

Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi

 ,
,
Abstract
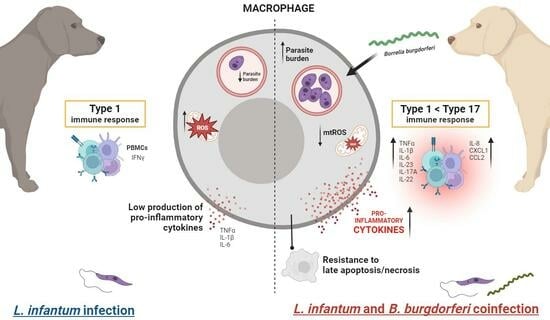
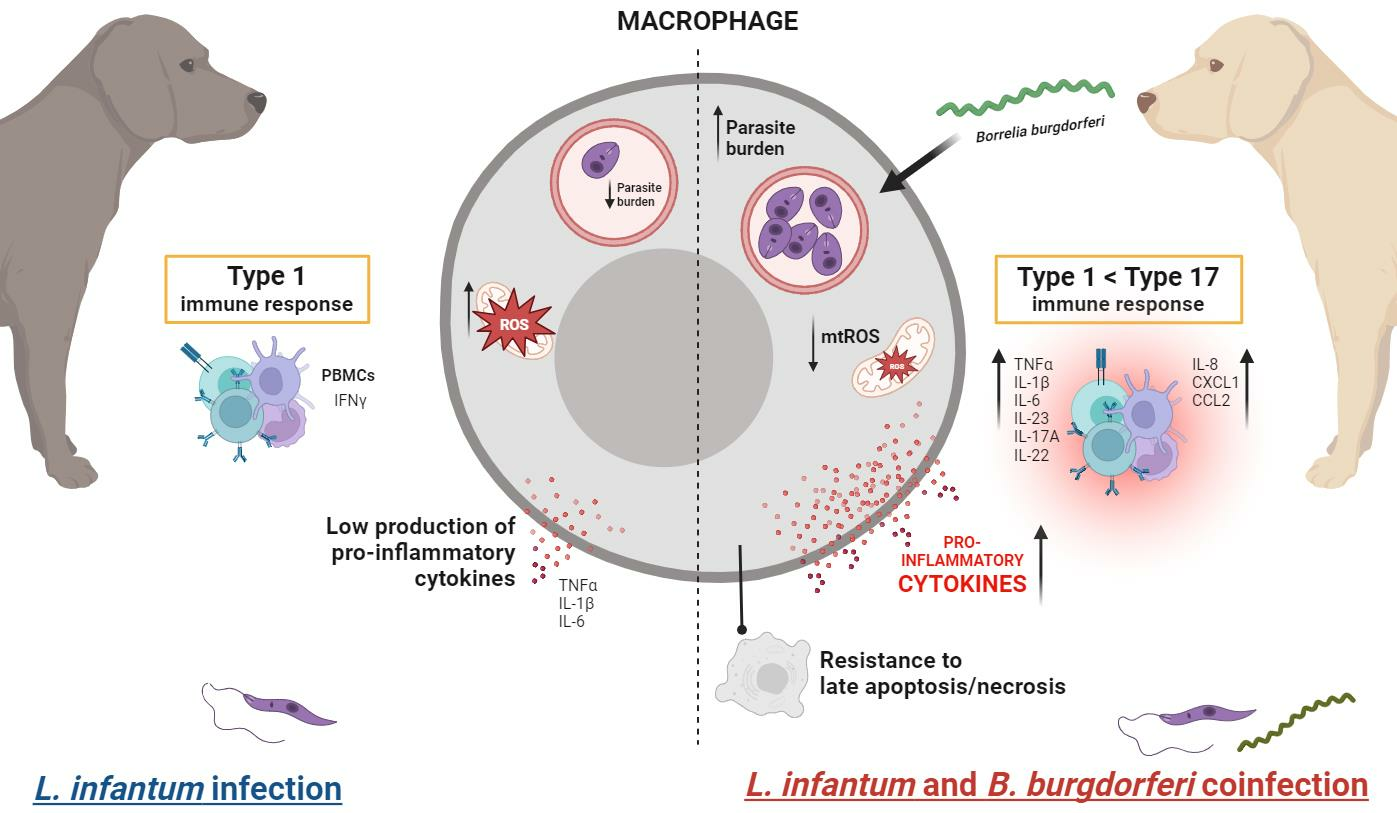
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Animals
2.3. Parasites and Bacteria
2.4. PBMC Stimulation Assay
2.5. Assessment of Leishmania In Vitro Burden
2.6. Apoptosis Assay
2.7. Reactive Oxygen Species (ROS) Detection
2.8. RNA Isolation and Quantitative PCR
2.9. Cytokine BioPlex and ELISA
2.10. Statistical Analyses
3. Results
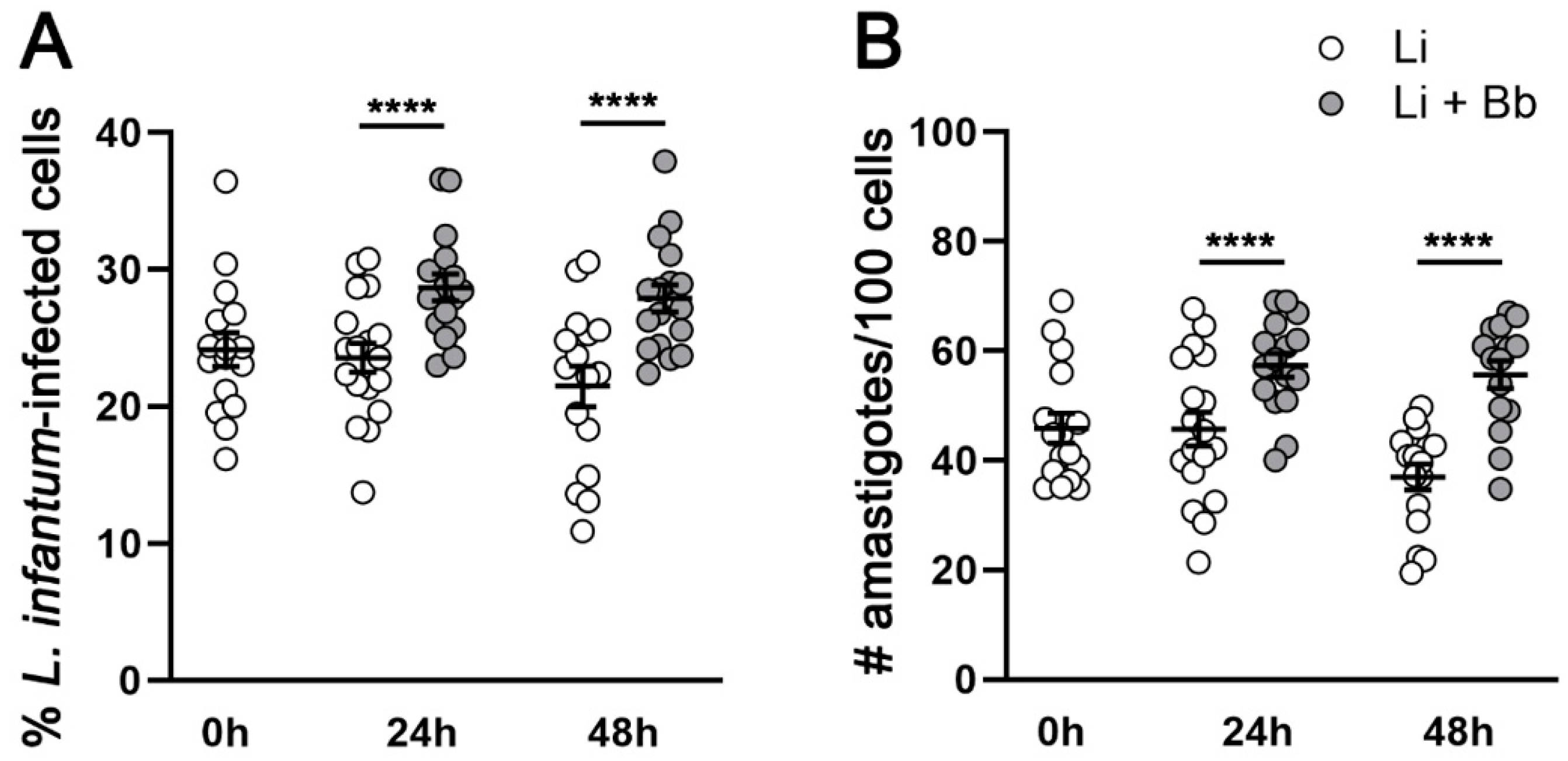
3.1. B. burgdorferi Coinfection Promotes L. infantum Infection in Macrophages
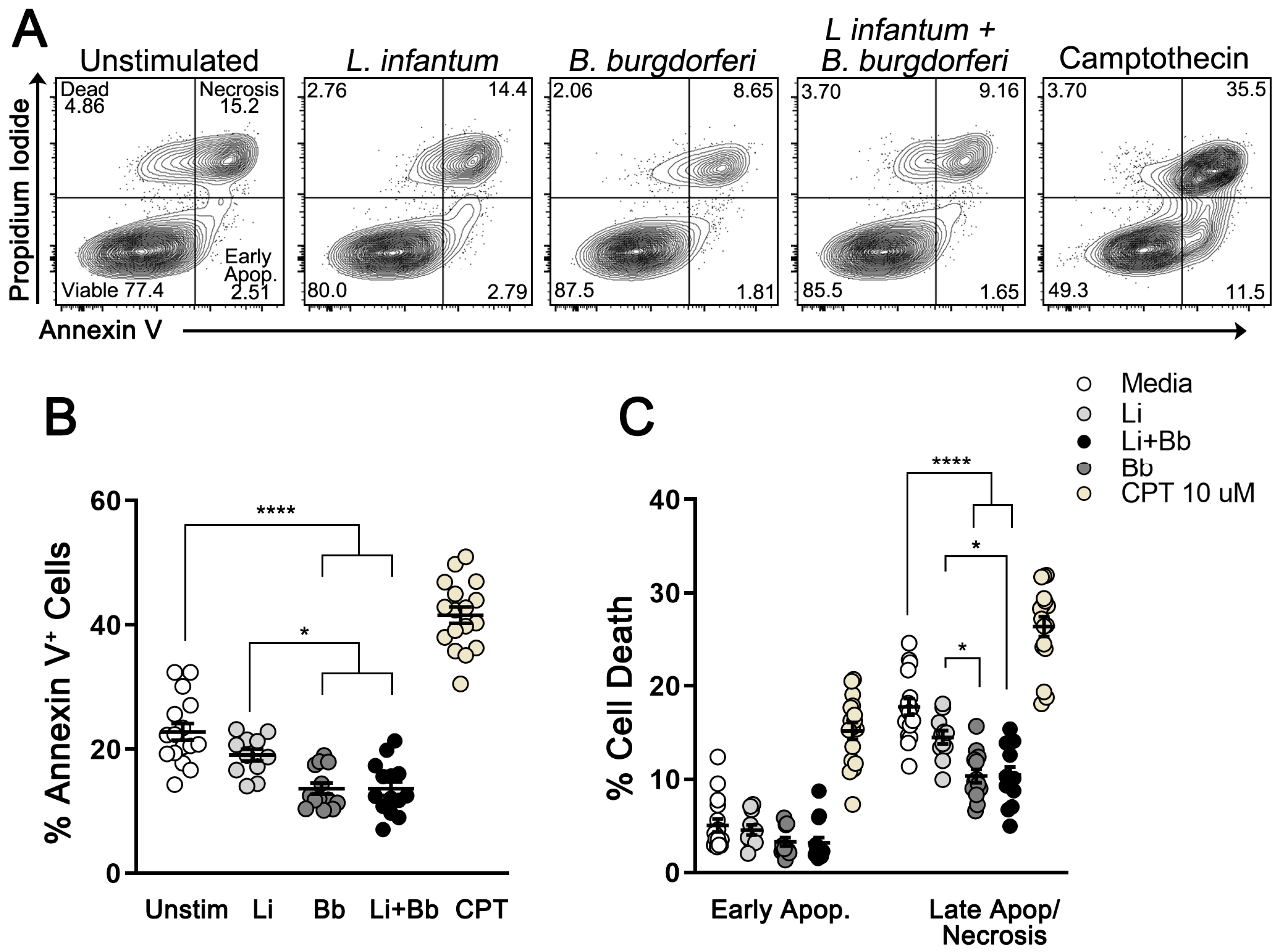
3.2. B. burgdorferi Coinfection Decreases Cell Death in L. infantum-Infected Macrophages
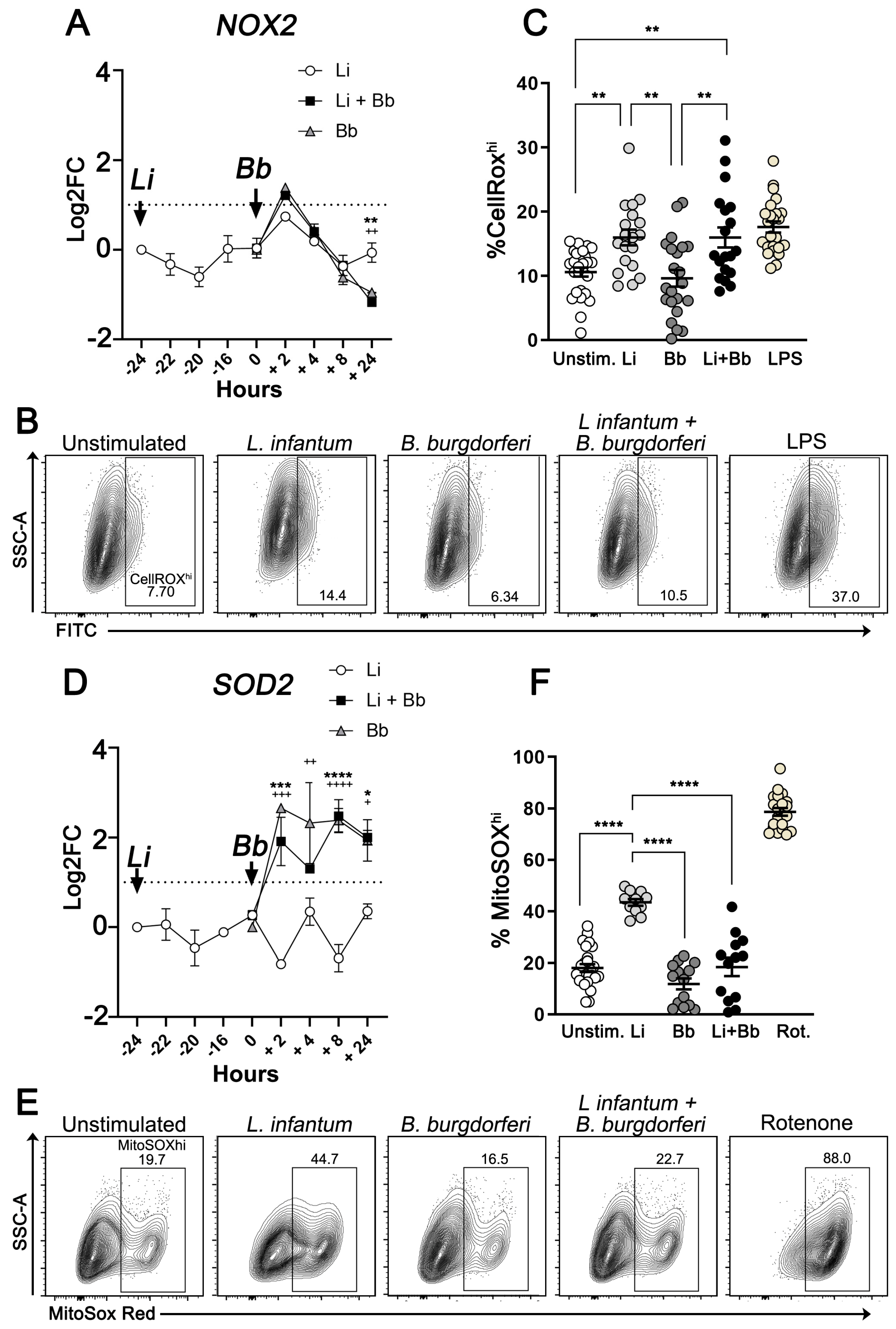
3.3. B. burgdorferi Coinfection Suppresses Mitochondrial ROS Production in L. infantum-Infected Macrophages
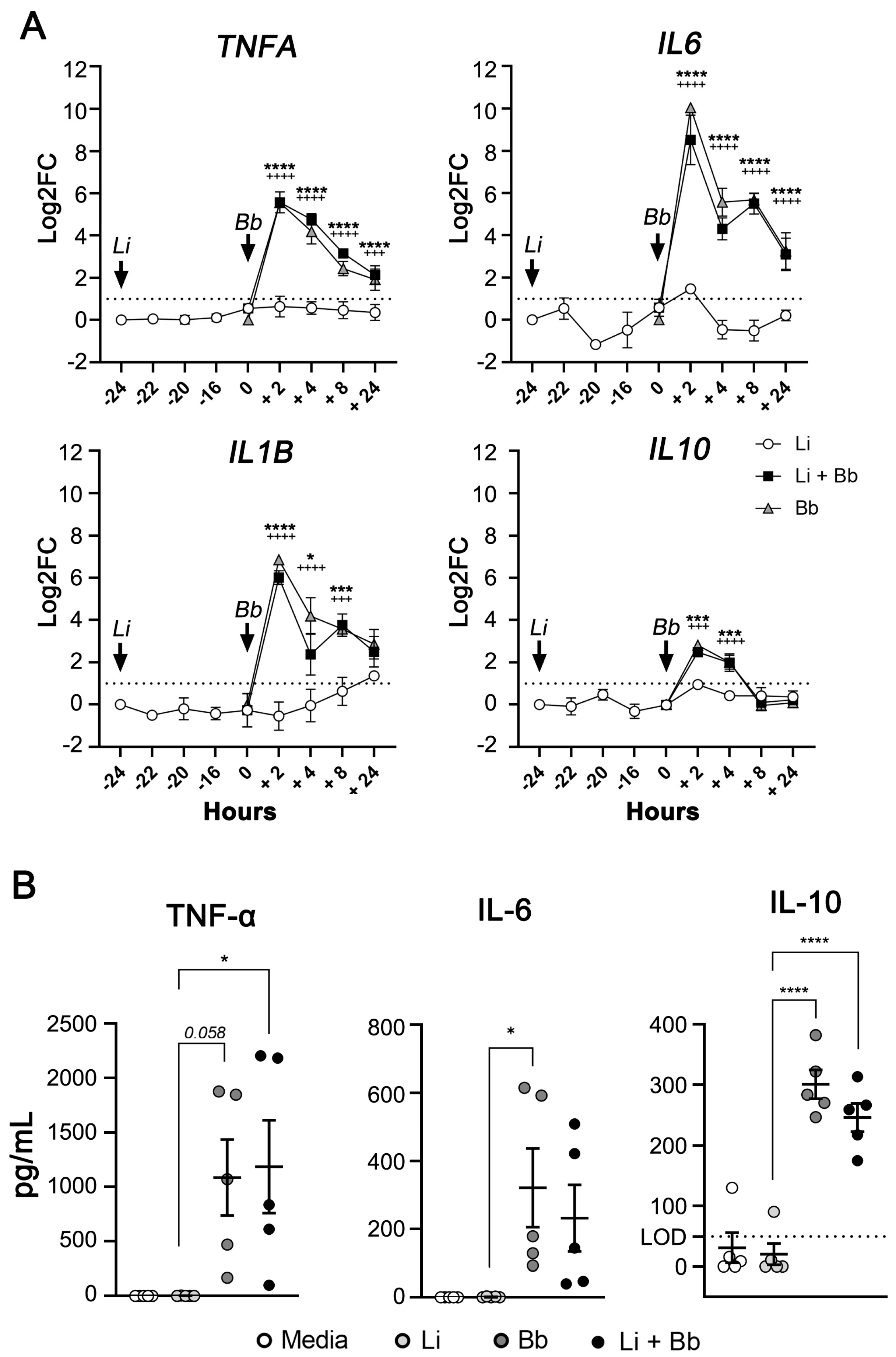
3.4. B. burgdorferi Infection Induces Transcription and Secretion of Pro-Inflammatory Cytokines in L. infantum-Infected Macrophages
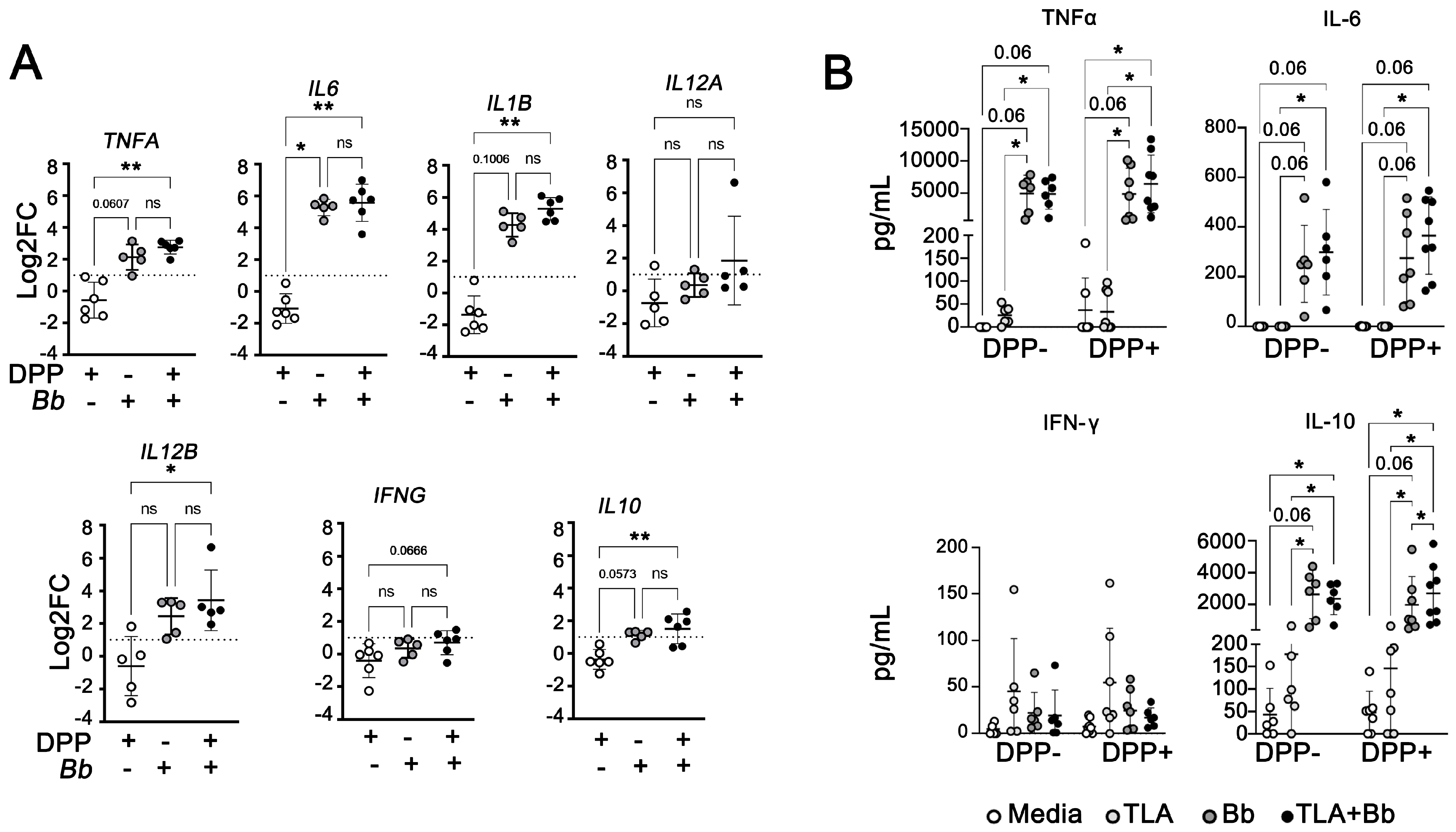
3.5. B. burgdorferi Induces Innate Pro-Inflammatory Responses in PBMCs from Dogs with Subclinical or Mild Canl
3.6. B. burgdorferi Induces Type 17 Cytokine Expression in PBMCs from L. infantum-Seropositive Dogs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Bogdan, C. Macrophages as host, effector and immunoregulatory cells in leishmaniasis: Impact of tissue micro-environment and metabolism. Cytokine X 2020, 2, 100041. [Google Scholar] [CrossRef]
- Kumar, R.; Nylén, S. Immunobiology of visceral leishmaniasis. Front. Immunol. 2012, 3, 251. [Google Scholar] [CrossRef]
- Gavgani, A.S.M.; Hodjati, M.; Mohite, H.; Davies, C. Effect of insecticide-impregnated dog collars on incidence of zoonotic visceral leishmaniasis in Iranian children: A matchedcluster randomised trial. Lancet Lond. Engl. 2002, 360, 374–379. [Google Scholar] [CrossRef]
- Dantas-Torres, F. The role of dogs as reservoirs of Leishmania parasites, with emphasis on Leishmania (Leishmania) infantum and Leishmania (Viannia) braziliensis. Vet. Parasitol. 2007, 149, 139–146. [Google Scholar] [CrossRef]
- Travi, B.L.; Tabares, C.J.; Cadena, H.; Ferro, C.; Osorio, Y. Canine visceral leishmaniasis in Colombia: Relationship between clinical and parasitologic status and infectivity for sand flies. Am. J. Trop. Med. Hyg. 2001, 64, 119–124. [Google Scholar] [CrossRef]
- Courtenay, O.; Quinnell, R.J.; Garcez, L.M.; Shaw, J.J.; Dye, C. Infectiousness in a Cohort of Brazilian Dogs: Why Culling Fails to Control Visceral Leishmaniasis in Areas of High Transmission. J. Infect. Dis. 2002, 186, 1314–1320. [Google Scholar] [CrossRef]
- Laurenti, M.D.; Rossi, C.N.; da Matta, V.L.R.; Tomokane, T.Y.; Corbett, C.E.P.; Secundino, N.F.C.; Pimenta, P.F.P.; Marcondes, M. Asymptomatic dogs are highly competent to transmit Leishmania (Leishmania) infantum chagasi to the natural vector. Vet. Parasitol. 2013, 196, 296–300. [Google Scholar] [CrossRef]
- Scorza, B.M.; Mahachi, K.G.; Cox, A.C.; Toepp, A.J.; Leal-Lima, A.; Kumar Kushwaha, A.; Kelly, P.; Meneses, C.; Wilson, G.; Gibson-Corley, K.N.; et al. Leishmania infantum xenodiagnosis from vertically infected dogs reveals significant skin tropism. PLoS Neglected Trop. Dis. 2021, 15, e0009366. [Google Scholar] [CrossRef] [PubMed]
- Boggiatto, P.M.; Gibson-Corley, K.N.; Metz, K.; Gallup, J.M.; Hostetter, J.M.; Mullin, K.; Petersen, C.A. Transplacental Transmission of Leishmania infantum as a Means for Continued Disease Incidence in North America. PLoS Neglected Trop. Dis. 2011, 5, e1019. [Google Scholar] [CrossRef]
- Toepp, A.J.; Schaut, R.G.; Scott, B.D.; Mathur, D.; Berens, A.J.; Petersen, C.A. Leishmania incidence and prevalence in U.S. hunting hounds maintained via vertical transmission. Vet. Parasitol. Reg. Stud. Rep. 2017, 10, 75–81. [Google Scholar] [CrossRef]
- Hlavacova, J.; Votypka, J.; Volf, P. The Effect of Temperature on Leishmania (Kinetoplastida: Trypanosomatidae) Development in Sand Flies. J. Med. Entomol. 2013, 50, 955–958. [Google Scholar] [CrossRef]
- Chalghaf, B.; Chemkhi, J.; Mayala, B.; Harrabi, M.; Benie, G.B.; Michael, E.; Ben Salah, A. Ecological niche modeling predicting the potential distribution of Leishmania vectors in the Mediterranean basin: Impact of climate change. Parasites Vectors 2018, 11, 461. [Google Scholar] [CrossRef]
- Curtin, J.M.; Aronson, N.E. Leishmaniasis in the United States: Emerging Issues in a Region of Low Endemicity. Microorganisms 2021, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Beasley, E.A.; Mahachi, K.G.; Petersen, C.A. Possibility of Leishmania Transmission via Lutzomyia spp. Sand Flies within the USA and Implications for Human and Canine Autochthonous Infection. Curr. Trop. Med. Rep. 2022, 9, 160–168. [Google Scholar] [CrossRef]
- Attipa, C.; Solano-Gallego, L.; Papasouliotis, K.; Soutter, F.; Morris, D.; Helps, C.; Carver, S.; Tasker, S. Association between canine leishmaniosis and Ehrlichia canis co-infection: A prospective case-control study. Parasites Vectors 2018, 11, 184. [Google Scholar] [CrossRef]
- Baxarias, M.; Álvarez-Fernández, A.; Martínez-Orellana, P.; Montserrat-Sangrà, S.; Ordeix, L.; Rojas, A.; Nachum-Biala, Y.; Baneth, G.; Solano-Gallego, L. Does co-infection with vector-borne pathogens play a role in clinical canine leishmaniosis? Parasites Vectors 2018, 11, 135. [Google Scholar] [CrossRef]
- Toepp, A.J.; Monteiro, G.R.G.; Coutinho, J.F.V.; Lima, A.L.; Larson, M.; Wilson, G.; Grinnage-Pulley, T.; Bennett, C.; Mahachi, K.; Anderson, B.; et al. Comorbid infections induce progression of visceral leishmaniasis. Parasites Vectors 2019, 12, 54. [Google Scholar] [CrossRef]
- Mahachi, K.; Kontowicz, E.; Anderson, B.; Toepp, A.J.; Lima, A.L.; Larson, M.; Wilson, G.; Grinnage-Pulley, T.; Bennett, C.; Ozanne, M.; et al. Predominant risk factors for tick-borne co-infections in hunting dogs from the USA. Parasites Vectors 2020, 13, 247. [Google Scholar] [CrossRef]
- Miró, G.; Wright, I.; Michael, H.; Burton, W.; Hegarty, E.; Rodón, J.; Buch, J.; Pantchev, N.; von Samson-Himmelstjerna, G. Seropositivity of main vector-borne pathogens in dogs across Europe. Parasites Vectors 2022, 15, 189. [Google Scholar] [CrossRef]
- Benjamin, S.J.; Hawley, K.L.; Vera-Licona, P.; La Vake, C.J.; Cervantes, J.L.; Ruan, Y.; Radolf, J.D.; Salazar, J.C. Macrophage mediated recognition and clearance of Borrelia burgdorferi elicits MyD88-dependent and -independent phagosomal signals that contribute to phagocytosis and inflammation. BMC Immunol. 2021, 22, 32. [Google Scholar] [CrossRef]
- Rossi, M.; Fasel, N. How to master the host immune system? Leishmania parasites have the solutions! Int. Immunol. 2018, 30, 103–111. [Google Scholar] [CrossRef]
- Anderson, C.; Brissette, C.A. The Brilliance of Borrelia: Mechanisms of Host Immune Evasion by Lyme Disease-Causing Spirochetes. Pathogens 2021, 10, 281. [Google Scholar] [CrossRef]
- van Zandbergen, G.; Klinger, M.; Mueller, A.; Dannenberg, S.; Gebert, A.; Solbach, W.; Laskay, T. Cutting Edge: Neutrophil Granulocyte Serves as a Vector for Leishmania Entry into Macrophages. J. Immunol. 2004, 173, 6521–6525. [Google Scholar] [CrossRef] [PubMed]
- Peters, N.C.; Egen, J.G.; Secundino, N.; Debrabant, A.; Kimblin, N.; Kamhawi, S.; Lawyer, P.; Fay, M.P.; Germain, R.N.; Sacks, D. In Vivo Imaging Reveals an Essential Role for Neutrophils in Leishmaniasis Transmitted by Sand Flies. Science 2008, 321, 970–974. [Google Scholar] [CrossRef]
- Gueirard, P.; Laplante, A.; Rondeau, C.; Milon, G.; Desjardins, M. Trafficking of Leishmania donovani promastigotes in non-lytic compartments in neutrophils enables the subsequent transfer of parasites to macrophages. Cell. Microbiol. 2007, 10, 100–111. [Google Scholar] [CrossRef]
- Lisi, S.; Sisto, M.; Acquafredda, A.; Spinelli, R.; Schiavone, M.; Mitolo, V.; Brandonisio, O.; Panaro, M.A. Infection with Leishmania infantum Inhibits Actinomycin D-Induced Apoptosis of Human Monocytic Cell Line U-937. J. Eukaryot. Microbiol. 2005, 52, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, A.; Leal, N.; Kima, P.E. Leishmania promastigotes activate PI3K/Akt signalling to confer host cell resistance to apoptosis. Cell. Microbiol. 2007, 9, 84–96. [Google Scholar] [CrossRef]
- Das, S.; Ghosh, A.K.; Singh, S.; Saha, B.; Ganguly, A.; Das, P. Unmethylated CpG motifs in the L. donovani DNA regulate TLR9-dependent delay of programmed cell death in macrophages. J. Leukoc. Biol. 2015, 97, 363–378. [Google Scholar] [CrossRef]
- Solano-Gálvez, S.-G.; Álvarez-Hernández, D.-A.; Gutiérrez-Kobeh, L.; Vázquez-López, R. Leishmania: Manipulation of signaling pathways to inhibit host cell apoptosis. Ther. Adv. Infect. Dis. 2021, 8, 20499361211014976. [Google Scholar] [CrossRef]
- Privé, C.; Descoteaux, A. Leishmania donovani promastigotes evade the activation of mitogen-activated protein kinases p38, c-Jun N-terminal kinase, and extracellular signal-regulated kinase-1/2 during infection of naive macrophages. Eur. J. Immunol. 2000, 30, 2235–2244. [Google Scholar] [CrossRef]
- Junghae, M.; Raynes, J.G. Activation of p38 Mitogen-Activated Protein Kinase Attenuates Leishmania donovani Infection in Macrophages. Infect. Immun. 2002, 70, 5026–5035. [Google Scholar] [CrossRef]
- Pandey, R.K.; Mehrotra, S.; Sharma, S.; Gudde, R.S.; Sundar, S.; Shaha, C. Leishmania donovani-Induced Increase in Macrophage Bcl-2 Favors Parasite Survival. Front. Immunol. 2016, 7, 456. [Google Scholar] [CrossRef]
- Cruz, A.R.; Moore, M.W.; La Vake, C.J.; Eggers, C.H.; Salazar, J.C.; Radolf, J.D. Phagocytosis of Borrelia burgdorferi, the Lyme Disease Spirochete, Potentiates Innate Immune Activation and Induces Apoptosis in Human Monocytes. Infect. Immun. 2008, 76, 56–70. [Google Scholar] [CrossRef]
- Karvonen, K.; Nykky, J.; Marjomäki, V.; Gilbert, L. Distinctive Evasion Mechanisms to Allow Persistence of Borrelia burgdorferi in Different Human Cell Lines. Front. Microbiol. 2021, 12, 711291. [Google Scholar] [CrossRef] [PubMed]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Saha, S.; Basu, M.; Guin, S.; Gupta, P.; Mitterstiller, A.-M.; Weiss, G.; Jana, K.; Ukil, A. Leishmania donovani Exploits Macrophage Heme Oxygenase-1 To Neutralize Oxidative Burst and TLR Signaling–Dependent Host Defense. J. Immunol. 2019, 202, 827–840. [Google Scholar] [CrossRef]
- Lodge, R.; Diallo, T.O.; Descoteaux, A. Leishmania donovani lipophosphoglycan blocks NADPH oxidase assembly at the phagosome membrane. Cell. Microbiol. 2006, 8, 1922–1931. [Google Scholar] [CrossRef]
- Boylan, J.A.; Lawrence, K.A.; Downey, J.S.; Gherardini, F.C. Borrelia burgdorferi membranes are the primary targets of reactive oxygen species. Mol. Microbiol. 2008, 68, 786–799. [Google Scholar] [CrossRef]
- Esteve-Gassent, M.D.; Elliott, N.L.; Seshu, J. sodA is essential for virulence of Borrelia burgdorferi in the murine model of Lyme disease. Mol. Microbiol. 2009, 71, 594–612. [Google Scholar] [CrossRef]
- Troxell, B.; Xu, H.; Yang, X.F. Borrelia burgdorferi, a Pathogen That Lacks Iron, Encodes Manganese-dependent Superoxide Dismutase Essential for Resistance to Streptonigrin. J. Biol. Chem. 2012, 287, 19284–19293. [Google Scholar] [CrossRef]
- Kerstholt, M.; Vrijmoeth, H.; Lachmandas, E.; Oosting, M.; Lupse, M.; Flonta, M.; Dinarello, C.A.; Netea, M.G.; Joosten, L.A.B. Role of glutathione metabolism in host defense against Borrelia burgdorferi infection. Proc. Natl. Acad. Sci. USA 2018, 115, E2320–E2328. [Google Scholar] [CrossRef]
- Kerstholt, M.; Brouwer, M.; te Vrugt, M.; Oosting, M.; Netea, M.G.; Joosten, L.A.B. Borrelia burgdorferi inhibits NADPH-mediated reactive oxygen species production through the mTOR pathway. Ticks Tick-Borne Dis. 2022, 13, 101943. [Google Scholar] [CrossRef]
- Basu Ball, W.; Kar, S.; Mukherjee, M.; Chande, A.G.; Mukhopadhyaya, R.; Das, P.K. Uncoupling Protein 2 Negatively Regulates Mitochondrial Reactive Oxygen Species Generation and Induces Phosphatase-Mediated Anti-Inflammatory Response in Experimental Visceral Leishmaniasis. J. Immunol. 2011, 187, 1322–1332. [Google Scholar] [CrossRef]
- Mukherjee, M.; Ball, W.B.; Das, P.K. Leishmania donovani activates SREBP2 to modulate macrophage membrane cholesterol and mitochondrial oxidants for establishment of infection. Int. J. Biochem. Cell Biol. 2014, 55, 196–208. [Google Scholar] [CrossRef]
- Gonçalves-De-Albuquerque, S.d.C.; Pessoa-E-Silva, R.; Trajano-Silva, L.A.M.; de Goes, T.C.; de Morais, R.C.S.; Oliveira, C.N.d.C.; de Lorena, V.M.B.; de Paiva-Cavalcanti, M. The Equivocal Role of Th17 Cells and Neutrophils on Immunopathogenesis of Leishmaniasis. Front. Immunol. 2017, 8, 1437. [Google Scholar] [CrossRef]
- Henningsson, A.J.; Tjernberg, I.; Malmvall, B.-E.; Forsberg, P.; Ernerudh, J. Indications of Th1 and Th17 responses in cerebrospinal fluid from patients with Lyme neuroborreliosis: A large retrospective study. J. Neuroinflamm. 2011, 8, 36. [Google Scholar] [CrossRef]
- Strle, K.; Sulka, K.B.; Pianta, A.; Crowley, J.T.; Arvikar, S.L.; Anselmo, A.; Sadreyev, R.; Steere, A.C. T-Helper 17 Cell Cytokine Responses in Lyme Disease Correlate with Borrelia burgdorferi Antibodies during Early Infection and with Autoantibodies Late in the Illness in Patients with Antibiotic-Refractory Lyme Arthritis. Clin. Infect. Dis. 2017, 64, 930–938. [Google Scholar] [CrossRef]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef]
- Hosein, S.; Rodríguez-Cortés, A.; Blake, D.P.; Allenspach, K.; Alberola, J.; Solano-Gallego, L. Transcription of Toll-like Receptors 2, 3, 4 and 9, FoxP3 and Th17 Cytokines in a Susceptible Experimental Model of Canine Leishmania infantum Infection. PLoS ONE 2015, 10, e0140325. [Google Scholar] [CrossRef]
- Nascimento, M.S.L.; Albuquerque, T.D.R.; Nascimento, A.F.S.; Caldas, I.S.; Do-Valle-Matta, M.A.; Souto, J.T.; Talvani, A.; Bahia, M.; Galvão, L.; Câmara, A.; et al. Impairment of Interleukin-17A Expression in Canine Visceral Leishmaniosis is Correlated with Reduced Interferon-γ and Inducible Nitric Oxide Synthase Expression. J. Comp. Pathol. 2015, 153, 197–205. [Google Scholar] [CrossRef]
- Boggiatto, P.M.; Ramer-Tait, A.E.; Metz, K.; Kramer, E.E.; Gibson-Corley, K.; Mullin, K.; Hostetter, J.M.; Gallup, J.M.; Jones, D.E.; Petersen, C.A. Immunologic Indicators of Clinical Progression during Canine Leishmania infantum Infection. Clin. Vaccine Immunol. 2010, 17, 267–273. [Google Scholar] [CrossRef]
- Solano-Gallego, L.; Miró, G.; Koutinas, A.; Cardoso, L.; Pennisi, M.G.; Ferrer, L.; Bourdeau, P.; Oliva, G.; Baneth, G. LeishVet guidelines for the practical management of canine leishmaniosis. Parasites Vectors 2011, 4, 86. [Google Scholar] [CrossRef]
- Nadaes, N.R.; da Costa, L.S.; Santana, R.C.; LaRocque-de-Freitas, I.F.; Vivarini, A.C.; Soares, D.C.; Wardini, A.B.; Lopes, U.G.; Saraiva, E.M.; Freire-de-Lima, C.G.; et al. DH82 Canine and RAW264.7 Murine Macrophage Cell Lines Display Distinct Activation Profiles upon Interaction with Leishmania infantum and Leishmania amazonensis. Front. Cell. Infect. Microbiol. 2020, 10, 247. [Google Scholar] [CrossRef]
- Flynn, J.M.; Melov, S. SOD2 in mitochondrial dysfunction and neurodegeneration. Free. Radic. Biol. Med. 2013, 62, 4–12. [Google Scholar] [CrossRef]
- Petzke, M.M.; Brooks, A.; Krupna, M.A.; Mordue, D.; Schwartz, I. Recognition of Borrelia burgdorferi, the Lyme Disease Spirochete, by TLR7 and TLR9 Induces a Type I IFN Response by Human Immune Cells. J. Immunol. 2009, 183, 5279–5292. [Google Scholar] [CrossRef]
- Shio, M.T.; Hassani, K.; Isnard, A.; Ralph, B.; Contreras, I.; Gomez, M.A.; Abu-Dayyeh, I.; Olivier, M. Host Cell Signalling and Leishmania Mechanisms of Evasion. J. Trop. Med. 2012, 2012, 819512. [Google Scholar] [CrossRef]
- Gupta, G.; Oghumu, S.; Satoskar, A.R. Mechanisms of Immune Evasion in Leishmaniasis. Adv. Appl. Microbiol. 2013, 82, 155–184. [Google Scholar] [CrossRef]
- Gurjar, D.; Kumar Patra, S.; Bodhale, N.; Lenka, N.; Saha, B. Leishmania intercepts IFN-γR signaling at multiple levels in macrophages. Cytokine 2022, 157, 155956. [Google Scholar] [CrossRef]
- Alexander, J.; Brombacher, F. T Helper1/T Helper2 Cells and Resistance/Susceptibility to Leishmania Infection: Is This Paradigm Still Relevant? Front. Immunol. 2012, 3, 80. [Google Scholar] [CrossRef]
- Zhou, F. Molecular Mechanisms of IFN-γ to Up-Regulate MHC Class I Antigen Processing and Presentation. Int. Rev. Immunol. 2009, 28, 239–260. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Jeon, R.; Vuckovic, I.; Jiang, X.; Lerman, A.; Folmes, C.D.; Dzeja, P.D.; Herrmann, J. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine 2018, 30, 303–316. [Google Scholar] [CrossRef]
- Espígol-Frigolé, G.; Planas-Rigol, E.; Lozano, E.; Corbera-Bellalta, M.; Terrades-García, N.; Prieto-González, S.; García-Martínez, A.; Hernández-Rodríguez, J.; Grau, J.M.; Cid, M.C. Expression and Function of IL12/23 Related Cytokine Subunits (p35, p40, and p19) in Giant-Cell Arteritis Lesions: Contribution of p40 to Th1- and Th17-Mediated Inflammatory Pathways. Front. Immunol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Zenobia, C.; Hajishengallis, G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol. 2000 2015, 69, 142–159. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Glickstein, L.J.; Coburn, J.L. Short report: Association of macrophage inflammatory response and cell death after in vitro Borrelia burgdorferi infection with arthritis resistance. Am. J. Trop. Med. Hyg. 2006, 75, 964–967. [Google Scholar] [CrossRef]
- Myers, T.A.; Kaushal, D.; Philipp, M.T. Microglia Are Mediators of Borrelia burgdorferi–Induced Apoptosis in SH-SY5Y Neuronal Cells. PLoS Pathog. 2009, 5, e1000659. [Google Scholar] [CrossRef]
- Livengood, J.A.; Gilmore, R.D. Invasion of human neuronal and glial cells by an infectious strain of Borrelia burgdorferi. Microbes Infect. 2006, 8, 2832–2840. [Google Scholar] [CrossRef]
- Karvonen, K.; Tammisto, H.; Nykky, J.; Gilbert, L. Borrelia burgdorferi Outer Membrane Vesicles Contain Antigenic Proteins, but Do Not Induce Cell Death in Human Cells. Microorganisms 2022, 10, 212. [Google Scholar] [CrossRef]
- Schramm, F.; Kern, A.; Barthel, C.; Nadaud, S.; Meyer, N.; Jaulhac, B.; Boulanger, N. Microarray Analyses of Inflammation Response of Human Dermal Fibroblasts to Different Strains of Borrelia burgdorferi Sensu Stricto. PLoS ONE 2012, 7, e40046. [Google Scholar] [CrossRef]
- Wawrzeniak, K.; Gaur, G.; Sapi, E.; Senejani, A.G. Effect of Borrelia burgdorferi Outer Membrane Vesicles on Host Oxidative Stress Response. Antibiotics 2020, 9, 275. [Google Scholar] [CrossRef]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef]
- Geng, J.; Sun, X.; Wang, P.; Zhang, S.; Wang, X.; Wu, H.; Hong, L.; Xie, C.; Li, X.; Zhao, H.; et al. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol. 2015, 16, 1142–1152. [Google Scholar] [CrossRef] [PubMed]
- Abuaita, B.H.; Schultz, T.L.; O’riordan, M.X. Mitochondria-Derived Vesicles Deliver Antimicrobial Reactive Oxygen Species to Control Phagosome-Localized Staphylococcus aureus. Cell Host Microbe 2018, 24, 625–636.e5. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Kim, Y.J.; Jo, E.-K. Mitochondrial Reactive Oxygen Species: Double-Edged Weapon in Host Defense and Pathological Inflammation during Infection. Front. Immunol. 2020, 11, 1649. [Google Scholar] [CrossRef]
- Ashida, H.; Mimuro, H.; Ogawa, M.; Kobayashi, T.; Sanada, T.; Kim, M.; Sasakawa, C. Cell death and infection: A double-edged sword for host and pathogen survival. J. Cell Biol. 2011, 195, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Toepp, A.J.; Petersen, C.A. The balancing act: Immunology of leishmaniosis. Res. Veter. Sci. 2020, 130, 19–25. [Google Scholar] [CrossRef]
- Esch, K.J.; Juelsgaard, R.; Martinez, P.A.; Jones, D.E.; Petersen, C.A. Programmed Death 1–Mediated T Cell Exhaustion during Visceral Leishmaniasis Impairs Phagocyte Function. J. Immunol. 2013, 191, 5542–5550. [Google Scholar] [CrossRef]
- van de Schoor, F.R.; Vrijmoeth, H.D.; Brouwer, M.A.E.; ter Hofstede, H.J.M.; Lemmers, H.L.M.; Dijkstra, H.; Boahen, C.K.; Oosting, M.; Kullberg, B.-J.; Hovius, J.W.; et al. Borrelia burgdorferi Is a Poor Inducer of Gamma Interferon: Amplification Induced by Interleukin-12. Infect. Immun. 2022, 90, e0055821. [Google Scholar] [CrossRef]
- Malovrh, T.; Skoberne, M.; Gruntar, I.; Kotnik, V. The cell-mediated immune response to Borrelia afzelii, garinii and burgdorferi in C57BL/6 mice is dependent on antigen specificity. FEMS Immunol. Med. Microbiol. 2000, 28, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, Y.; Tupin, E.; Wu, D.; Fujio, M.; Garcia-Navarro, R.; Benhnia, M.R.-E.; Zajonc, D.M.; Ben-Menachem, G.; Ainge, G.D.; Painter, G.F.; et al. Natural killer T cells recognize diacylglycerol antigens from pathogenic bacteria. Nat. Immunol. 2006, 7, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Codolo, G.; Amedei, A.; Steere, A.C.; Papinutto, E.; Cappon, A.; Polenghi, A.; Benagiano, M.; Paccani, S.R.; Sambri, V.; Del Prete, G.; et al. Borrelia burgdorferi NapA-driven Th17 cell inflammation in lyme arthritis. Arthritis Rheum. 2008, 58, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Oosting, M.; van de Veerdonk, F.L.; Kanneganti, T.-D.; Sturm, P.; Verschueren, I.; Berende, A.; van der Meer, J.W.M.; Kullberg, B.-J.; Netea, M.G.; Joosten, L.A.B. Borrelia species induce inflammasome activation and IL-17 production through a caspase-1-dependent mechanism. Eur. J. Immunol. 2011, 41, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Grygorczuk, S.; Świerzbińska, R.; Moniuszko, A.; Kondrusik, M.; Zajkowska, J.; Czupryna, P.; Dunaj, J.; Pancewicz, S. Synthesis of Th17 cytokines in the culture of peripheral blood mononuclear cells stimulated with Borrelia burgdorferi sensu lato. Ann. Agric. Environ. Med. 2016, 23, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Gyllemark, P.; Forsberg, P.; Ernerudh, J.; Henningsson, A.J. Intrathecal Th17- and B cell-associated cytokine and chemokine responses in relation to clinical outcome in Lyme neuroborreliosis: A large retrospective study. J. Neuroinflamm. 2017, 14, 27. [Google Scholar] [CrossRef]
- Pitta, M.G.R.; Romano, A.; Cabantous, S.; Henri, S.; Hammad, A.; Kouriba, B.; Argiro, L.; el Kheir, M.; Bucheton, B.; Mary, C.; et al. IL-17 and IL-22 are associated with protection against human kala azar caused by Leishmania donovani. J. Clin. Investig. 2009, 119, 2379–2387. [Google Scholar] [CrossRef]
- Nascimento, M.S.L.; Carregaro, V.; Lima-Júnior, D.S.; Costa, D.L.; Ryffel, B.; Duthie, M.S.; de Jesus, A.; de Almeida, R.P.; da Silva, J.S. Interleukin 17A Acts Synergistically with Interferon γ to Promote Protection against Leishmania infantum Infection. J. Infect. Dis. 2014, 211, 1015–1026. [Google Scholar] [CrossRef]
- Sheel, M.; Beattie, L.; Frame, T.C.M.; de Labastida Rivera, F.; Faleiro, R.J.; Bunn, P.T.; de Oca, M.M.; Edwards, C.L.; Ng, S.S.; Kumar, R.; et al. IL-17A–Producing γδ T Cells Suppress Early Control of Parasite Growth by Monocytes in the Liver. J. Immunol. 2015, 195, 5707–5717. [Google Scholar] [CrossRef]
- Terrazas, C.; Varikuti, S.; Kimble, J.; Moretti, E.; Boyaka, P.N.; Satoskar, A.R. IL-17A promotes susceptibility during experimental visceral leishmaniasis caused by Leishmania donovani. FASEB J. 2016, 30, 1135–1143. [Google Scholar] [CrossRef]
- Khatonier, R.; Ahmed, G.; Sarmah, P.; Narain, K.; Khan, A.M. Immunomodulatory role of Th17 pathway in experimental visceral leishmaniasis. Immunobiology 2021, 226, 152148. [Google Scholar] [CrossRef]
- Beasley, E.A.; Pessôa-Pereira, D.; Scorza, B.M.; Petersen, C.A. Epidemiologic, Clinical and Immunological Consequences of Co-Infections during Canine Leishmaniosis. Animals 2021, 11, 3206. [Google Scholar] [CrossRef] [PubMed]
- Montoya-Alonso, J.A.; Morchón, R.; Costa-Rodríguez, N.; Matos, J.I.; Falcón-Cordón, Y.; Carretón, E. Current Distribution of Selected Vector-Borne Diseases in Dogs in Spain. Front. Veter. Sci. 2020, 7, 564429. [Google Scholar] [CrossRef]
- Oliveira, V.D.C.; Junior, A.A.V.M.; Ferreira, L.C.; Calvet, T.M.Q.; dos Santos, S.A.; Figueiredo, F.B.; Campos, M.P.; Rodrigues, F.d.C.D.C.; Oliveira, R.d.V.C.D.; de Lemos, E.R.S.; et al. Frequency of co-seropositivities for certain pathogens and their relationship with clinical and histopathological changes and parasite load in dogs infected with Leishmania infantum. PLoS ONE 2021, 16, e0247560. [Google Scholar] [CrossRef] [PubMed]
- Bitsaktsis, C.; Huntington, J.; Winslow, G. Production of IFN-γ by CD4 T Cells Is Essential for Resolving Ehrlichia Infection. J. Immunol. 2004, 172, 6894–6901. [Google Scholar] [CrossRef]
- Lina, T.T.; Farris, T.; Luo, T.; Mitra, S.; Zhu, B.; McBride, J.W. Hacker within! Ehrlichia chaffeensis Effector Driven Phagocyte Reprogramming Strategy. Front. Cell. Infect. Microbiol. 2016, 6, 58. [Google Scholar] [CrossRef]
- Zückert, W.R. Laboratory Maintenance of Borrelia burgdorferi. Curr. Protoc. Microbiol. 2007, 4, 12C-1. [Google Scholar] [CrossRef]
- Cheng, C.; Ganta, R.R. Laboratory Maintenance of Ehrlichia chaffeensis and Ehrlichia canis and Recovery of Organisms for Molecular Biology and Proteomics Studies. Curr. Protoc. Microbiol. 2008, 9, 3A-1. [Google Scholar] [CrossRef]
- Nadelman, R.B.; Wormser, G.P. Reinfection in Patients with Lyme Disease. Clin. Infect. Dis. 2007, 45, 1032–1038. [Google Scholar] [CrossRef]
- Beall, M.J.; Alleman, A.R.; Breitschwerdt, E.B.; Cohn, L.A.; Couto, C.G.; Dryden, M.W.; Guptill, L.C.; Iazbik, C.; Kania, S.A.; Lathan, P.; et al. Seroprevalence of Ehrlichia canis, Ehrlichia chaffeensis and Ehrlichia ewingii in dogs in North America. Parasites Vectors 2012, 5, 29. [Google Scholar] [CrossRef]
- Herrin, B.H.; Beall, M.J.; Feng, X.; Papeş, M.; Little, S.E. Canine and human infection with Borrelia burgdorferi in the New York City metropolitan area. Parasites Vectors 2018, 11, 187. [Google Scholar] [CrossRef]
- Little, S.; Braff, J.; Place, J.; Buch, J.; Dewage, B.G.; Knupp, A.; Beall, M. Canine infection with Dirofilaria immitis, Borrelia burgdorferi, Anaplasma spp., and Ehrlichia spp. in the United States, 2013–2019. Parasites Vectors 2021, 14, 10. [Google Scholar] [CrossRef]
- Dantas-Torres, F.; Figueredo, L.A.; Sales, K.G.d.S.; Miranda, D.E.d.O.; Alexandre, J.L.d.A.; da Silva, Y.Y.; da Silva, L.G.; Valle, G.R.; Ribeiro, V.M.; Otranto, D.; et al. Prevalence and incidence of vector-borne pathogens in unprotected dogs in two Brazilian regions. Parasites Vectors 2011, 4, 141. [Google Scholar] [CrossRef] [PubMed]
- Abd Rani, P.A.M.; Irwin, P.J.; Coleman, G.T.; Gatne, M.; Traub, R.J. A survey of canine tick-borne diseases in India. Parasites Vectors 2011, 4, 141. [Google Scholar] [CrossRef] [PubMed]
- Otranto, D.; Iatta, R.; Baneth, G.; Cavalera, M.A.; Bianco, A.; Parisi, A.; Dantas-Torres, F.; Colella, V.; McMillan-Cole, A.C.; Chomel, B. High prevalence of vector-borne pathogens in domestic and wild carnivores in Iraq. Acta Trop. 2019, 197, 105058. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Leishmania Seroreactivity (DPP Test) | ||
|---|---|---|
| Negative (N = 6) | Positive (N = 8) | |
| Age | ||
| Mean (SD) | 3.67 (1.63) | 3.62 (1.06) |
| Sex | ||
| Female | 2 (33.3%) | 3 (37.5%) |
| Male | 4 (66.7%) | 5 (62.5%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pessôa-Pereira, D.; Scorza, B.M.; Cyndari, K.I.; Beasley, E.A.; Petersen, C.A. Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi. Pathogens 2023, 12, 1128. https://doi.org/10.3390/pathogens12091128
Pessôa-Pereira D, Scorza BM, Cyndari KI, Beasley EA, Petersen CA. Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi. Pathogens. 2023; 12(9):1128. https://doi.org/10.3390/pathogens12091128
Chicago/Turabian StylePessôa-Pereira, Danielle, Breanna M. Scorza, Karen I. Cyndari, Erin A. Beasley, and Christine A. Petersen. 2023. "Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi" Pathogens 12, no. 9: 1128. https://doi.org/10.3390/pathogens12091128
APA StylePessôa-Pereira, D., Scorza, B. M., Cyndari, K. I., Beasley, E. A., & Petersen, C. A. (2023). Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi. Pathogens, 12(9), 1128. https://doi.org/10.3390/pathogens12091128