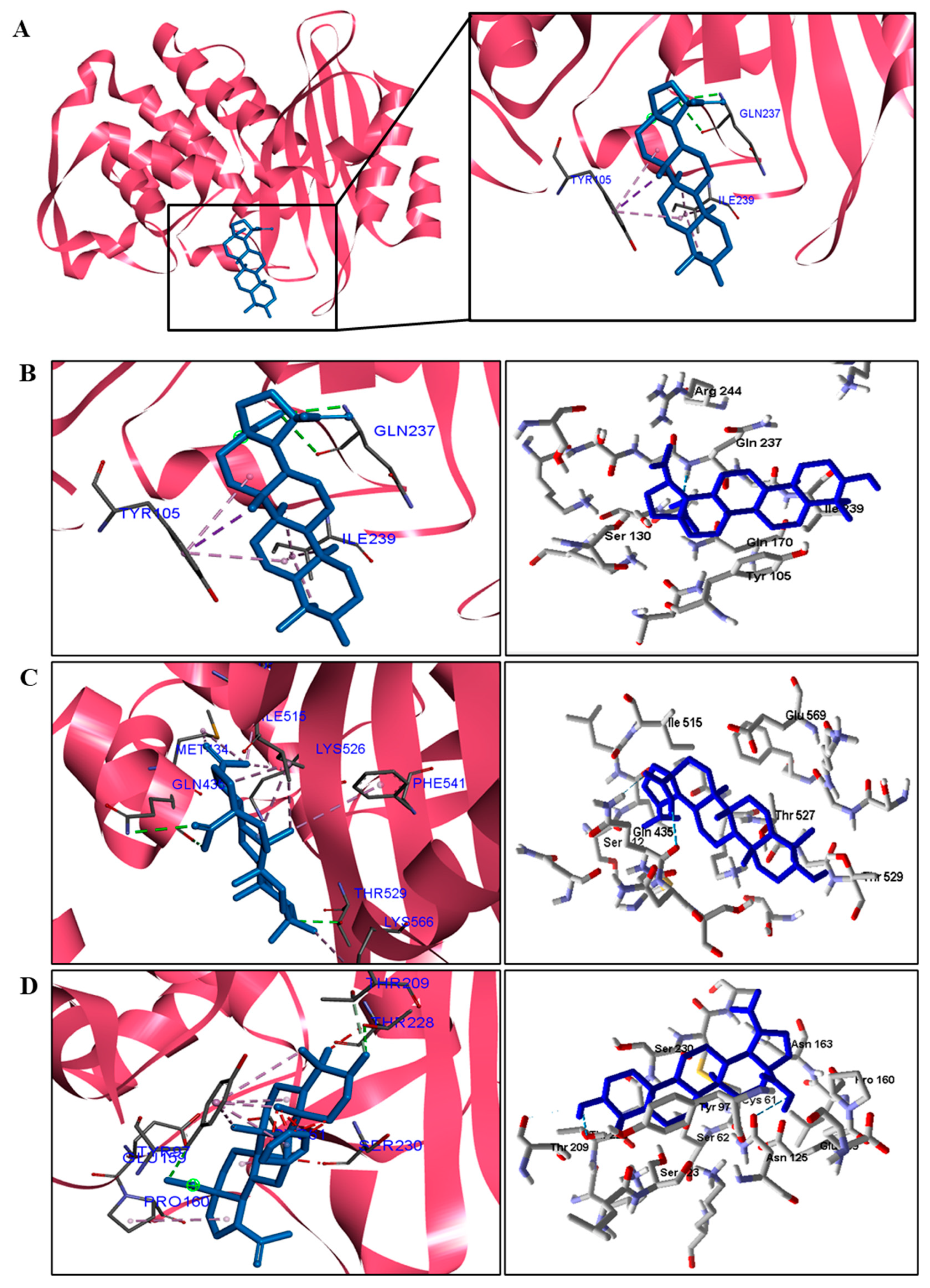
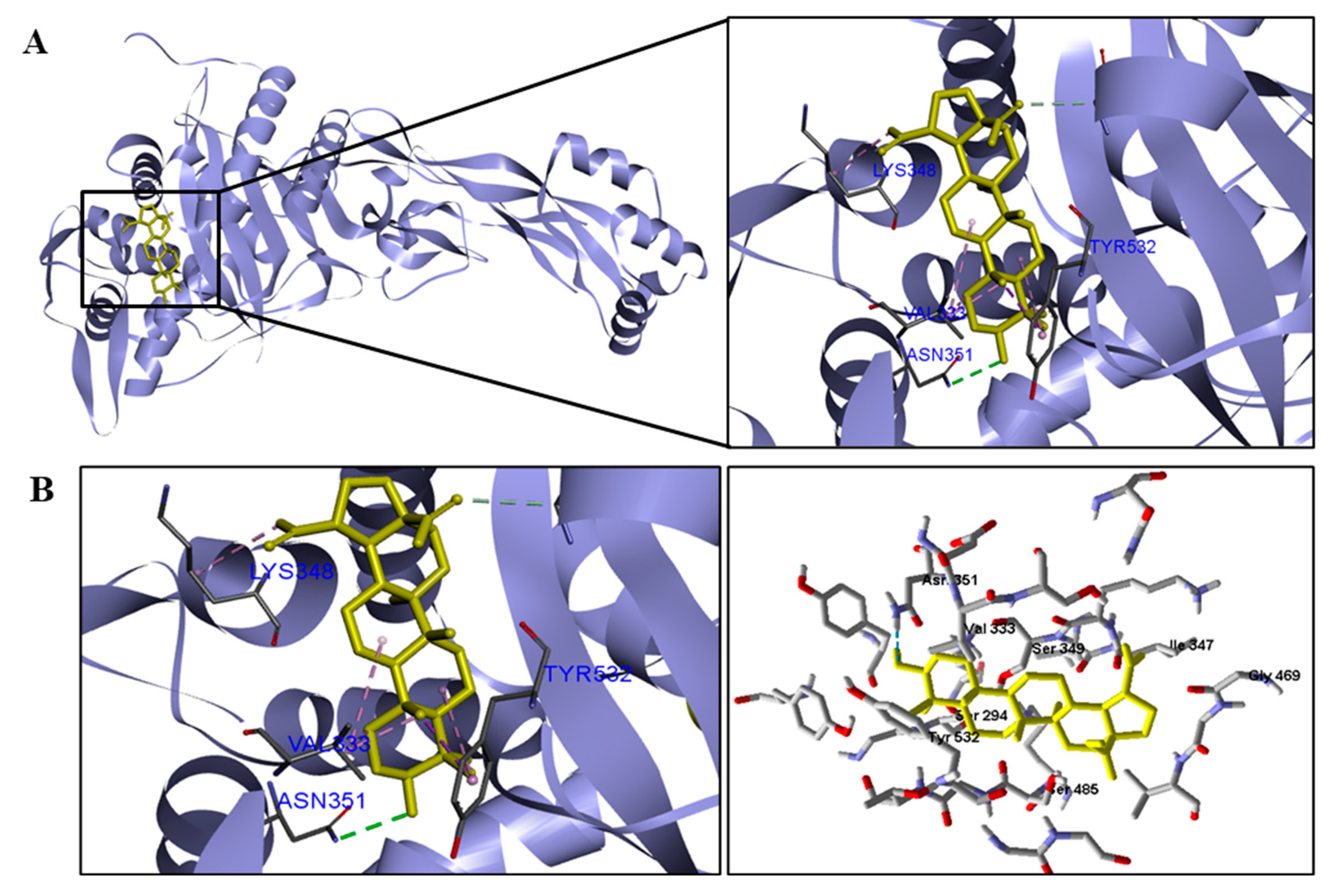
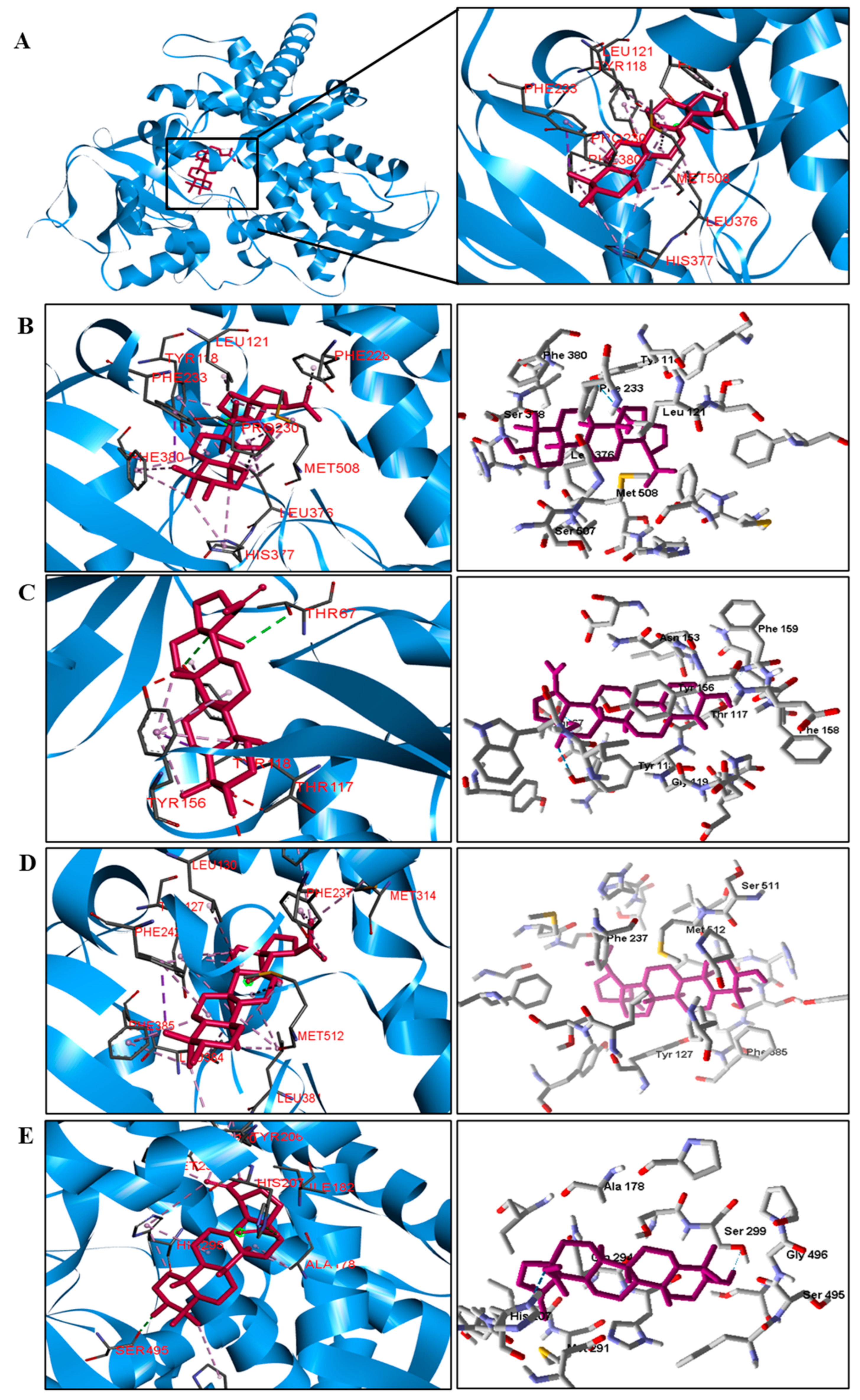
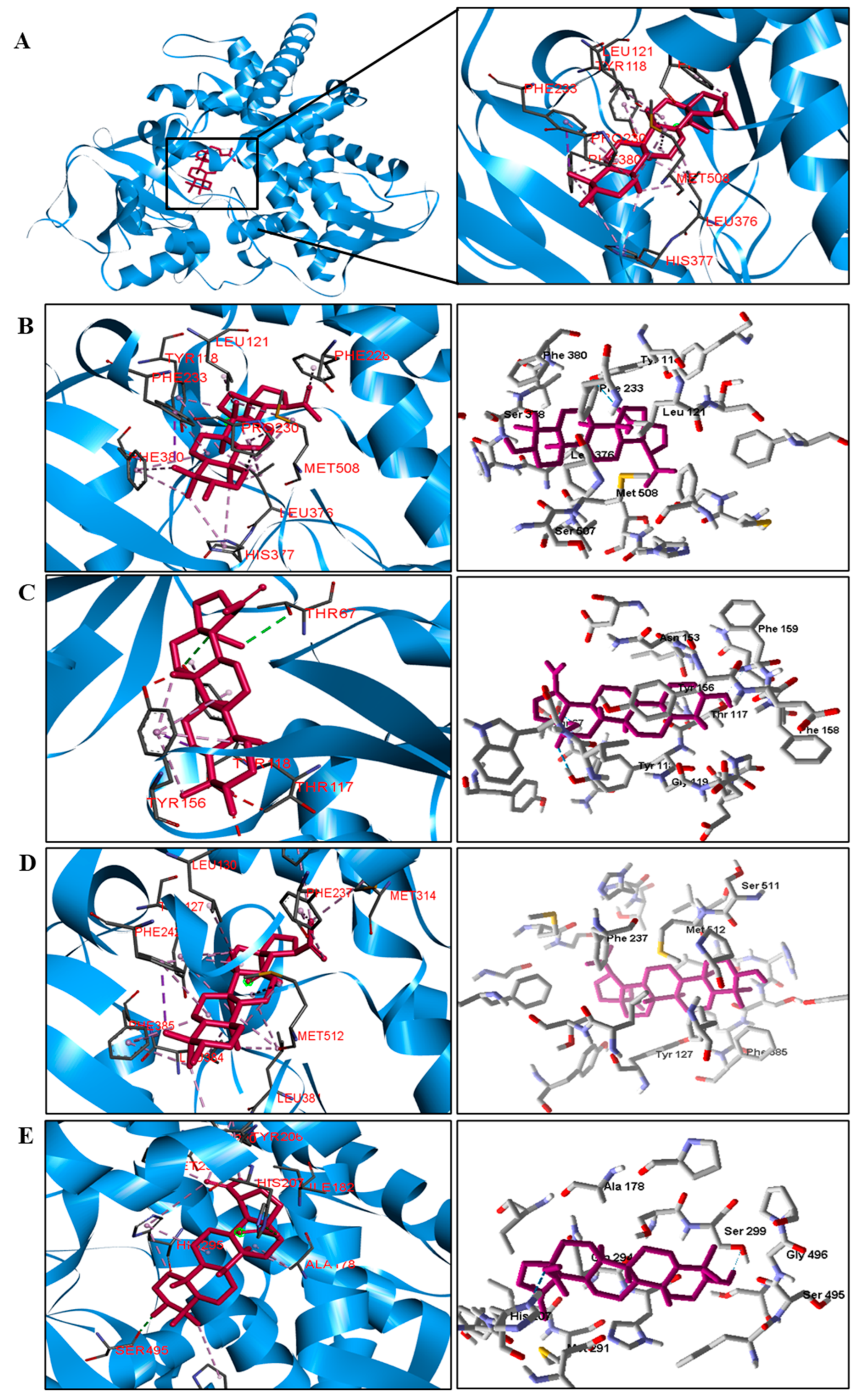
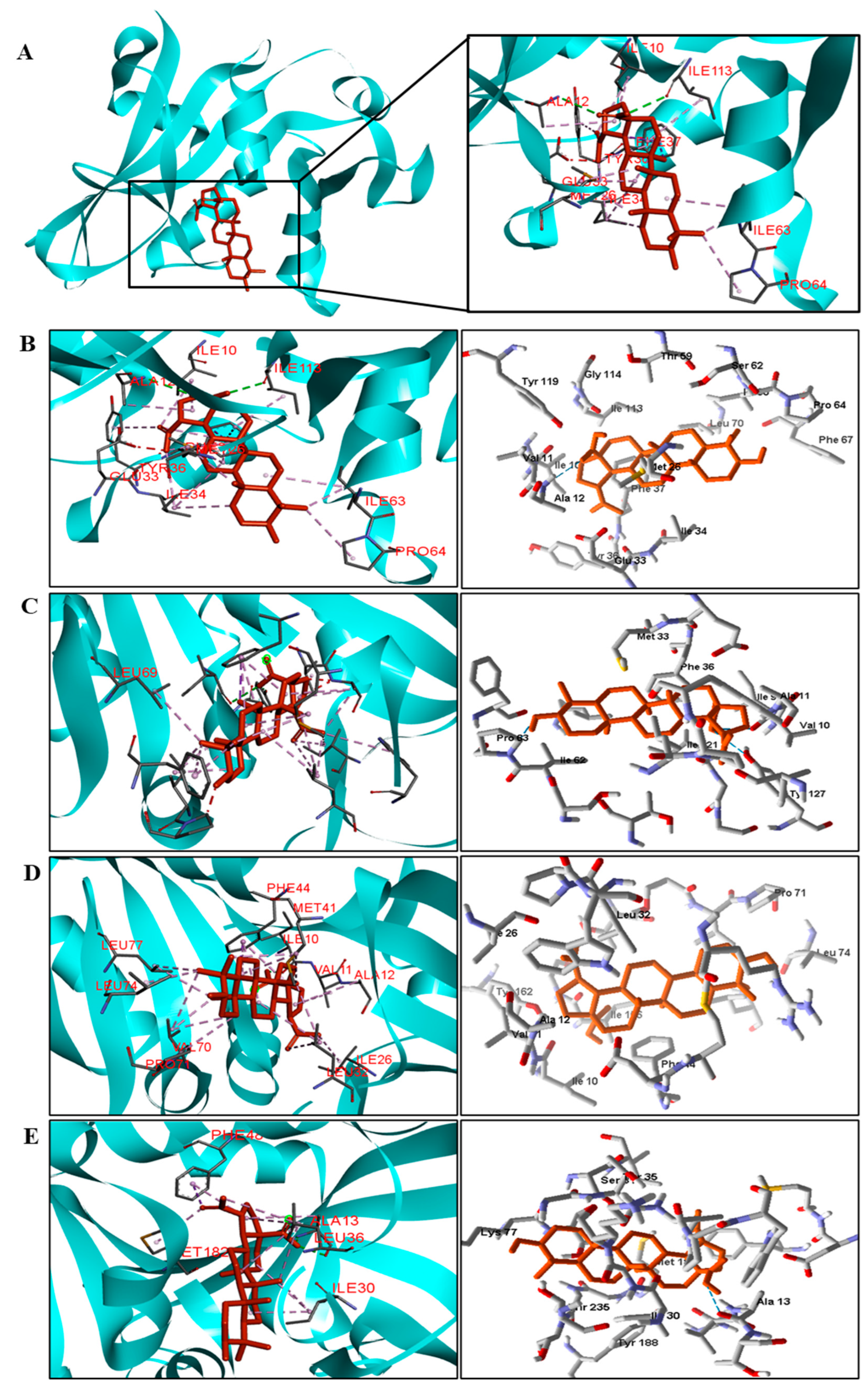
Table 5). We observed that the binding affinity of BA was better for the DNA gyrase and beta-lactamase enzymes. The beta-lactamase of all bacteria was able to present higher binding affinity values than the drug used as a control. According to these results, we suggest that BA has DNA gyrase and beta-lactamase as its mechanism of action.
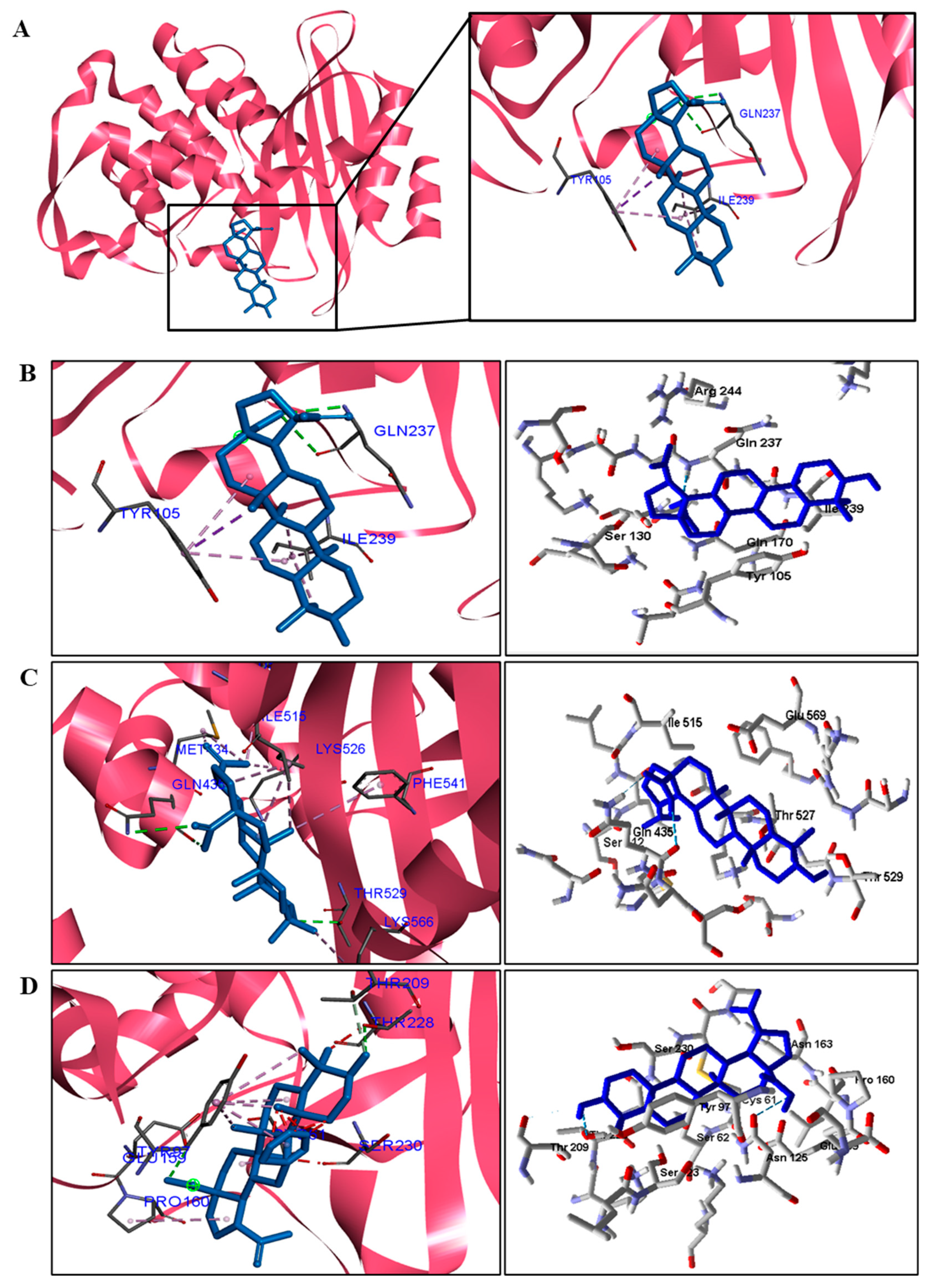
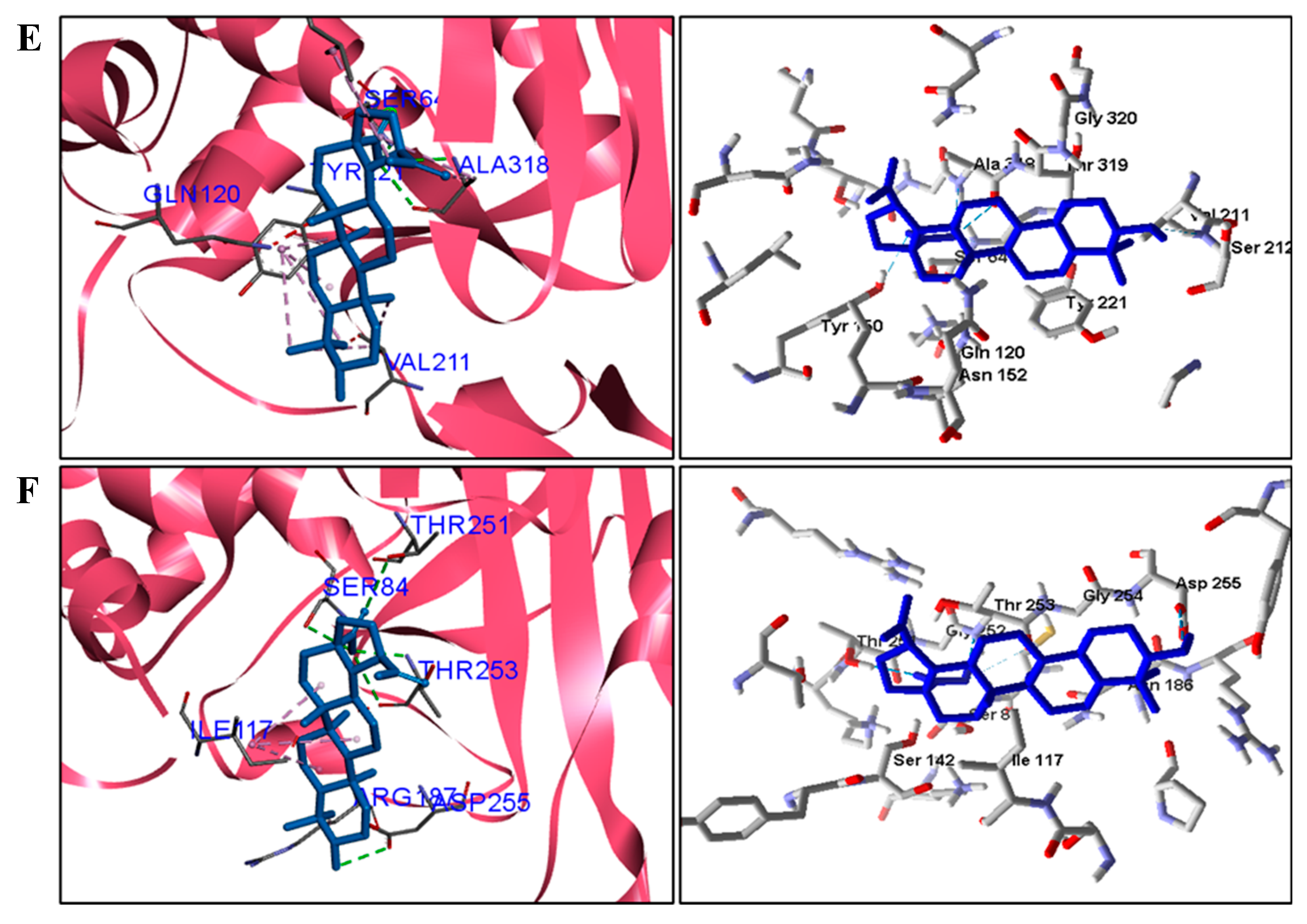
When analyzing the docking results for the fungal species, we observed that BA was able to interact with CYP and DHFR (
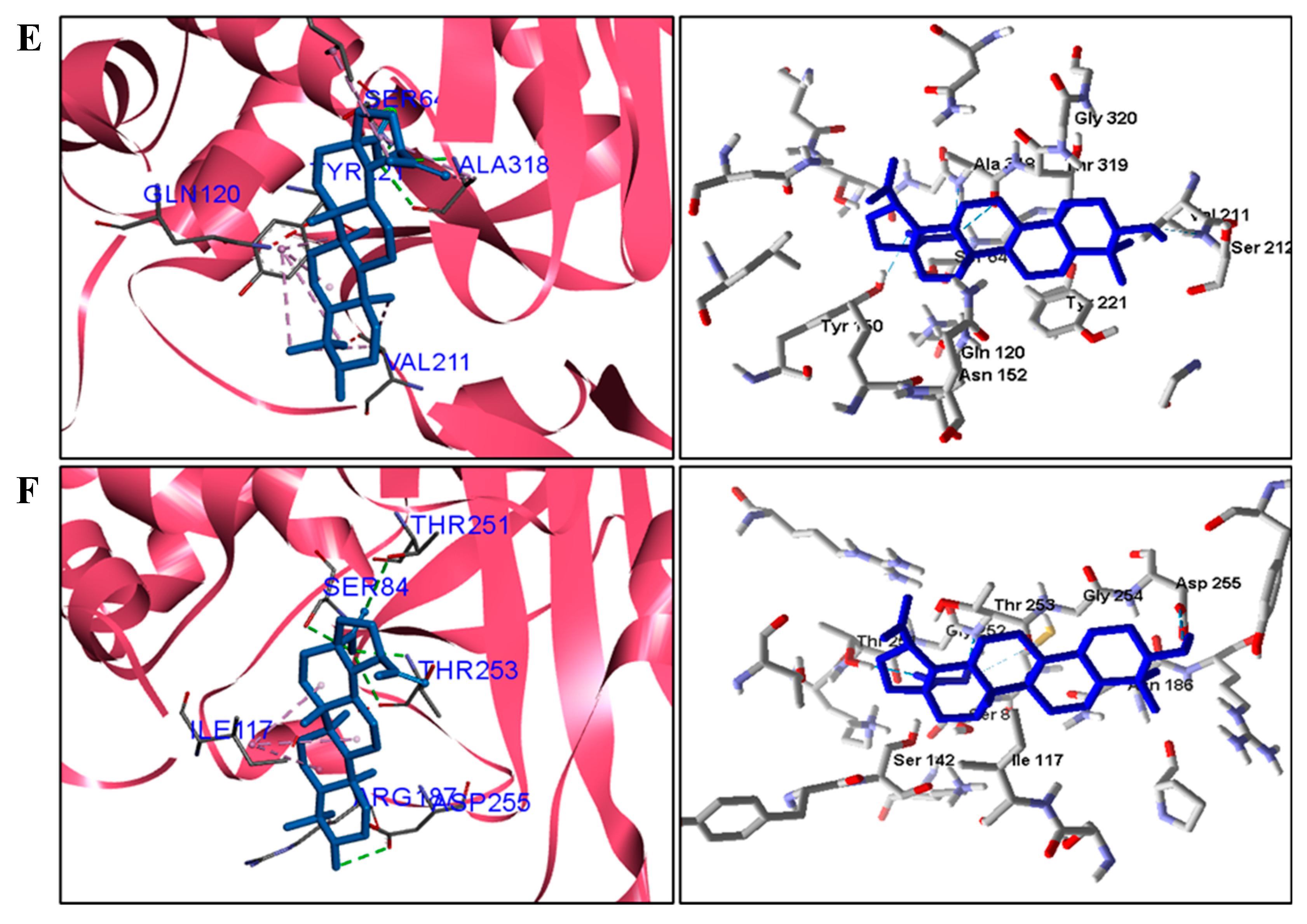
Table 6). BA showed higher binding energy values than the standard drug, except for the DHFR of
C. albicans. These results suggest that the mechanism of action of BA in fungi is on CYP51 and DHFR.
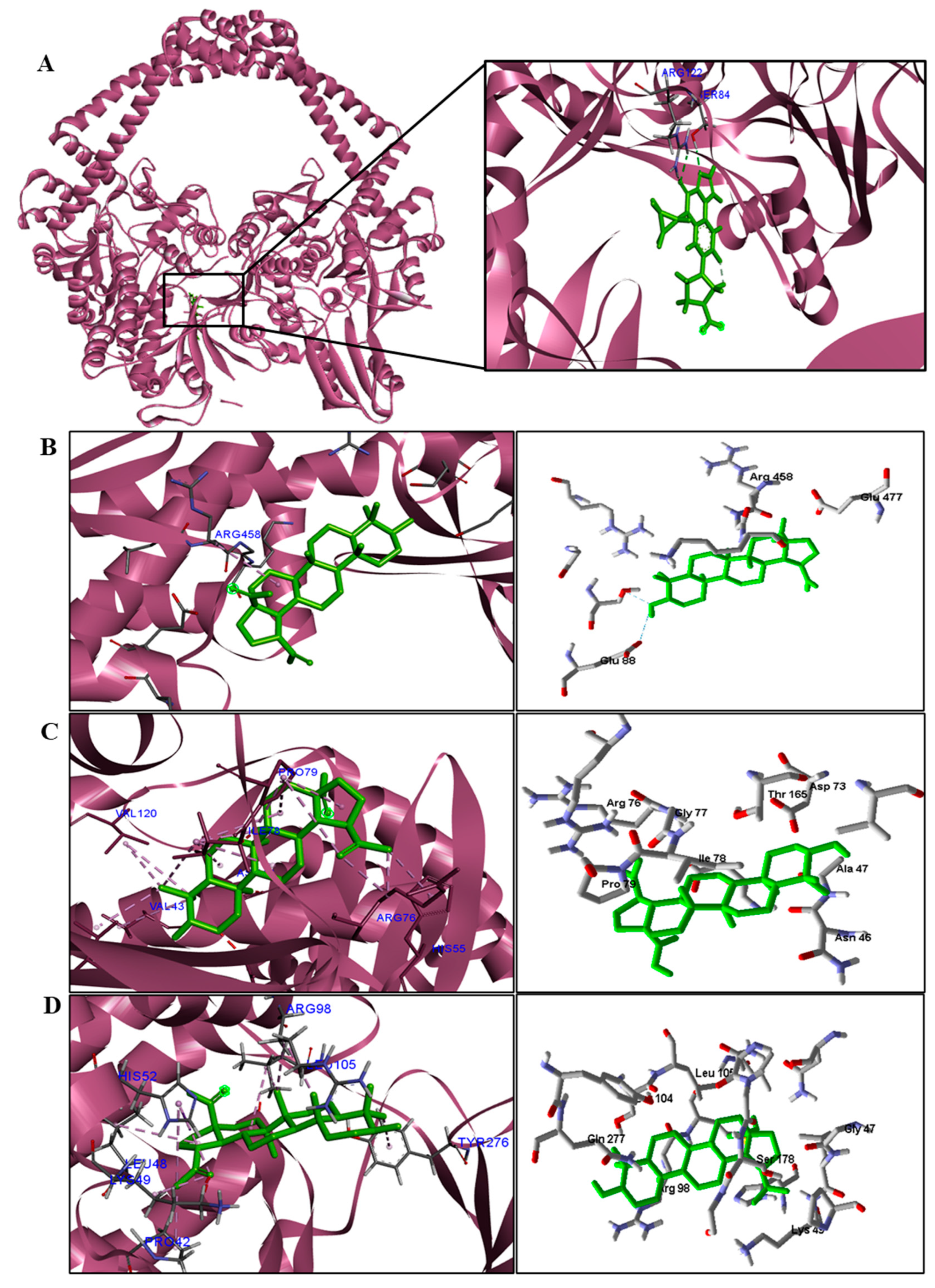
We analyzed in detail the interactions formed by BA with the proteins in which this compound obtained superior binding affinity compared to the standard drug. For the DNA gyrase enzyme, three species obtained excellent results. BA showed binding energy values of −109.22 kcal/mol−1 in S. aureus DNA gyrase, while the standard drug showed −98.76 kcal/mol−1. A hydrophobic interaction of BA with the amino acid Arg458 of the DNA gyrase active site was observed. The bacteria E. coli and M. tuberculosis also showed higher binding affinity values than the drug.
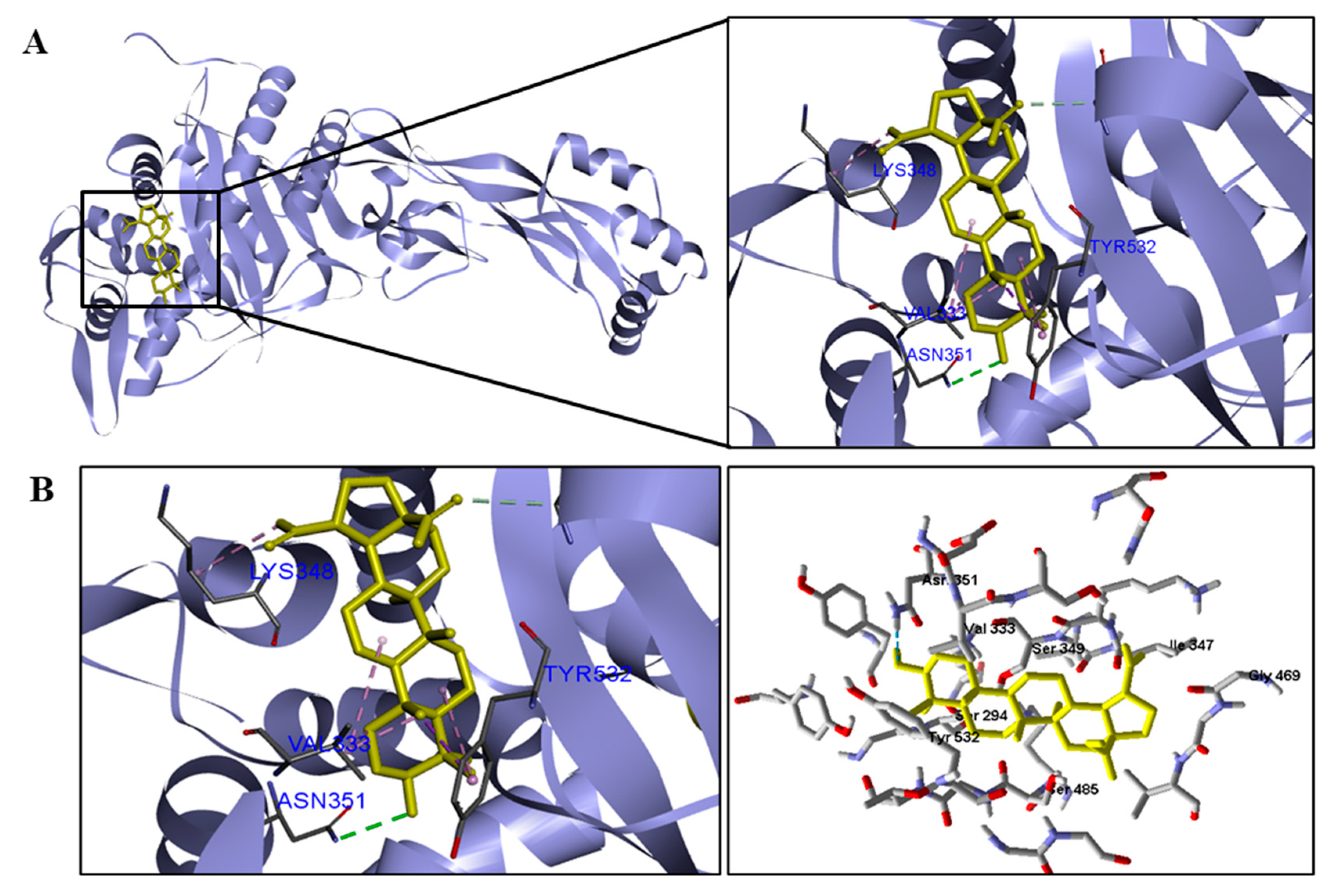
The docking results showed that BA presented higher binding energy values than the drug used as a control in all beta−lactamases of the investigated bacteria. We report here the bindings and interactions that contributed to the binding affinity of the compound with the active site of the enzymes. This was the only species among the investigated bacteria that obtained a binding energy value considered satisfactory for a probability of effect through this mechanism of action. The result showed that BA had a binding energy value (−107.59 kcal/mol−1) close to the energy value of the standard drug (−113.99 kcal/mol−1).
For fungi, the docking results showed that BA has CYP51 and DHFR as its mechanism of action. Among the fungal species investigated in this study that had genomic and structural data for these enzymes for docking, four species showed excellent docking results for each of the analyzed enzymes, but not SAP−2. The CYP51 enzymes from C. albicans, C. tropicalis, C. glabrata, and A. flavus were able to interact well with BA, presenting binding energy values higher or close to the energy value of the drug used as a control. We analyzed the interactions of BA with the DHFR enzyme of the fungal species that obtained excellent molecular docking results.
4. Discussion
Triterpenoids are widely distributed in the plant kingdom and are known to have many beneficial effects such as anti−inflammatory, immunomodulatory, antiproliferative, and antimicrobial activity [
50].
Studies have reported the use of triterpenoids as anti−staphylococcal compounds but also as resistance−modifying agents when combined with common antibiotics. Among these studies, we highlight the work of Júnior et al. [
51], who verified the ability of BA to reduce the activation of the SOS response and its associated phenotypic changes, induced by ciprofloxacin in
S. aureus. The result showed that there was no antimicrobial activity against
S. aureus; however, it was able to reduce ciprofloxacin−induced recA expression.
Furthermore, it was observed that BA inhibited the progress of tolerance and mutagenesis induced by this drug. Another study, by Chung [
52], showed that
S. aureus is sensitive to pentacyclic triterpenoids, particularly α−amyrin (AM), BA, and betulinaldehyde (BE), acting synergistically and through different targets of conventional antibiotics. Thus, triterpenoids can be considered therapeutic alternatives against infections caused by
S. aureus.
A study by Haque et al. [
53] sought to investigate betulin and 51 derivatives thereof against five strains, including
Enterobacter aerogenes,
E. coli,
Enterococcus faecalis,
P. aeruginosa, and
S. aureus, and the fungal strain
C. albicans, using broth microdilution assays. Primary antimicrobial screening at a concentration of 50 mM led to the identification of five triterpenoids with antimicrobial properties (0.70% growth inhibition against one or more microbial strains). However, betulinic acid was not able to inhibit the growth of any of the strains when compared to the controls used in the study. Procedures such as the type of strains and control drugs used can contribute to the production of contradictory results.
Our study also evaluated the mechanism of action on important bacterial and fungal targets that are already considered clinically relevant for the proliferation and survival of these microorganisms. According to Germe et al. [
54], DNA gyrases are composed of two subunits forming heterotetrameric complexes that establish a transient double−stranded DNA break and facilitate the passage of DNA polymerase across the break. They are considered clinically validated targets for antibiotics and are essential for the transcription and segregation of the genome in bacterial cells. According to Pozzi et al. [
55], beta−lactamases represent the main mechanism of resistance to β−lactam antibiotics, conferring resistance to penicillins and cephalosporins. The role of β−lactamases in bacterial resistance is complex and extensive, but the development of inhibitors of this target is important to obtain the antimicrobial effect.
In another study, carried out by Innocente et al. [
56], derivatives obtained from ursolic and betulinic acid were tested against various strains of filamentous fungi and yeasts (
C. albicans,
C. krusei,
C. glabrata,
C. tropicalis,
C. parapsilosis, and
Epidermophyton floccosum, among others). The results of the expressed susceptibility assays showed that compound 10 was active against yeast and fungal filaments, while compound 9 showed antifungal activity only against yeasts. The researchers observed that the most active derivatives had the piperazinyl C−28 substituent. Furthermore, based on the MIC and MFC,
C. glabrata and
C. tropicalis were more susceptible than other tested species. In addition, based on the MIC and MFC,
C. glabrata and
C. tropicalis were more susceptible than other species tested. According to Hargrove et al. [
57],
C. albicans CYP51 prefers its natural substrate lanosterol but is more strongly inhibited by clinical azoles that have a longer side chain arm. The authors further conclude that the interaction with the enzyme is strengthened by hydrogen bonding with His−377, which is conserved in all CYP51 enzymes of the genus
Candida.
Infections caused by bacteria and fungi have been worrying millions of people around the world due to the large number infected, opportunistic diseases, and the several cases of antimicrobial resistance. Thus, it is necessary to develop new compounds with potential, selective, and effective antimicrobial activity. For this, natural products are important sources of new therapeutic agents due to their structural diversity.
5. Conclusions
Our study showed that bacteria share at least 45.39% of their DNA gyrase amino acids. A lower degree of identity was found for the beta−lactamase and PBP enzymes, with at least 14.66 and 19.31% identity between the bacteria, respectively. As for fungi, we observed a minimum degree of identity of 47.14% for the CYP51 enzyme, 47.21% for the SAP−2 enzyme, and 27.84% for the DHFR enzyme. In general, for species of the same genus, the degree of conserved regions exceeds 80%, except for the species C. glabrata, which has protein sequences very different from the enzymes of other species of the genus Candida. These data show that despite these enzymes being present in several microorganisms, they differ sequentially and structurally as the degree of identity and similarity between the sequences decreases. This indicates that it is necessary to plan and develop selective drugs for each target species.
Two predictive models were built against M. tuberculosis and E. coli. The compound presented a prediction of biological activity of 63 and 65% against M. tuberculosis and E. coli species, respectively. Given these results, it was possible to investigate the in vitro activity, which was confirmed and extended to other species.
BA at a concentration of 561 to 1122 μM produced growth inhibition of 50%/bacteria and 75%/fungi. When evaluating the bactericidal and fungicidal effect, the results showed that BA had a bacteriostatic effect against E. coli and no fungistatic effect for the investigated strains. Thus, we suggest that BA has potential antimicrobial activity.
The construction of protein models and molecular docking made it possible to investigate the mechanism of action of BA against the investigated microorganisms. From the results, we suggest that the BA compound has a mechanism of action against DNA gyrase and beta−lactamase targets for most investigated bacteria, and CYP51 and DHFR for most fungi.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}