


Designing and Validation of a Droplet Digital PCR Procedure for Diagnosis and Accurate Quantification of Nervous Necrosis Virus in the Mediterranean Area


Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral Strains, Cell Culture and Viral Titration
2.2. Optimization of the Procedure
2.3. Reference Templates
2.4. cDNA Synthesis
2.5. Real-Time Quantitative PCR (rt-qPCR)
2.6. Droplet Digital PCR (ddPCR)
2.7. Limit of Detection, Quantification and Dynamic Range
2.8. Reliability of the Procedure
2.9. Performance of the Procedure with Field Samples
2.10. Statistical Analysis
3. Results
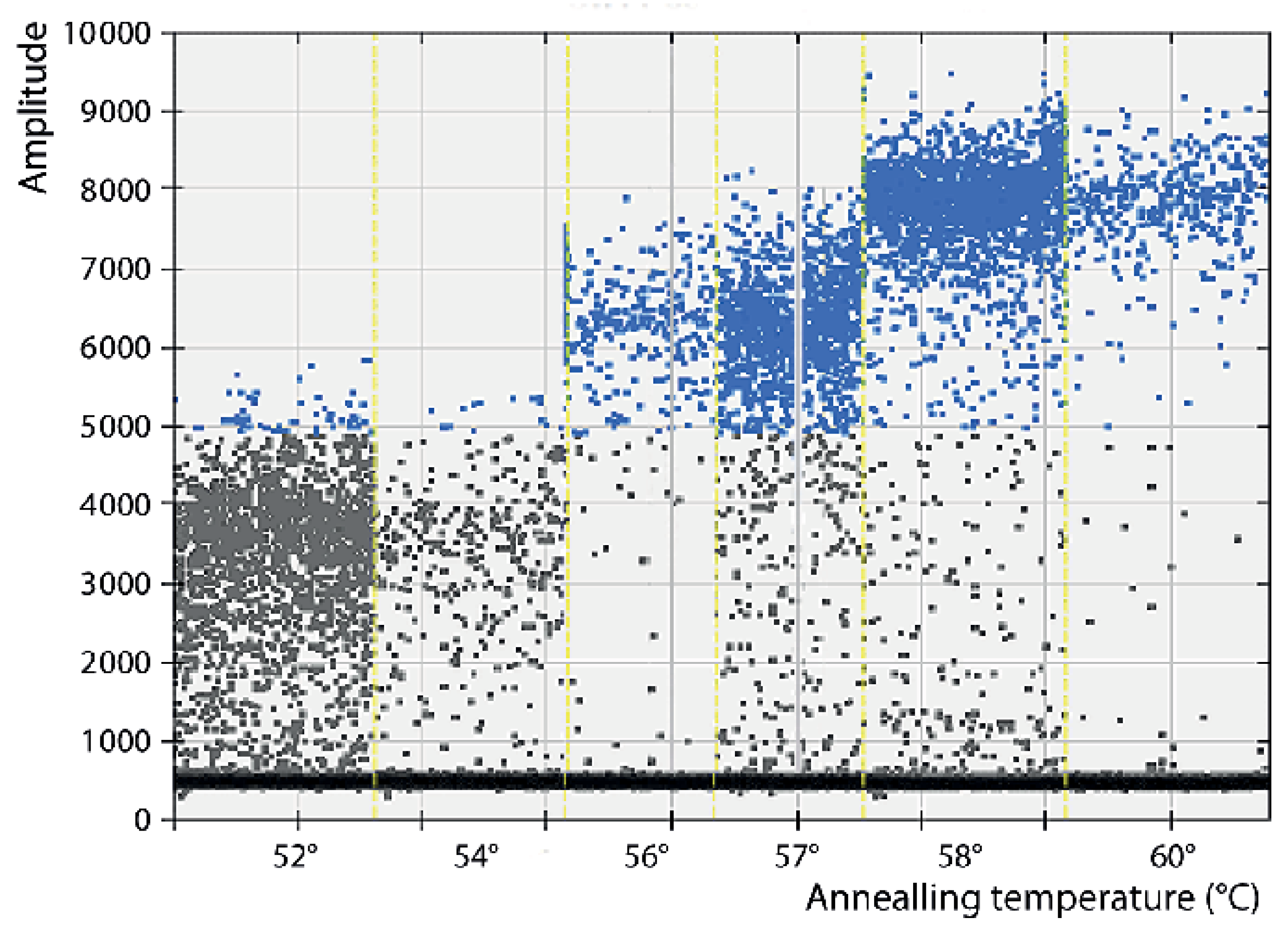
3.1. Optimization of the Conditions
3.2. Repeatability, Reproducibility and Specificity
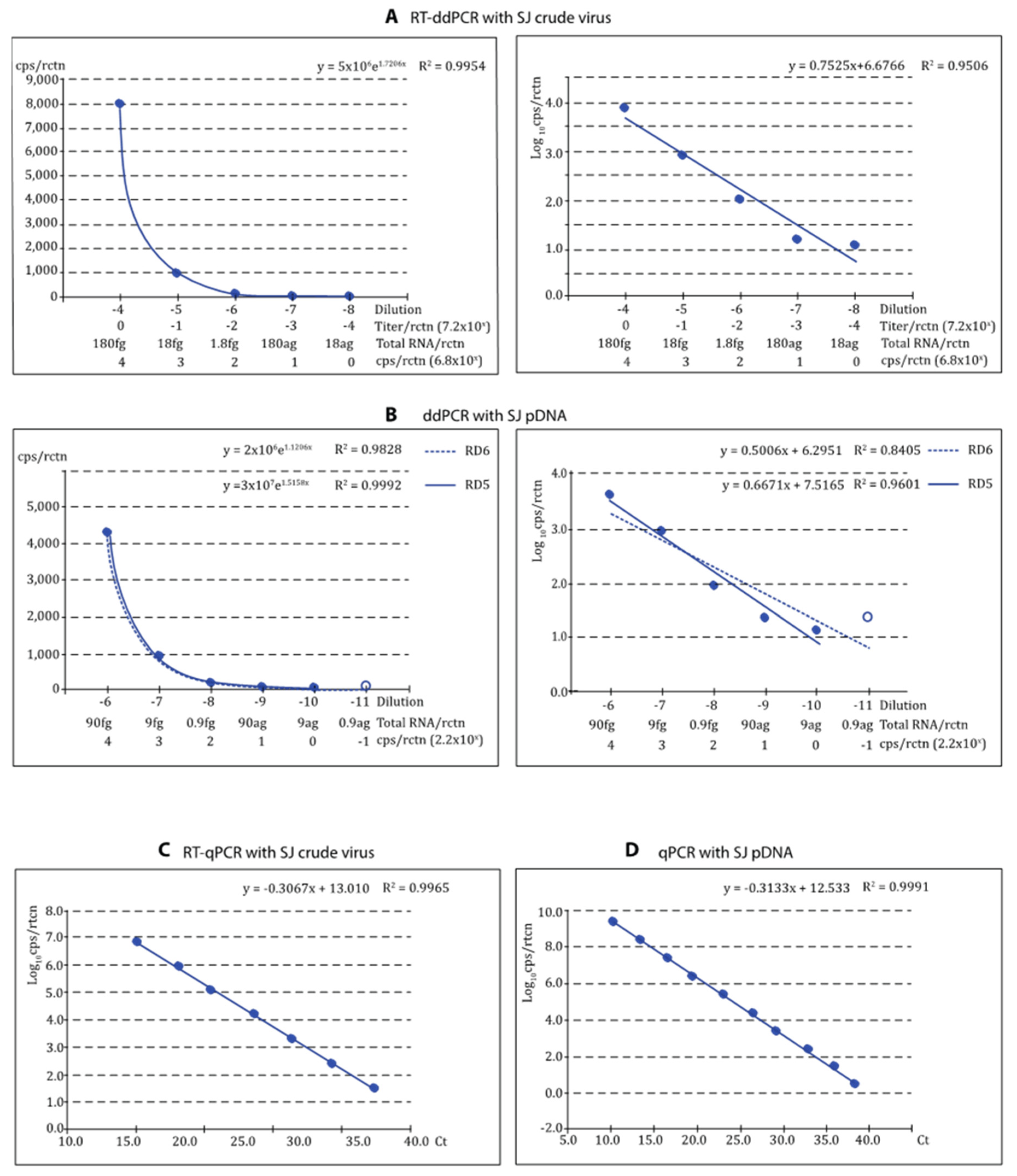
3.3. Dynamic Range (DR); Limit of Detection (LOD) and Limit of Quantification (LOQ)
3.4. Reliability of the Quantification
3.5. Correlation between ddPCR and qPCR Quantification Methods
3.6. Performance of the RT-ddPCR Procedure in Field Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bandín, I.; Souto, S. Betanodavirus and VER Disease: A 30-year Research Review. Pathogens 2020, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- World Organization of Animal Health (WOAH). Viral Encephalopathy and Retinopathy. In Manual of Diagnostic Tests for Aquatic Animals; World Organization of Animal Health: Paris, France, 2019; Available online: https://www.woah.org/fileadmin/Home/eng/Health_standards/aahm/current/chapitre_viral_encephalopathy_retinopathy.pdf (accessed on 9 July 2023).
- Panzarin, V.; Patarnello, P.; Mori, A.; Rampazzo, E.; Cappellozza, E.; Bovo, G.; Cattoli, G. Development and validation of a real-time TaqMan PCR assay for the detection of Betanodavirus in clinical specimens. Arch. Virol. 2010, 155, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Olveira, J.G.; Souto, S.; Bandín, I.; Dopazo, C.P. Development and validation of a SYBR Green real time PCR protocol for detection and quantification of nervous necrosis virus (NNV) using different standards. Animals 2021, 11, 1100. [Google Scholar] [CrossRef]
- Gezer, U.; Bronkhorst, A.J.; Holdenrieder, S. The Clinical Utility of Droplet Digital PCR for Profiling Circulating Tumor DNA in Breast Cancer Patients. (Special Issue Cell-Free Nucleic Acids—New Insights into Physico-Chemical Properties, Analytical Considerations, and Clinical Applications). Diagnostics 2022, 12, 3042. [Google Scholar] [CrossRef] [PubMed]
- Huerta, M.; Roselló, S.; Sabater, L.; Ferrer, A.; Tarazona, N.; Roda, D.; Gambardella, V.; Alfaro-Cervelló, C.; Garcés-Albir, M.; Cervantes, A.; et al. Circulating Tumor DNA Detection by Digital-Droplet PCR in Pancreatic Ductal Adenocarcinoma: A Systematic Review. Cancers 2021, 13, 994. [Google Scholar] [CrossRef]
- Salipante, S.J.; Jerome, K.R. Digital PCR—An Emerging Technology with Broad Applications in Microbiology. Clin. Chemist. 2020, 66, 117–123. [Google Scholar] [CrossRef]
- Chen, B.; Jiang, Y.; Cao, X.; Liu, C.; Zhang, N.; Shi, D. Droplet digital PCR as an emerging tool in detecting pathogens nucleic acids in infectious diseases. Clin. Chim. Acta. 2021, 517, 156–161. [Google Scholar] [CrossRef]
- Morley, A.A. Digital PCR: A brief history. Biomol. Detect. Quantif. 2014, 1, 1–2. [Google Scholar] [CrossRef]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The Digital MIQE Guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chemist. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- European Network of GMO Laboratories (ENGL). Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing. 2015. Available online: https://gmo-crl.jrc.ec.europa.eu/doc/Min_Perf_Requirements_Analytical_methods.pdf (accessed on 9 July 2023).
- Olveira, J.G.; Soares, F.; Engrola, S.; Dopazo, C.P.; Bandín, I. Antemortem versus postmortem methods for detection of betanodavirus in Senegalese sole (Solea senegalensis). J. Vet. Diagn. Investig. 2008, 20, 215–219. [Google Scholar] [CrossRef]
- Dopazo, C.P.; Moreno, P.; Olveira, J.G.; Borrego, J.J. The theoretical reliability of PCRbased fish viral diagnostic methods is critically affected when they are applied to fish populations with low prevalence and virus loads. J. Appl. Microbiol. 2017, 124, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Chen, S.; Zhong, Q. Digital PCR for accurate quantification of pathogens: Principles, applications, challenges and future prospects. Int. J. Biol. Macromol. 2021, 184, 750–759. [Google Scholar] [CrossRef]
- Jia, P.; Purcell, M.K.; Pan, G.; Wang, J.; Kan, S.; Liu, Y.; Zheng, X.; Shi, X.; He, J.; Yu, L.; et al. Analytical validation of a reverse transcriptase droplet digital PCR (RTddPCR) for quantitative detection of infectious hematopoietic necrosis virus. J. Virol. Methods 2017, 245, 73–80. [Google Scholar] [CrossRef]
- Lin, Q.; Fu, X.; Liu, L.; Liang, H.; Niu, Y.; Wen, Y.; Huang, Z.; Li, N. Development and application of a sensitive droplet digital PCR (ddPCR) for the detection of infectious spleen and kidney necrosis virus. Aquaculture 2020, 529, 735697. [Google Scholar] [CrossRef]
- Wang, N.; Zhang, Z.; Jing, H.; Zhang, M.; Wu, S.; Lin, X. Development of a novel droplet digital PCR assay for the sensitive detection of carp edema virus. Aquaculture 2021, 545, 737162. [Google Scholar] [CrossRef]
- Jiang, N.; Shen, J.; Zhou, Y.; Liu, W.; Meng, Y.; Li, Y.; Xue, M.; Xu, C.; Fan, Y. Development of a droplet digital PCR method for the sensitive detection and quantification of largemouth bass ranavirus. J. Fish Dis. 2022, 46, 91–98. [Google Scholar] [CrossRef]
- Shahi, N.; Prasartset, T.; Surachetpong, W. A specific and sensitive droplet digital polymerase chain reaction assay for the detection of tilapia lake virus in fish tissue and environmental samples. J. Fish Dis. 2023, 46, 957–966. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, Y.; Fan, Y.; Jiang, N.; Meng, Y.; Li, Y.; Xue, M.; Xu, C.; Guo, W.; Liu, W. Development and application of a sensitive droplet digital PCR-based method to detect tilapia parvovirus. J. Fish Dis. 2023, 46, 239–245. [Google Scholar] [CrossRef]
- Han, Y.; Wang, J.; Zhang, S.; Yang, S.; Wang, X.; Han, Y.; Shen, Z.; Xu, X. Simultaneous quantification of hepatitis A virus and norovirus genogroup I and II by triplex droplet digital PCR. Food Microbiol. 2022, 103, 103933. [Google Scholar] [CrossRef]
- Mairiang, D.; Songjaeng, A.; Hansuealueang, P.; Malila, Y.; Lertsethtakarn, P.; Silapong, S.; Poolpanichupatam, Y.; Klungthong, C.; Chin-Inmanu, C.; Thiemmeca, S.; et al. Application of One-Step Reverse Transcription Droplet Digital PCR for Dengue Virus Detection and Quantification in Clinical Specimens. Diagnostics 2021, 11, 639. [Google Scholar] [CrossRef]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Yang, Q.; Xi, J.; Chen, X.; Hu, S.; Chen, N.; Qiao, S.; Wan, S.; Bao, D. The development of a sensitive droplet digital PCR for quantitative detection of porcine reproductive and respiratory syndrome virus. Int. J. Biol. Macromol. 2017, 104, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Rački, R.; Morisset, D.; Gutierrez-Aguirre, I.; Ravnikar, M. One-step RT-droplet digital PCR: A breakthrough in the quantification of waterborne RNA viruses. Anal. Bioanal. Chem. 2014, 406, 661–667. [Google Scholar] [CrossRef] [PubMed]
- World Organization of Animal Health (WOAH). Manual of Diagnostic Tests for Aquatic Animals; World Organization of Animal Health: Paris, France, 2023; Available online: https://www.woah.org/en/what-we-do/standards/codes-and-manuals/aquatic-manual-online-access/ (accessed on 9 July 2023).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pr/Pb 1 | Name | S/As 2 | Sequence (5′ to 3′) | Position 3 | Ampl. Size 4 |
|---|---|---|---|---|---|
| Pr | T_NodR2_330 | S | TACGCTGTTGAAACACTG | 330–347 | 100 bp |
| Pr | T_NodR2_430 | As | CGTTGTCAGTTGGATCAG | 429–412 | |
| Pb | TQM_NodR2_359 | S | ATTCAGCCAATGTG | 357–370 |
| Limit of Detection | Dynamic Range for Detection | |||||||
|---|---|---|---|---|---|---|---|---|
| Plasmid | Crude Virus | Plasmid | Crude Virus | |||||
| qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | |
| Dilution 1 | −10 | −11 | −7 | −8 | −2 to −10 | −6 to −11 | −2 to −7 | −4 to −8 |
| w/reaction 2 | 9 ag | 0.9 ag | 0.18 fg | 18 ag | 0.9 ng–9 ag | 90 fg–0.9 ag | 18 pg–0.18 fg | 0.18 pg–18 ag |
| cps/reaction 3 | 2.2 | 2.2 × 10−1 | 68 | 6.8 | 2.19 × 108–2.19 | 2.19 × 104–2.19 × 10−1 | 6.8 × 106–6.8 × 101 | 6.8 × 104–6.8 × 100 |
| Tit/react 4 | N/A | N/A | 7.2 × 10−3 | 7.2 × 10−4 | N/A | N/A | 7.2 × 102–7.2 × 10−3 | 7.2 × 100–7.2 × 10−4 |
| Tit/mL 5 | N/A | N/A | 5.6 | 0.56 | N/A | N/A | 5.6 × 105–5.6 × 100 | 5.6 × 103–0.56 |
| Limit of Quantification | Dynamic range for quantification | |||||||
| Plasmid | Crude virus | Plasmid | Crude virus | |||||
| qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | |
| Dilution 1 | −9 | −10 | −7 | −8 | −2 to −9 | −6 to −10 | −2 to −7 | −4 to −8 |
| w/reaction 2 | 90 ag | 9 ag | 0.18 fg | 18 ag | 0.9 ng–90 ag | 90 fg–9 ag | 18 pg–0.18 fg | 0.18–pg 18 ag |
| cps/reaction 3 | 21.9 | 2.19 | 68 | 6.8 | 2.19 × 108–21.9 | 2.19 × 104–2.19 | 6.8 × 106–6.8 × 101 | 6.8 × 104–6.8 × 100 |
| Tit/react 4 | N/A | N/A | 7.2 × 10−3 | 7.2 × 10−4 | N/A | N/A | 7.2 × 102–7.2 × 10−3 | 7.2 × 100–7.2/10−4 |
| Tit/mL 5 | N/A | N/A | 5.6 | 0.56 | N/A | N/A | 5.6 × 105–5.6 × 100 | 5.6 × 103–0.56 |
| Limit of Detection | Dynamic Range for Detection | |||||||
|---|---|---|---|---|---|---|---|---|
| Plasmid | Crude Virus | Plasmid | Crude Virus | |||||
| qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | |
| Dilution 1 | −9 | −10 | −7 | −9 | −1 to −10 | −5 to −11 | −1 to −7 | −3 to −9 |
| w/reaction 2 | 46 ag | 4.6 ag | 0.26 fg | 2.6 ag | 4.6 ag–4.6 ng | 0.46 pg–0.46 ag | 0.26 fg–0.26 ng | 2.6 ag–2.6 pg |
| cps/reaction 3 | 9.5 | 0.95 | 10.4 | 0.1 | 0.946–9.46 × 108 | 9.46 × 10−2–9.46 × 104 | 10.4–1.04 × 107 | 0.1–1.04 × 105 |
| Tit/react 4 | N/A | N/A | 1.3 × 10−3 | 1.3 × 10−5 | N/A | N/A | 1.3 × 10−3–1.3 × 103 | 1.3 × 10−5–1.3 × 101 |
| Tit/mL 5 | N/A | N/A | 1 | 1 × 10−2 | N/A | N/A | 1–1 × 106 | 1 × 10−2–1 × 104 |
| Limit of Quantification | Dynamic range for quantification | |||||||
| Plasmid | Crude virus | Plasmid | Crude virus | |||||
| qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | qPCR | ddPCR | |
| Dilution 1 | −9 | −9 | −7 | −7 | −1 to −9 | −5 to −9 | −1 to −7 | −3 to −7 |
| w/reaction 2 | 46 ag | 46 ag | 0.26 fg | 0.26 fg | 4.6 ng–46 ag | 0.46 pg–46 ag | 0.26 ng–0.26 fg | 2.6 pg–0.26 fg |
| cps/reaction 3 | 9.46 | 9.46 | 10.4 | 10.4 | 9.46 × 108–9.46 | 9.46 × 104–9.46 | 1.04 × 107–10.4 | 1.04 × 105–10.4 |
| Tit/react 4 | N/A | N/A | 1.3 × 10−3 | 1.3 × 10−3 | N/A | N/A | 1.3 × 103–1.3 × 10−3 | 1.3 × 101–1.3 × 10−3 |
| Tit/mL 5 | N/A | N/A | 1 | 1 | N/A | N/A | 1 × 106–1 | 1 × 104–1 |
| RT-ddPCR | RT-qPCR | ||
|---|---|---|---|
| Sample ID | Quantf 1 | Ct | Quantf |
| 273.22 | 8.0 | ≥40 | 0.0 |
| 274.22 | 1.4 | 39.5 | 1.3 |
| 275.22 | ND | ≥40 | 0.0 |
| 281.22 | ND | ≥40 | 0.0 |
| 282.22 | 27.8 | ≥40 | 0.0 |
| 283.22 | 14.3 | ≥40 | 0.0 |
| 290.22 | ND | ≥40 | 0.0 |
| 291.22 | 19.4 | 37.7 | 4.2 |
| 292.22 | 20.6 | ≥40 | 0.0 |
| 297.22 | ND | 39.7 | 1.1 |
| 298.22 | 4.4 | 35.1 | 23.5 |
| 299.22 | 5.2 | ≥40 | 0.0 |
| 305.22 | ND | ≥40 | 0.0 |
| 306.22 | 7.0 | 35.7 | 16.2 |
| 307.22 | 16.8 | 39.1 | 1.6 |
| 313.22 | ND | ≥40 | 0.0 |
| 314.22 | 7.8 | 38.9 | 1.8 |
| 315.22 | ND | ≥40 | 0.0 |
| 321.22 | ND | 39.8 | 1.0 |
| 322.22 | 4.3 | ≥40 | 0.0 |
| 323.22 | 0.0 | ≥40 | 0.0 |
| 329.22 | 0.0 | ≥40 | 0.0 |
| 330.22 | 0.0 | ≥40 | 0.0 |
| 331.22 | 0.0 | ≥40 | 0.0 |
| A/Considering Positive Ct ≤ 40 | B/Considering Positive Ct ≤ 39.5 | |||
|---|---|---|---|---|
| Gold Standard: RT-qPCR | Gold Standard: RT-qPCR | |||
| Test: RT-ddPCR | cSs = 0.75 | PPV = 0.50 | cSs = 1 | PPV = 0.50 |
| cSp = 0.63 | NPV = 0.83 | cSp = 0.67 | NPV = 1 | |
| Gold Standard: RT-ddPCR | Gold Standard: RT-ddPCR | |||
| Test: RT-qPCR | cSs = 0.5 | PPV = 0.75 | cSs = 0.5 | PPV = 1.00 |
| cSp = 0.83 | NPV = 0.63 | cSp = 1 | NPV = 0.67 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souto, S.; Olveira, J.G.; López-Vázquez, C.; Bandín, I.; Dopazo, C.P. Designing and Validation of a Droplet Digital PCR Procedure for Diagnosis and Accurate Quantification of Nervous Necrosis Virus in the Mediterranean Area. Pathogens 2023, 12, 1155. https://doi.org/10.3390/pathogens12091155
Souto S, Olveira JG, López-Vázquez C, Bandín I, Dopazo CP. Designing and Validation of a Droplet Digital PCR Procedure for Diagnosis and Accurate Quantification of Nervous Necrosis Virus in the Mediterranean Area. Pathogens. 2023; 12(9):1155. https://doi.org/10.3390/pathogens12091155
Chicago/Turabian StyleSouto, Sandra, José G. Olveira, Carmen López-Vázquez, Isabel Bandín, and Carlos P. Dopazo. 2023. "Designing and Validation of a Droplet Digital PCR Procedure for Diagnosis and Accurate Quantification of Nervous Necrosis Virus in the Mediterranean Area" Pathogens 12, no. 9: 1155. https://doi.org/10.3390/pathogens12091155
APA StyleSouto, S., Olveira, J. G., López-Vázquez, C., Bandín, I., & Dopazo, C. P. (2023). Designing and Validation of a Droplet Digital PCR Procedure for Diagnosis and Accurate Quantification of Nervous Necrosis Virus in the Mediterranean Area. Pathogens, 12(9), 1155. https://doi.org/10.3390/pathogens12091155