

Timeline of SARS-CoV-2 Transmission in Sabah, Malaysia: Tracking the Molecular Evolution

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Acquisition
2.2. Epidemiology of Coronavirus Disease 2019 in Sabah
2.3. Whole Genome Sequencing of SARS-CoV-2
2.4. Reporting
2.5. Analysis of SARS-CoV-2 Sequences from Sabah/Phylogenetic and Mutational Analysis
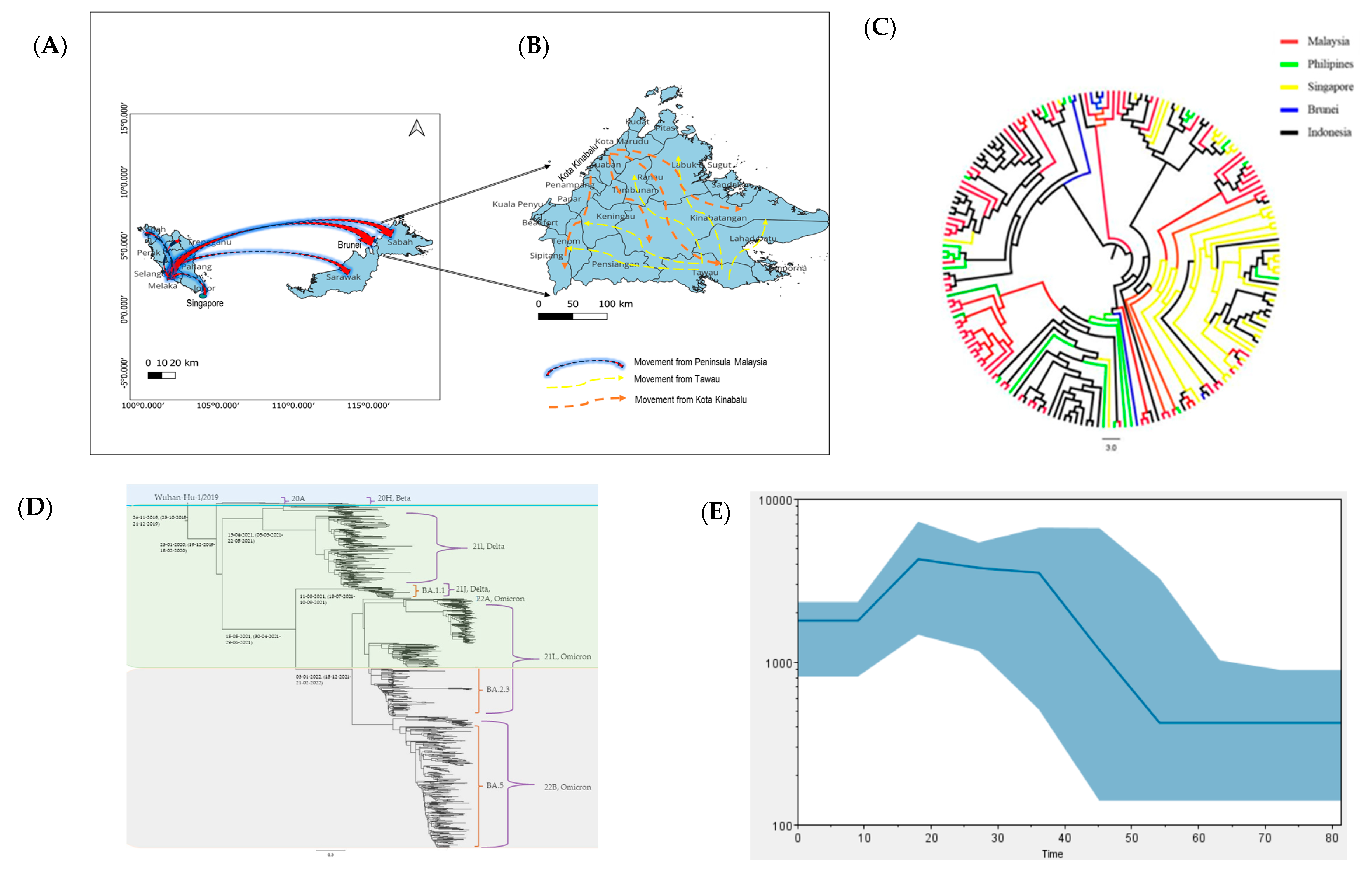
2.6. Phylogeographic and Phylodynamic Reconstructions
3. Results
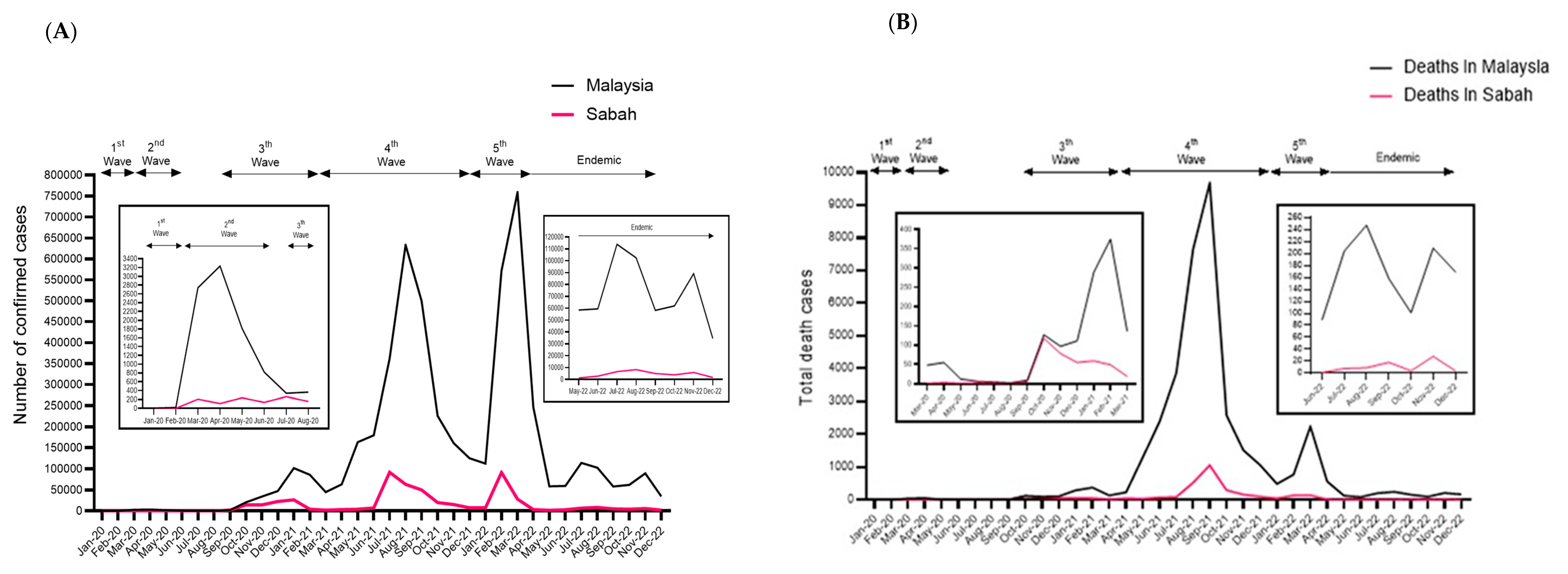
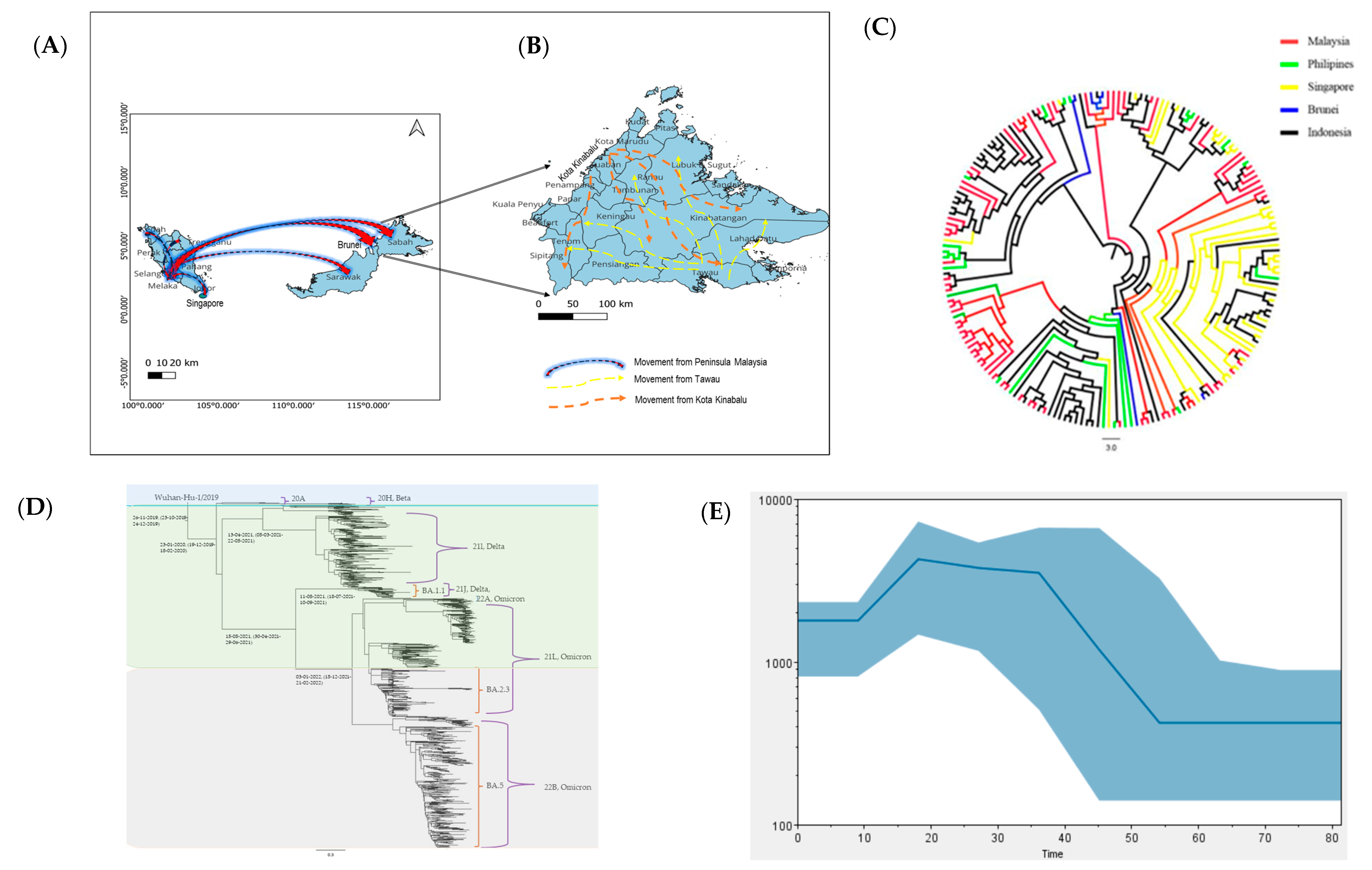
3.1. The Status of COVID-19 in Sabah
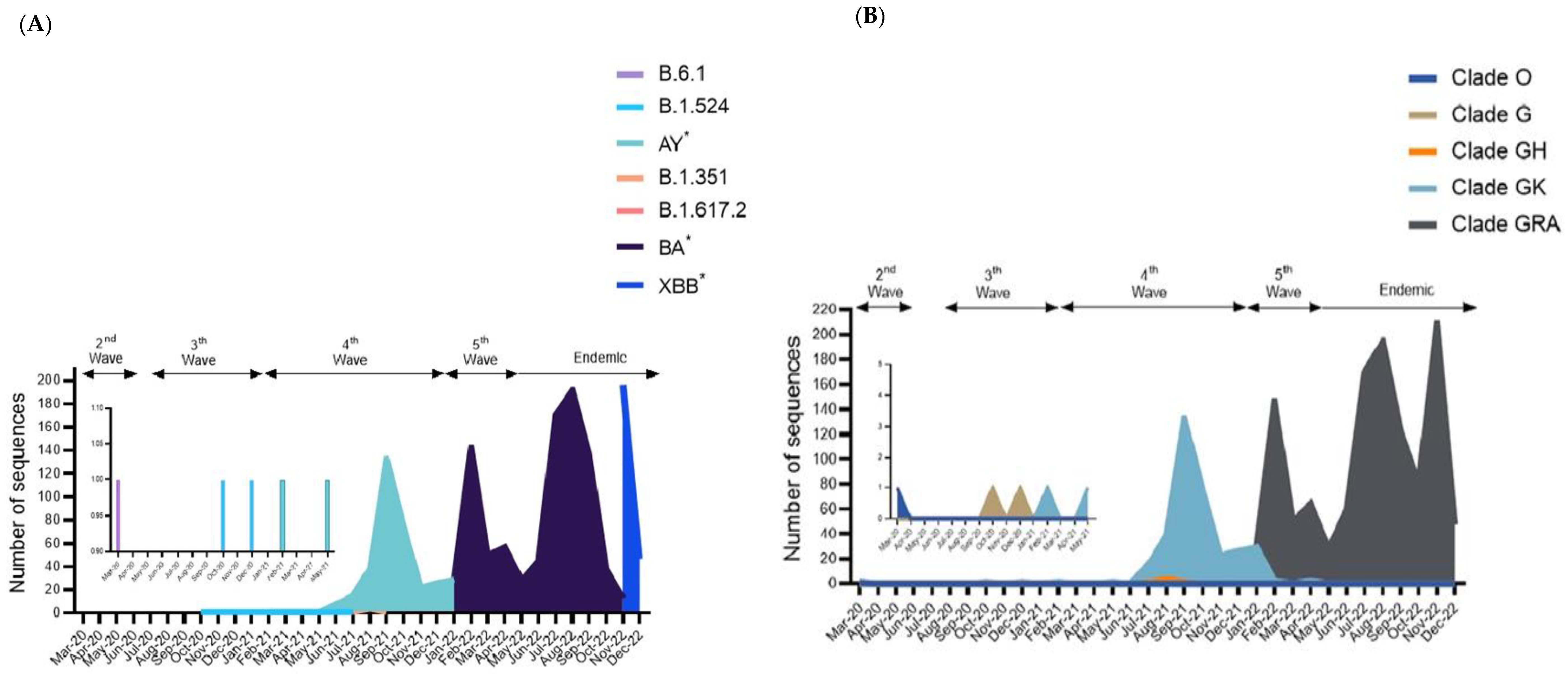
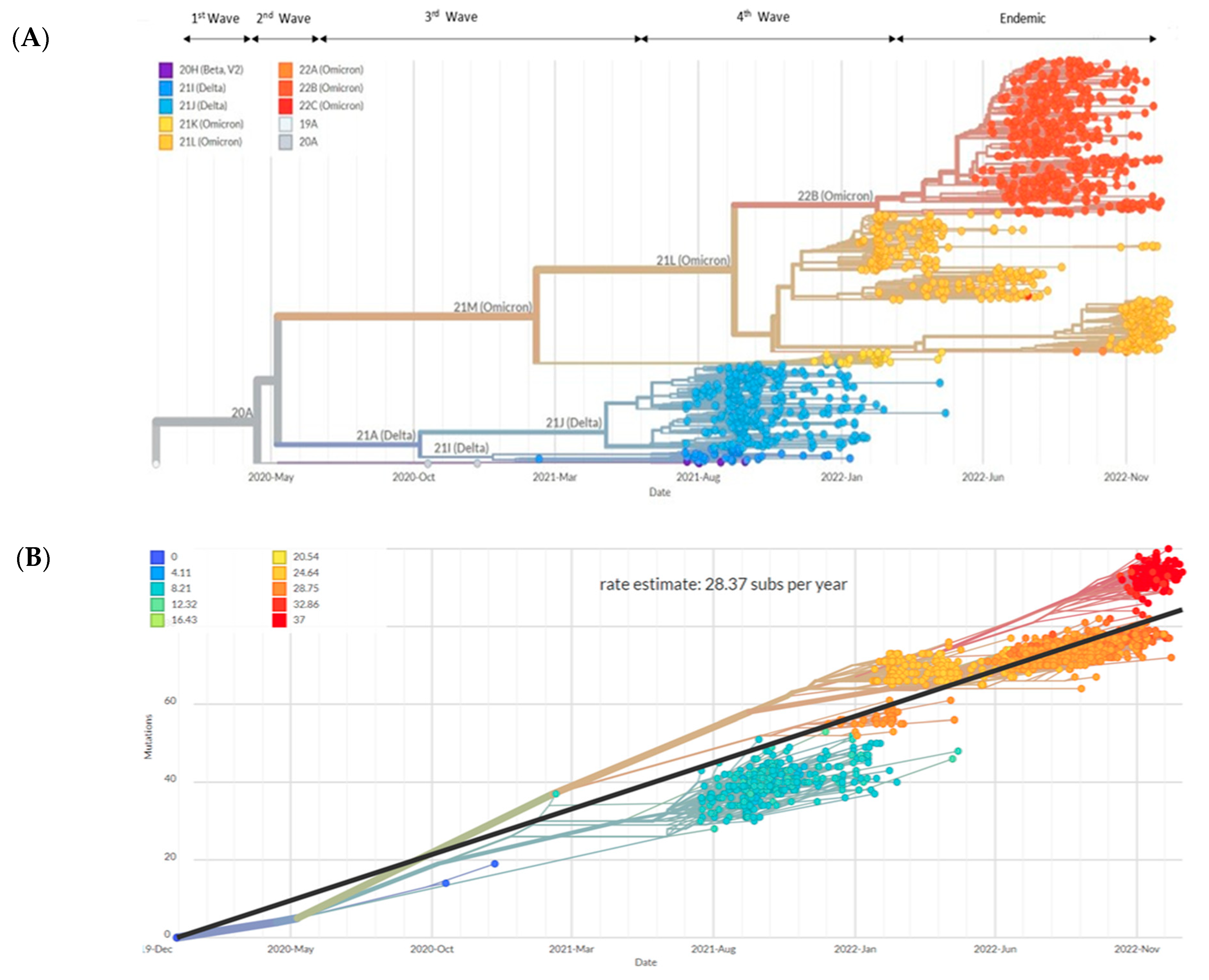
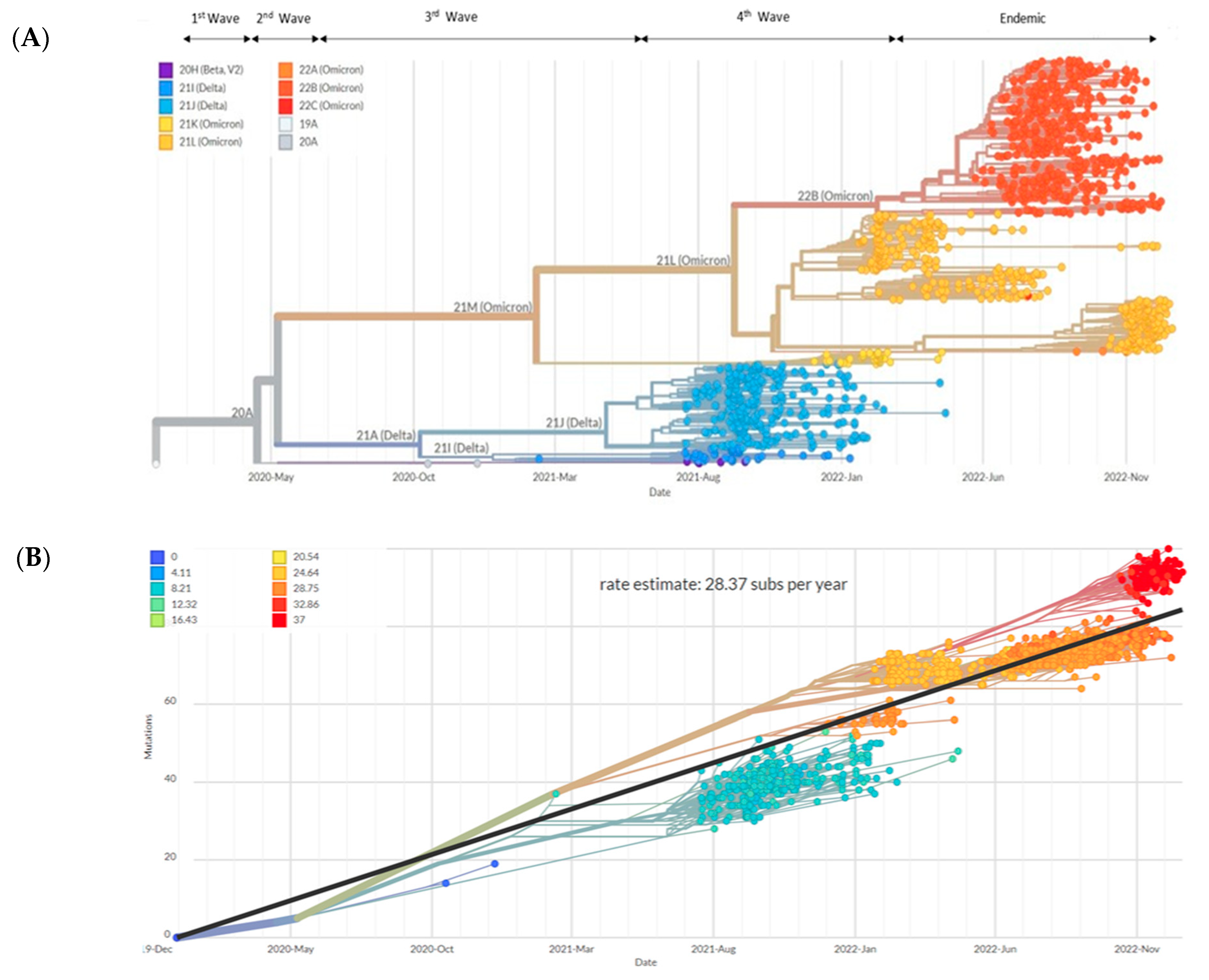
3.2. Analysis of SARS-CoV-2 Sequences from Sabah
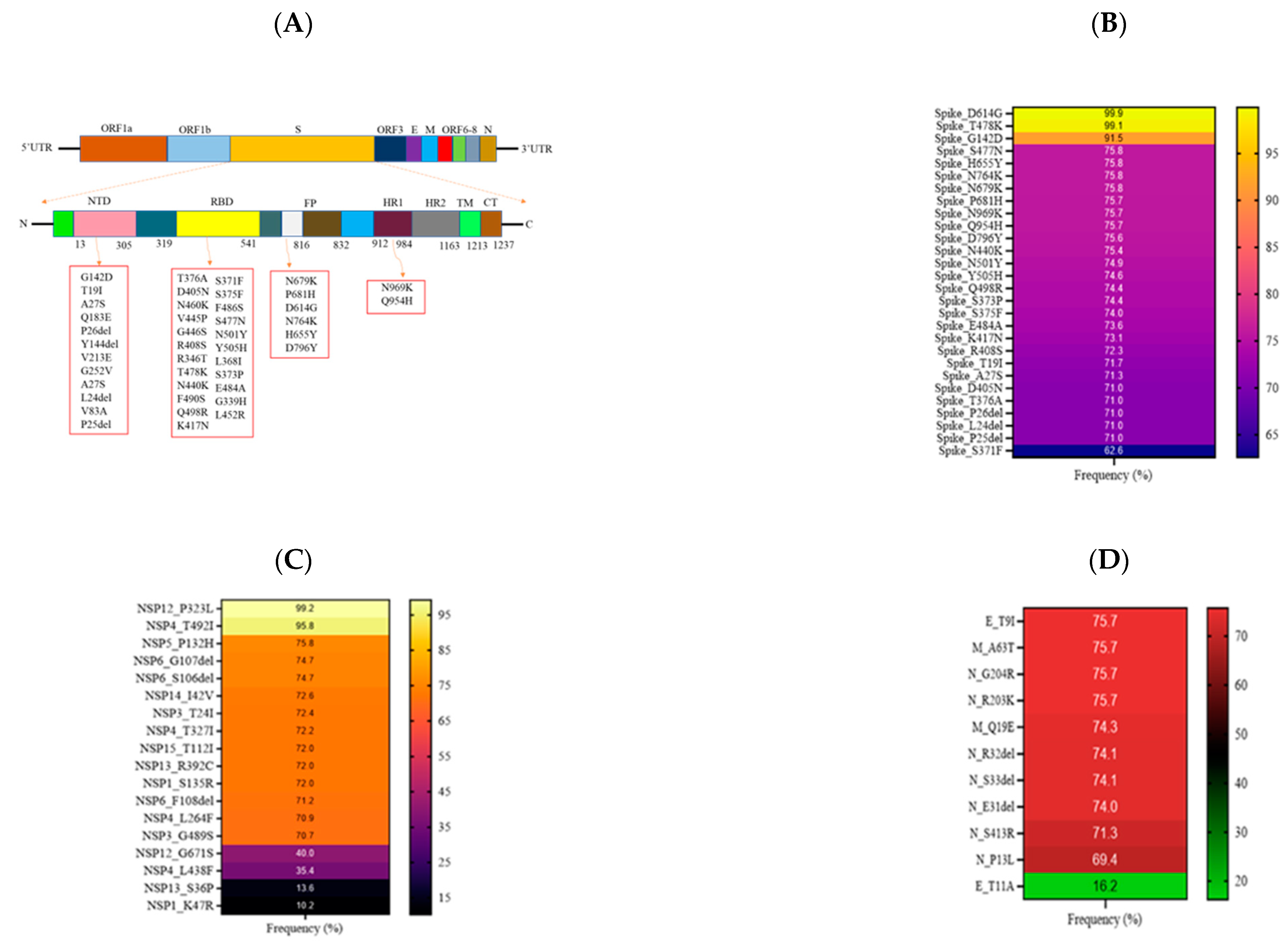
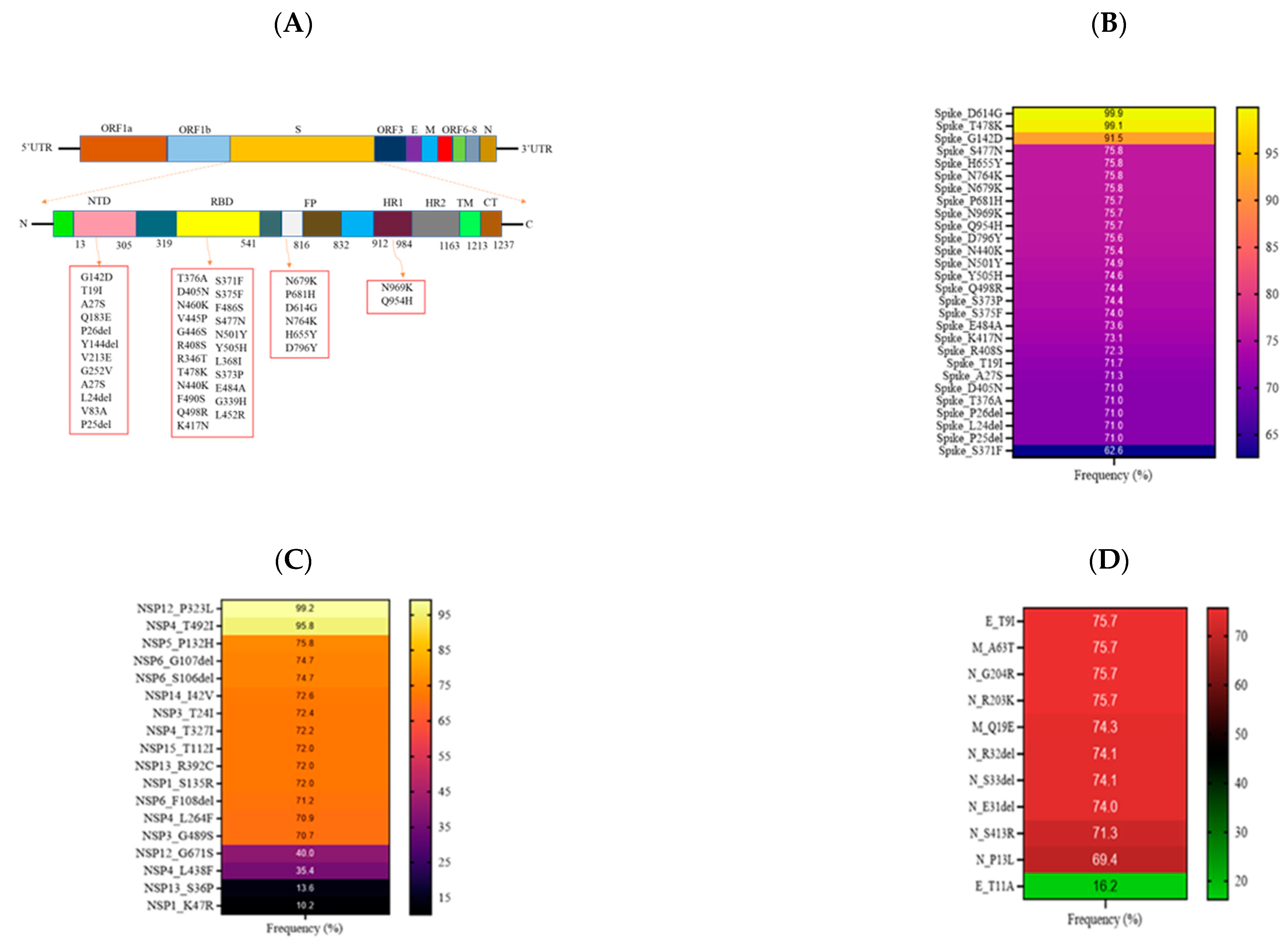
3.3. Mutational Analysis in Sabah SARS-CoV-2 Isolates
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- WHO. World Health Organization (WHO) Coronavirus (COVID-19) Dashboard (2023); World Health Organization: Geneva, Switzerland, 2023; Available online: https://covid19.who.int/ (accessed on 20 February 2023).
- Ang, Z.Y.; Cheah, K.Y.; Shakirah, M.S.; Fun, W.H.; Anis-Syakira, J.; Kong, Y.L.; Sararaks, S. Malaysia’s Health Systems Response to COVID-19. Int. J. Environ. Res. Public Health 2021, 18, 11109. [Google Scholar] [CrossRef] [PubMed]
- Al-Tawfiq, J.A.; Hoang, V.-T.; Le Bui, N.; Chu, D.-T.; Memish, Z.A. The emergence of the omicron (B.1.1.529) SARS-CoV-2 variant: What is the impact on the continued pandemic? J. Epidemiol. Glob. Health 2022, 12, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.K.; Tan, J.Y.; Wong, J.E.; Teoh, B.T.; Tiong, V.; Abd-Jamil, J.; Nor’e, S.S.; Khor, C.S.; Johari, J.; Yaacob, C.N.; et al. Emergence of B.1.524(G) SARS-CoV-2 in Malaysia during the third COVID-19 epidemic wave. Sci. Rep. 2021, 11, 22105. [Google Scholar] [CrossRef] [PubMed]
- Chakkour, M.; Salami, A.; Olleik, D.; Kamal, I.; Noureddine, F.Y.; El Roz, A.; Ghssein, G. Risk Markers of COVID-19, a Study from South-Lebanon. COVID 2022, 2, 867–876. [Google Scholar] [CrossRef]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.W.; Ardern, Z.; Goldberg, T.L.; Meng, C.; Kuo, C.-H.; Ludwig, C.; Kolokotronis, S.-O.; Wei, X. Dynamically evolving novel overlapping gene as a factor in the SARS-CoV-2 pandemic. eLife 2020, 9, e59633. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef] [PubMed]
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.W.; Hakmi, M.; Essabbar, A.; Laamarti, M.; Hlali, H.; Bendani, H.; Boumajdi, N.; et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. PLoS ONE 2020, 15, e0240345. [Google Scholar] [CrossRef] [PubMed]
- Wen, F.; Yu, H.; Guo, J.; Li, Y.; Luo, K.; Huang, S. Identification of the hyper-variable genomic hotspot for the novel coronavirus SARS-CoV-2. J. Infect. 2020, 80, 671–693. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.I.; Correia, I.; Seagal, J.; DeGoey, D.A.; Schrimpf, M.R.; Hardee, D.J.; Noey, E.L.; Kati, W.M. Antiviral Drug Discovery for the Treatment of COVID-19 Infections. Viruses 2022, 14, 961. [Google Scholar] [CrossRef]
- Ho, C.; Lee, P.C. COVID-19 Treatment—Current Status, Advances, and Gap. Pathogens 2022, 11, 1201. [Google Scholar] [CrossRef]
- WHO. Tracking SARS-CoV-2 Variants. 2022. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 20 February 2023).
- O’Toole, A.; Pybus, O.G.; Abram, M.E.; Kelly, E.J.; Rambaut, A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genom. 2022, 23, 121. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [Green Version]
- Sellin Jeffries, M.K.; Kiss, A.J.; Smith, A.W.; Oris, J.T. A comparison of commercially-available automated and manual extraction kits for the isolation of total RNA from small tissue samples. BMC Biotechnol. 2014, 14, 94. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.Y.; Wong, S.Y.; Liew, N.W.C.; Joseph, N.; Zakaria, Z.; Nurulfiza, I.; Soe, H.J.; Kairon, R.; Amin-Nordin, S.; Chee, H.Y. Whole genome sequencing analysis of SARS-CoV-2 from Malaysia: From alpha to Omicron. Front. Med. 2022, 9, 1001022. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nextstrain. Nextstrain SARS-CoV-2 Resources, on Nextstrain. 2021. Available online: https://nextstrain.org/sars-cov-2/ (accessed on 31 January 2023).
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- QGIS, Development Team. QGIS Geographic Information System, Open Source Geospatial Foundation Project. 2015.
- Yusof, A. Timeline: How the COVID-19 pandemic has unfolded in Malaysia since January 2020. Channel News Asia, 5 August 2021. [Google Scholar]
- Masidi: High COVID-19 cases in Sabah due to backlog of 18,000 sample results. The Star, 18 October 2020.
- Kaos, J.J. COVID-19: 50 More Cases Linked to Benteng Lahad Datu Cluster, All Prisoners, Says Health DG. 2020. Available online: https://www.nst.com.my/news/nation/2020/03/575560/how-sri-petaling-tablighbecame-southeast-asias-covid-19-hotspot (accessed on 20 February 2023).
- Ng, J.W.; Chong, E.T.J.; Tan, Y.A.; Lee, H.G.; Chan, L.L.; Lee, Q.Z.; Saw, Y.T.; Wong, Y.; Zakaria, A.A.B.; Amin, Z.B. Prevalence of Coronavirus Disease 2019 (COVID-19) in Different Clinical Stages before the National COVID-19 Vaccination Programme in Malaysia: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 2216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Health Malaysia. Situasi dan Maklumat Terkini Mengenai Mutasi Spike Protein COVID-19. 2021. Available online: https://www.ukm.my/umbi/news/situasi-dan-maklumatterkini-mengenai-mutasi-spike-protein-COVID-19-di-malaysia-3/ (accessed on 22 January 2023).
- Shiehzadegan, S.; Alaghemand, N.; Fox, M.; Venketaraman, V. Analysis of the Delta Variant B.1.617.2 COVID-19. Clin. Pract. 2021, 11, 778–784. [Google Scholar] [CrossRef]
- Chen, K.-W.K.; Huang, D.T.-N.; Huang, L.-M. SARS-CoV-2 variants–evolution, spike protein, and vaccines. Biomed. J. 2022, 45, 573–579. [Google Scholar] [CrossRef]
- Lum, R. COVID-19: Malaysia hit by record cases despite prolonged lockdown. BMJ 2021, 374, n2155. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C. Crushed by COVID, Malaysia now has one of the world’s fastest vaccination rates. The Sydney Morning Herald, 14 July 2021. [Google Scholar]
- Jafar, A.; Mapa, M.T.; Sakke, N.; Dollah, R.; Joko, E.P.; Atang, C.; Ahmad, S.A.; Hung, C.V.; Geogre, F. Vaccine hesitancy in East Malaysia (Sabah): A survey of the national COVID-19 immunisation programme. Geospat. Health 2022, 17, 1037. [Google Scholar] [CrossRef]
- Yang, S.L.; Teh, H.S.; Suah, J.L.; Husin, M.; Hwong, W.Y. SARS-CoV-2 in Malaysia: A surge of reinfection during the predominantly Omicron period. Lancet Reg. Health West. Pac. 2022, 26, 100572. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.R. 73 Infected in Yet Another Sabah School Cluster. Free Malaysia Today. 2022. Available online: https://www.freemalaysiatoday.com/category/nation/2022/01/19/73-infected-in-yet-another-sabah-school-cluster/ (accessed on 19 January 2022).
- Fong, D.R. 24 Students Infected in Sabah School Cluster. 2021. Available online: https://www.freemalaysiatoday.com/category/nation/2021/12/01/24-students-infected-in-sabah-school-cluster/ (accessed on 25 February 2023).
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef] [PubMed]
- Nextstrain. Genomic Epidemiology of Novel Coronavirus—Global Subsampling. 2023. Available online: https://nextstrain.org/ncov/global (accessed on 26 July 2023).
- Suppiah, J.; Kamel, K.; Mohd-Zawawi, Z.; Afizan, M.; Yahya, H.; Md-Hanif, S.; Thayan, R. Short Communication Phylogenomic analysis of SARS-CoV-2 from third wave clusters in Malaysia reveals dominant local lineage B.1.524 and persistent spike mutation A701V. Trop. Biomed. 2021, 38, 289–293. [Google Scholar]
- Cahyani, I.; Putro, E.W.; Ridwanuloh, A.M.; Wibowo, S.; Hariyatun, H.; Syahputra, G.; Akbariani, G.; Utomo, A.R.; Ilyas, M.; Loose, M.; et al. Genome Profiling of SARS-CoV-2 in Indonesia, ASEAN and the Neighbouring East Asian Countries: Features, Challenges and Achievements. Viruses 2022, 14, 778. [Google Scholar] [CrossRef]
- Azami, N.A.M.; Perera, D.; Thayan, R.; AbuBakar, S.; Sam, I.-C.; Salleh, M.Z.; Isa, M.N.M.; Ab Mutalib, N.S.; Aik, W.K.; Suppiah, J. SARS-CoV-2 genomic surveillance in Malaysia: Displacement of B.1.617.2 with AY lineages as the dominant Delta variants and the introduction of Omicron during the fourth epidemic wave. Int. J. Infect. Dis. 2022, 125, 216–226. [Google Scholar] [CrossRef]
- Franke, K.R.; Isett, R.; Robbins, A.; Paquette-Straub, C.; Shapiro, C.A.; Lee, M.M.; Crowgey, E.L. Genomic surveillance of SARS-CoV-2 in the state of Delaware reveals tremendous genomic diversity. PLoS ONE 2022, 17, e0262573. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balakrishnan, K.N.; Yew, C.W.; Chong, E.T.J.; Daim, S.; Mohamad, N.E.; Rodrigues, K.; Lee, P.-C. Timeline of SARS-CoV-2 Transmission in Sabah, Malaysia: Tracking the Molecular Evolution. Pathogens 2023, 12, 1047. https://doi.org/10.3390/pathogens12081047
Balakrishnan KN, Yew CW, Chong ETJ, Daim S, Mohamad NE, Rodrigues K, Lee P-C. Timeline of SARS-CoV-2 Transmission in Sabah, Malaysia: Tracking the Molecular Evolution. Pathogens. 2023; 12(8):1047. https://doi.org/10.3390/pathogens12081047
Chicago/Turabian StyleBalakrishnan, Krishnan Nair, Chee Wei Yew, Eric Tzyy Jiann Chong, Sylvia Daim, Nurul Elyani Mohamad, Kenneth Rodrigues, and Ping-Chin Lee. 2023. "Timeline of SARS-CoV-2 Transmission in Sabah, Malaysia: Tracking the Molecular Evolution" Pathogens 12, no. 8: 1047. https://doi.org/10.3390/pathogens12081047
APA StyleBalakrishnan, K. N., Yew, C. W., Chong, E. T. J., Daim, S., Mohamad, N. E., Rodrigues, K., & Lee, P. -C. (2023). Timeline of SARS-CoV-2 Transmission in Sabah, Malaysia: Tracking the Molecular Evolution. Pathogens, 12(8), 1047. https://doi.org/10.3390/pathogens12081047