
Roles of ADP-Ribosylation during Infection Establishment by Trypanosomatidae Parasites

{kind=link}
Abstract
:1. ADP-Ribosylation in Infection
1.1. ADP-Ribosylation in Viral Pathogens
1.2. ADP-Ribosylation in Bacterial Pathogens
2. Trypanosomatidae
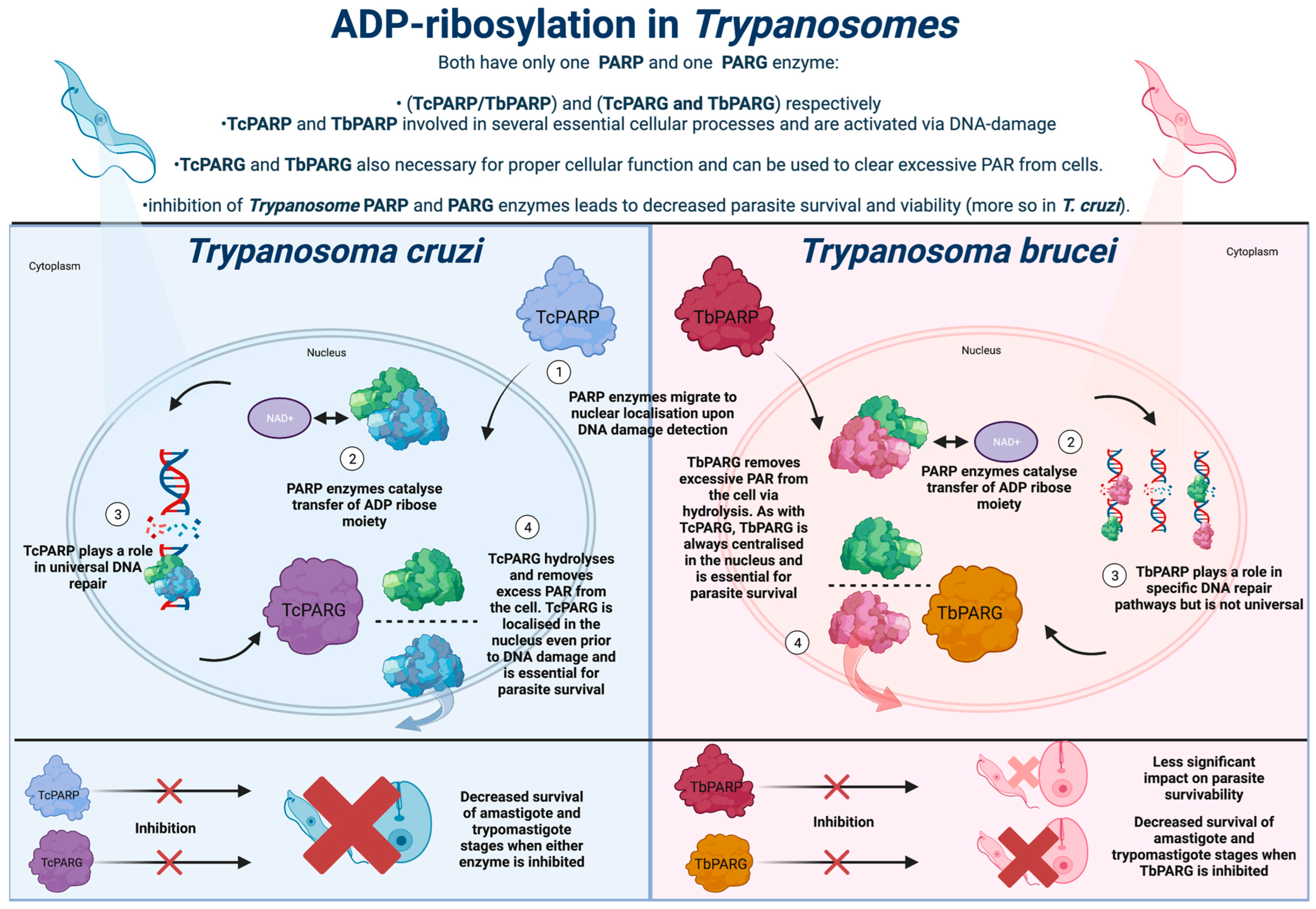
3. ADP-Ribosylation in Trypanosoma cruzi
3.1. Use of ADP-Ribosylation and Associated PARP and PARG Enzymes in Trypanosoma cruzi
3.2. The Potential of ADP-Ribosylation Targeted Therapy for Trypanosoma cruzi
4. ADP-Ribosylation in Trypanosoma brucei
4.1. PARP Enzymes in Trypanosoma brucei and Potential Therapies
4.2. PARG Enzymes in Trypanosoma brucei and Potential Therapies Targeting PARG Enzymes
5. ADP-Ribosylation in Leishmania
5.1. LdARL-3A Ribosylation Factor as a Therapeutic Target in Leishmania
5.2. LiSIR2RP1 as a Therapeutic Target in Leishmania
5.3. Targeting NAD+ Salvage Pathways as a Therapeutic Option in Leishmania
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Miwa, M.; Ishihara, M.; Takishima, S.; Takasuka, N.; Maeda, M.; Yamaizumi, Z.; Sugimura, T.; Yokoyama, S.; Miyazawa, T. The branching and linear portions of poly(adenosine diphosphate ribose) have the same alpha(1 leads to 2) ribose-ribose linkage. J. Biol. Chem. 1981, 256, 2916–2921. [Google Scholar] [CrossRef] [PubMed]
- Laing, S.; Koch-Nolte, F.; Haag, F.; Buck, F. Strategies for the identification of arginine ADP-ribosylation sites. J. Proteom. 2011, 75, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host–pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef]
- Kuny, C.V.; Sullivan, C.S. Virus–Host Interactions and the ARTD/PARP Family of Enzymes. PLoS Pathog. 2016, 12, e1005453. [Google Scholar] [CrossRef] [PubMed]
- Malgras, M.; Garcia, M.; Jousselin, C.; Bodet, C.; Lévêque, N. The Antiviral Activities of Poly-ADP-Ribose Polymerases. Viruses 2021, 13, 582. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Hauer, D.; McPherson, R.L.; Utt, A.; Kirby, I.T.; Cohen, M.S.; Merits, A.; Leung, A.K.L.; Griffin, D.E. ADP-ribosyl–binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proc. Natl. Acad. Sci. USA 2018, 115, E10457–E10466. [Google Scholar] [CrossRef]
- Zhu, H.; Zheng, C. When PARPs Meet Antiviral Innate Immunity. Trends Microbiol. 2021, 29, 776–778. [Google Scholar] [CrossRef]
- Chiu, H.-P.; Chiu, H.; Yang, C.-F.; Lee, Y.-L.; Chiu, F.-L.; Kuo, H.-C.; Lin, R.-J.; Lin, Y.-L. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PLoS Pathog. 2018, 14, e1007166. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-H.; Zhou, L.; Chen, G.; Krug, R.M. Battle between influenza A virus and a newly identified antiviral activity of the PARP-containing ZAPL protein. Proc. Natl. Acad. Sci. USA 2015, 112, 14048–14053. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Akhrymuk, M.; Frolova, E.I.; Frolov, I. New PARP Gene with an Anti-Alphavirus Function. J. Virol. 2012, 86, 8147–8160. [Google Scholar] [CrossRef]
- Todorova, T.; Bock, F.J.; Chang, P. PARP13 and RNA regulation in immunity and cancer. Trends Mol. Med. 2015, 21, 373–384. [Google Scholar] [CrossRef]
- Deng, L.; Ammosova, T.; Pumfery, A.; Kashanchi, F.; Nekhai, S. HIV-1 Tat Interaction with RNA Polymerase II C-terminal Domain (CTD) and a Dynamic Association with CDK2 Induce CTD Phosphorylation and Transcription from HIV-1 Promoter. J. Biol. Chem. 2002, 277, 33922–33929. [Google Scholar] [CrossRef] [PubMed]
- Parent, M.; Yung, T.M.C.; Rancourt, A.; Ho, E.L.Y.; Vispé, S.; Suzuki-Matsuda, F.; Uehara, A.; Wada, T.; Handa, H.; Satoh, M.S. Poly(ADP-ribose) Polymerase-1 Is a Negative Regulator of HIV-1 Transcription through Competitive Binding to TAR RNA with Tat·Positive Transcription Elongation Factor b (p-TEFb) Complex. J. Biol. Chem. 2005, 280, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-D.; Gong, J.; Gandhi, V.B.; Liu, X.; Shi, Y.; Johnson, E.F.; Donawho, C.K.; Ellis, P.A.; Bouska, J.J.; Osterling, D.J.; et al. Discovery and SAR of orally efficacious tetrahydropyridopyridazinone PARP inhibitors for the treatment of cancer. Bioorganic Med. Chem. 2012, 20, 4635–4645. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Zhang, A.; Du, Y.; Fang, M.; Minze, L.J.; Liu, Y.-J.; Li, X.C.; Zhang, Z. Identification of poly(ADP-ribose) polymerase 9 (PARP9) as a noncanonical sensor for RNA virus in dendritic cells. Nat. Commun. 2021, 12, 2681. [Google Scholar] [CrossRef]
- Yu, M.; Zhang, C.; Yang, Y.; Yang, Z.; Zhao, L.; Xu, L.; Wang, R.; Zhou, X.; Huang, P. The interaction between the PARP10 protein and the NS1 protein of H5N1 AIV and its effect on virus replication. Virol. J. 2011, 8, 546. [Google Scholar] [CrossRef]
- Li, L.; Zhao, H.; Liu, P.; Li, C.; Quanquin, N.; Ji, X.; Sun, N.; Du, P.; Qin, C.-F.; Lu, N.; et al. PARP12 suppresses Zika virus infection through PARP-dependent degradation of NS1 and NS3 viral proteins. Sci. Signal. 2018, 11, eaas9332. [Google Scholar] [CrossRef]
- Miettinen, M.; Vedantham, M.; Pulliainen, A.T. Host poly(ADP-ribose) polymerases (PARPs) in acute and chronic bacterial infections. Microbes Infect. 2019, 21, 423–431. [Google Scholar] [CrossRef]
- Soriano, F.G.; Liaudet, L.; Szabó, É.; Virág, L.; Mabley, J.G.; Pacher, P.; Szabó, C. Resistance to Acute Septic Peritonitis in Poly(ADP-ribose) Polymerase-1-Deficient Mice. Shock 2002, 17, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Enkhbaatar, P.; Shimoda, K.; Cox, R.A.; Burke, A.S.; Hawkins, H.K.; Traber, L.D.; Schmalstieg, F.C.; Salzman, A.L.; Mabley, J.G.; et al. Inhibition of Poly (ADP-ribose) Polymerase Attenuates Acute Lung Injury in an Ovine Model of Sepsis. Shock 2004, 21, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Yélamos, J.; Buendía, A.J.; Ortega, N.; Monreal, Y.; Gallego, M.C.; Sánchez, J.; Ramírez, P.; Parrilla, P.; Caro, M.R.; Aparicio, P.; et al. Genetic and pharmacological inhibition of poly(ADP-ribose) polymerase-1 interferes in the chlamydial life cycle. Biochem. Biophys. Res. Commun. 2004, 324, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Kunze, F.A.; Hottiger, M.O. Regulating Immunity via ADP-Ribosylation: Therapeutic Implications and Beyond. Trends Immunol. 2019, 40, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, S.; Caparon, M.G. The Streptococcus pyogenes NAD+ glycohydrolase modulates epithelial cell PARylation and HMGB1 release. Cell Microbiol. 2015, 17, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.-H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef] [PubMed]
- Oliver, F.J.; Ménissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef]
- Gupte, R.; Nandu, T.; Kraus, W.L. Nuclear ADP-ribosylation drives IFNγ-dependent STAT1α enhancer formation in macrophages. Nat. Commun. 2021, 12, 3931. [Google Scholar] [CrossRef] [PubMed]
- Mikolčević, P.; Hloušek-Kasun, A.; Ahel, I.; Mikoč, A. ADP-ribosylation systems in bacteria and viruses. Comput. Struct. Biotechnol. J. 2021, 19, 2366–2383. [Google Scholar] [CrossRef] [PubMed]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)—Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front. Mol. Biosci. 2020, 7, 191. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.-W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kim, N.S.; Haince, J.-F.; Kang, H.C.; David, K.K.; Andrabi, S.A.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-Ribose) (PAR) Binding to Apoptosis-Inducing Factor Is Critical for PAR Polymerase-1–Dependent Cell Death (Parthanatos). Sci. Signal. 2011, 4, ra20. [Google Scholar] [CrossRef]
- Barkauskaite, E.; Brassington, A.; Tan, E.S.; Warwicker, J.; Dunstan, M.S.; Banos, B.; Lafite, P.; Ahel, M.; Mitchison, T.J.; Ahel, I.; et al. Visualization of poly(ADP-ribose) bound to PARG reveals inherent balance between exo- and endo-glycohydrolase activities. Nat. Commun. 2013, 4, 2164. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Ferreira, M.T.; Gagné, J.-P.; Sharma, A.K.; Hendzel, M.J.; Masson, J.-Y.; Poirier, G.G. Emerging roles of eraser enzymes in the dynamic control of protein ADP-ribosylation. Nat. Commun. 2019, 10, 1182. [Google Scholar] [CrossRef] [PubMed]
- Pourfarjam, Y.; Kasson, S.; Tran, L.; Ho, C.; Lim, S.; Kim, I.-K. PARG has a robust endo-glycohydrolase activity that releases protein-free poly(ADP-ribose) chains. Biochem. Biophys. Res. Commun. 2020, 527, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.S.; Dharwal, V.; Naura, A.S. Poly(ADP-Ribose)Polymerase-1 in Lung Inflammatory Disorders: A Review. Front. Immunol. 2017, 8, 1172. [Google Scholar] [CrossRef] [PubMed]
- Wasyluk, W.; Zwolak, A. PARP Inhibitors: An Innovative Approach to the Treatment of Inflammation and Metabolic Disorders in Sepsis. J. Inflamm. Res. 2021, 14, 1827–1844. [Google Scholar] [CrossRef] [PubMed]
- Caprara, G.; Prosperini, E.; Piccolo, V.; Sigismondo, G.; Melacarne, A.; Cuomo, A.; Boothby, M.; Rescigno, M.; Bonaldi, T.; Natoli, G. PARP14 Controls the Nuclear Accumulation of a Subset of Type I IFN–Inducible Proteins. J. Immunol. 2018, 200, 2439–2454. [Google Scholar] [CrossRef]
- Ackley, C.; Elsheikh, M.; Zaman, S. Scoping review of Neglected Tropical Disease Interventions and Health Promotion: A framework for successful NTD interventions as evidenced by the literature. PLoS Negl. Trop. Dis. 2021, 15, e0009278. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, L.E.; Morillo, C.A. American Trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2019, 33, 119–134. [Google Scholar] [CrossRef]
- Sangenito, L.S.; Branquinha, M.H.; Santos, A.L.S. Funding for Chagas Disease: A 10-Year (2009–2018) Survey. Trop. Med. Infect. Dis. 2020, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, J.M.; Tarleton, R.L. Potential new clinical therapies for Chagas disease. Expert Rev. Clin. Pharmacol. 2014, 7, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
- Kennedy, P.G. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar] [CrossRef]
- Baker, N.; de Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Desjeux, P. The increase in risk factors for leishmaniasis worldwide. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 239–243. [Google Scholar] [CrossRef]
- Zulfiqar, B.; Shelper, T.B.; Avery, V.M. Leishmaniasis drug discovery: Recent progress and challenges in assay development. Drug Discov. Today 2017, 22, 1516–1531. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Jain, K. Molecular targets and pathways for the treatment of visceral leishmaniasis. Drug Discov. Today 2018, 23, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.S.; Marques, C.S.; de Sousa, D.P.; Andrade, L.N.; Fricks, A.T.; Jain, S.; Branquinha, M.H.; Souto, E.B.; Santos, A.L.; Severino, P. Analysis of the mechanisms of action of isopentenyl caffeate against Leishmania. Biochimie 2021, 189, 158–167. [Google Scholar] [CrossRef]
- Laniado-Laborín, R.; Cabrales-Vargas, M.N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 2009, 26, 223–227. [Google Scholar] [CrossRef]
- Schlesinger, M.; Larrea, S.C.V.; Haikarainen, T.; Narwal, M.; Venkannagari, H.; Flawiá, M.M.; Lehtiö, L.; Villamil, S.H.F. Disrupted ADP-ribose metabolism with nuclear Poly (ADP-ribose) accumulation leads to different cell death pathways in presence of hydrogen peroxide in procyclic Trypanosoma brucei. Parasites Vectors 2016, 9, 173. [Google Scholar] [CrossRef]
- Agnew, T.; Munnur, D.; Crawford, K.; Palazzo, L.; Mikoč, A.; Ahel, I. MacroD1 Is a Promiscuous ADP-Ribosyl Hydrolase Localized to Mitochondria. Front. Microbiol. 2018, 9, 20. [Google Scholar] [CrossRef]
- Haikarainen, T.; Lehtiö, L. Proximal ADP-ribose Hydrolysis in Trypanosomatids is Catalyzed by a Macrodomain. Sci. Rep. 2016, 6, 24213. [Google Scholar] [CrossRef] [PubMed]
- Larrea, S.C.V.; Alonso, G.D.; Schlesinger, M.; Torres, H.N.; Flawiá, M.M.; Villamil, S.H.F. Poly(ADP-ribose) polymerase plays a differential role in DNA damage-response and cell death pathways in Trypanosoma cruzi. Int. J. Parasitol. 2011, 41, 405–416. [Google Scholar] [CrossRef]
- Fernández Villamil, S.H.; Vilchez Larrea, S.C. Poly(ADP-ribose) metabolism in human parasitic protozoa. Acta Trop. 2020, 208, 105499. [Google Scholar] [CrossRef]
- Otto, H.; Reche, P.A.; Bazan, F.; Dittmar, K.; Haag, F.; Koch-Nolte, F. In silico characterization of the family of PARP-like poly(ADP-ribosyl)transferases (pARTs). BMC Genom. 2005, 6, 139. [Google Scholar] [CrossRef]
- Villamil, S.H.F.; Baltanás, R.; Alonso, G.D.; Larrea, S.C.V.; Torres, H.N.; Flawiá, M.M. TcPARP: A DNA damage-dependent poly(ADP-ribose) polymerase from Trypanosoma cruzi. Int. J. Parasitol. 2008, 38, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Amé, J.C.; Roe, S.M.; Good, V.; de Murcia, G.; Pearl, L.H. Crystal structure of the catalytic fragment of murine poly(ADP-ribose) polymerase-2. Nucleic Acids Res. 2004, 32, 456–464. [Google Scholar] [CrossRef]
- Gonçalves, V.M.; Matteucci, K.C.; Buzzo, C.L.; Miollo, B.H.; Ferrante, D.; Torrecilhas, A.C.; Rodrigues, M.M.; Alvarez, J.M.; Bortoluci, K.R. NLRP3 Controls Trypanosoma cruzi Infection through a Caspase-1-Dependent IL-1R-Independent NO Production. PLoS Negl. Trop. Dis. 2013, 7, e2469. [Google Scholar] [CrossRef]
- Stahl, P.; Ruppert, V.; Meyer, T.; Schmidt, J.; Campos, M.A.; Gazzinelli, R.T.; Maisch, B.; Schwarz, R.T.; Debierre-Grockiego, F. Trypomastigotes and amastigotes of Trypanosoma cruzi induce apoptosis and STAT3 activation in cardiomyocytes in vitro. Apoptosis 2013, 18, 653–663. [Google Scholar] [CrossRef]
- Priotto, S.; Sartori, M.; Repossi, G.; Valentich, M. Trypanosoma cruzi: Participation of cholesterol and placental alkaline phosphatase in the host cell invasion. Exp. Parasitol. 2009, 122, 70–73. [Google Scholar] [CrossRef]
- Nogueira, N.P.; Saraiva, F.M.S.; Sultano, P.E.; Cunha, P.R.B.B.; Laranja, G.A.T.; Justo, G.A.; Sabino, K.C.C.; Coelho, M.G.P.; Rossini, A.; Atella, G.C.; et al. Proliferation and Differentiation of Trypanosoma cruzi inside Its Vector Have a New Trigger: Redox Status. PLoS ONE 2015, 10, e0116712. [Google Scholar] [CrossRef] [PubMed]
- Poveda, C.; Fresno, M.; Gironès, N.; Martins-Filho, O.A.; Ramírez, J.D.; Santi-Rocca, J.; Marin-Neto, J.A.; Morillo, C.; Rosas, F.; Guhl, F. Cytokine Profiling in Chagas Disease: Towards Understanding the Association with Infecting Trypanosoma cruzi Discrete Typing Units (A BENEFIT TRIAL Sub-Study). PLoS ONE 2014, 9, e91154. [Google Scholar] [CrossRef]
- Ba, X.; Gupta, S.; Davidson, M.; Garg, N.J. Trypanosoma cruzi Induces the Reactive Oxygen Species-PARP-1-RelA Pathway for Up-regulation of Cytokine Expression in Cardiomyocytes. J. Biol. Chem. 2010, 285, 11596–11606. [Google Scholar] [CrossRef]
- Pinto, A.M.T.; Sales, P.C.M.; Camargos, E.R.S.; Silva, A.M. Tumour necrosis factor (TNF)-mediated NF-κB activation facilitates cellular invasion of non-professional phagocytic epithelial cell lines by Trypanosoma cruzi. Cell. Microbiol. 2011, 13, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef]
- Alonso, G.D.; Vilchez Larrea, S.C.; Fernández Villamil, S.H. Metabolism of poly-ADP-ribose in trypanosomatids. In Parasitology Research Trends; Le Novo Science Publishers Inc.: Buenos Aires, Argentina, 2010. [Google Scholar]
- Larrea, S.C.V.; Schlesinger, M.; Kevorkian, M.L.; Flawiá, M.M.; Alonso, G.D.; Villamil, S.H.F. Host Cell Poly(ADP-Ribose) Glycohydrolase Is Crucial for Trypanosoma cruzi Infection Cycle. PLoS ONE 2013, 8, e67356. [Google Scholar] [CrossRef]
- Wen, J.J.; Yin, Y.W.; Garg, N.J. PARP1 depletion improves mitochondrial and heart function in Chagas disease: Effects on POLG dependent mtDNA maintenance. PLoS Pathog. 2018, 14, e1007065. [Google Scholar] [CrossRef]
- Haikarainen, T.; Schlesinger, M.; Obaji, E.; Villamil, S.H.F.; Lehtiö, L. Structural and Biochemical Characterization of Poly-ADP-ribose Polymerase from Trypanosoma brucei. Sci. Rep. 2017, 7, 3642. [Google Scholar] [CrossRef]
- Niemirowicz, G.T.; Cazzulo, J.J.; Álvarez, V.E.; Bouvier, L.A. Simplified inducible system for Trypanosoma brucei. PLoS ONE 2018, 13, e0205527. [Google Scholar] [CrossRef]
- Kun, E.; Kirsten, E.; Mendeleyev, J.; Ordahl, C.P. Regulation of the Enzymatic Catalysis of Poly(ADP-ribose) Polymerase by dsDNA, Polyamines, Mg2+, Ca2+, Histones H1 and H3, and ATP. Biochemistry 2003, 43, 210–216. [Google Scholar] [CrossRef] [PubMed]
- van Beek, L.; McClay, É.; Patel, S.; Schimpl, M.; Spagnolo, L.; Maia de Oliveira, T. Parp power: A structural perspective on parp1, parp2, and parp3 in dna damage repair and nucleosome remodelling. Int. J. Mol. Sci. 2021, 2, 5112. [Google Scholar] [CrossRef]
- Klebanov-Akopyan, O.; Mishra, A.; Glousker, G.; Tzfati, Y.; Shlomai, J. Trypanosoma brucei UMSBP2 is a single-stranded telomeric DNA binding protein essential for chromosome end protection. Nucleic Acids Res. 2018, 46, 7757–7771. [Google Scholar] [CrossRef]
- Glover, L.; McCulloch, R.; Horn, D. Sequence homology and microhomology dominate chromosomal double-strand break repair in African trypanosomes. Nucleic Acids Res. 2008, 36, 2608–2618. [Google Scholar] [CrossRef]
- Welburn, S.C.; Maudlin, I. Control of Trypanosoma brucei brucei infections in tsetse, Glossina morsitans. Med. Veter. Entomol. 1997, 11, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Nepomuceno-Mejía, T.; Lara-Martínez, R.; Hernández, R.; De, M.; Segura-Valdez, L.; Jiménez-García, L.F. Nucleologenesis in Trypanosoma cruzi. Microsc. Microanal. 2016, 22, 621–629. [Google Scholar] [CrossRef]
- Nenarokova, A.; Záhonová, K.; Krasilnikova, M.; Gahura, O.; McCulloch, R.; Zíková, A.; Yurchenko, V.; Lukeš, J. Causes and Effects of Loss of Classical Nonhomologous End Joining Pathway in Parasitic Eukaryotes. mBio 2019, 10, e01541-19. [Google Scholar] [CrossRef]
- Cuvillier, A.; Miranda, J.C.; Ambit, A.; Barral, A.; Merlin, G. Abortive infection of Lutzomyia longipalpis insect vectors by aflagellated LdARL-3A-Q70L overexpressing Leishmania amazonensis parasites. Cell. Microbiol. 2003, 5, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Sahin, A.; Espiau, B.; Tetaud, E.; Cuvillier, A.; Lartigue, L.; Ambit, A.; Robinson, D.R.; Merlin, G. The Leishmania ARL-1 and Golgi Traffic. PLoS ONE 2008, 3, e1620. [Google Scholar] [CrossRef]
- Tavares, J.; Ouaissi, A.; Kongâthooâlin, P.; Loureiro, I.; Kaur, S.; Roy, N.; Cordeiro-Da-Silva, A. Bisnaphthalimidopropyl Derivatives as Inhibitors of Leishmania SIR2 Related Protein 1. ChemBioChem 2009, 5, 140–147. [Google Scholar] [CrossRef]
- Ronin, C.; Costa, D.M.; Tavares, J.; Faria, J.; Ciesielski, F.; Ciapetti, P.; Smith, T.K.; MacDougall, J.; Cordeiro-Da-Silva, A.; Pemberton, I.K. The crystal structure of the Leishmania infantum Silent Information Regulator 2 related protein 1: Implications to protein function and drug design. PLoS ONE 2018, 13, e0193602. [Google Scholar] [CrossRef] [PubMed]
- Gazanion, E.; Garcia, D.; Silvestre, R.; Gérard, C.; Guichou, J.F.; Labesse, G.; Seveno, M.; Cordeiro-Da-Silva, A.; Ouaissi, A.; Sereno, D.; et al. The Leishmania nicotinamidase is essential for NAD+ production and parasite proliferation. Mol. Microbiol. 2011, 82, 21–38. [Google Scholar] [CrossRef]
- Reidl, J.; Schlör, S.; Kraiß, A.; Schmidt-Brauns, J.; Kemmer, G.; Soleva, E. NADP and NAD utilization in Haemophilus influenzae. Mol. Microbiol. 2000, 35, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Rodrigues, V.; Abengozar, M.; Rivas, L.; Rial, E.; Laforge, M.; Li, X.; Foretz, M.; Viollet, B.; Estaquier, J.; et al. Leishmania infantum Modulates Host Macrophage Mitochondrial Metabolism by Hijacking the SIRT1-AMPK Axis. PLoS Pathog. 2015, 11, e1004684. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dowling, J.; Doig, C.L. Roles of ADP-Ribosylation during Infection Establishment by Trypanosomatidae Parasites. Pathogens 2023, 12, 708. https://doi.org/10.3390/pathogens12050708
Dowling J, Doig CL. Roles of ADP-Ribosylation during Infection Establishment by Trypanosomatidae Parasites. Pathogens. 2023; 12(5):708. https://doi.org/10.3390/pathogens12050708
Chicago/Turabian StyleDowling, Joshua, and Craig L. Doig. 2023. "Roles of ADP-Ribosylation during Infection Establishment by Trypanosomatidae Parasites" Pathogens 12, no. 5: 708. https://doi.org/10.3390/pathogens12050708
APA StyleDowling, J., & Doig, C. L. (2023). Roles of ADP-Ribosylation during Infection Establishment by Trypanosomatidae Parasites. Pathogens, 12(5), 708. https://doi.org/10.3390/pathogens12050708