
Is Short-Read 16S rRNA Sequencing of Oral Microbiome Sampling a Suitable Diagnostic Tool for Head and Neck Cancer?

 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
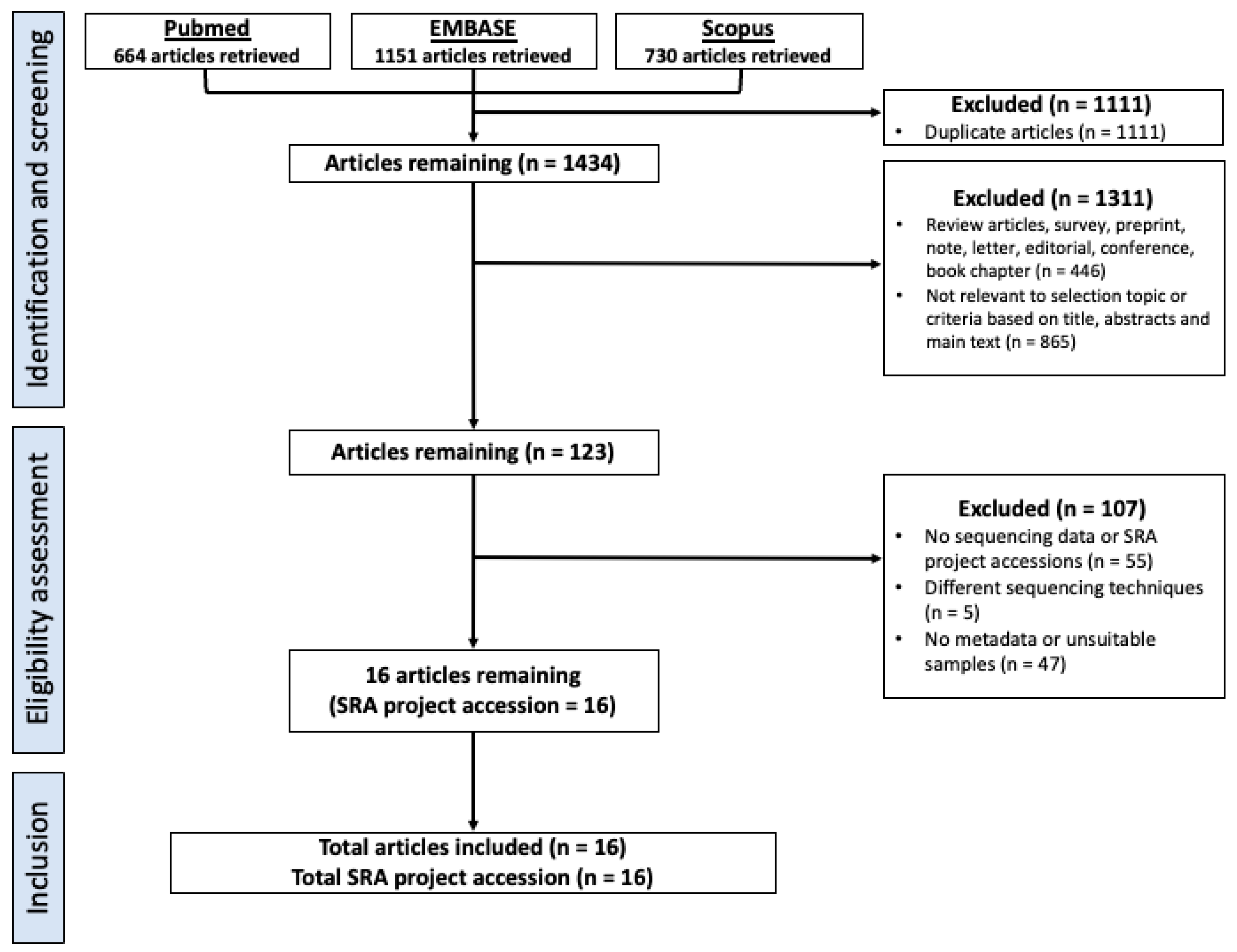
2.1. Selection Criteria, Database Search and Study Design
2.2. Downloading, Pre-Processing and Metadata Collection of 16S rRNA Datasets
2.3. Evaluation and Correcting for Batch Effects
2.4. Alpha and Beta Diversity Analysis at the Genus Level
2.5. Differential Abundance Analysis between HNC, Healthy and Premalignant Oral Samples
2.6. Multivariate Sparse Partial Least Squares Discriminant Analysis (sPLS-DA) Classification to Discriminate Oral Sampling
3. Results
3.1. Study Characteristics and Dataset
3.2. Patient Type, 16S rRNA Amplification Primer Sets, and Study Batch Affect Overall Microbial Composition of Saliva and Oral Rinse Samples
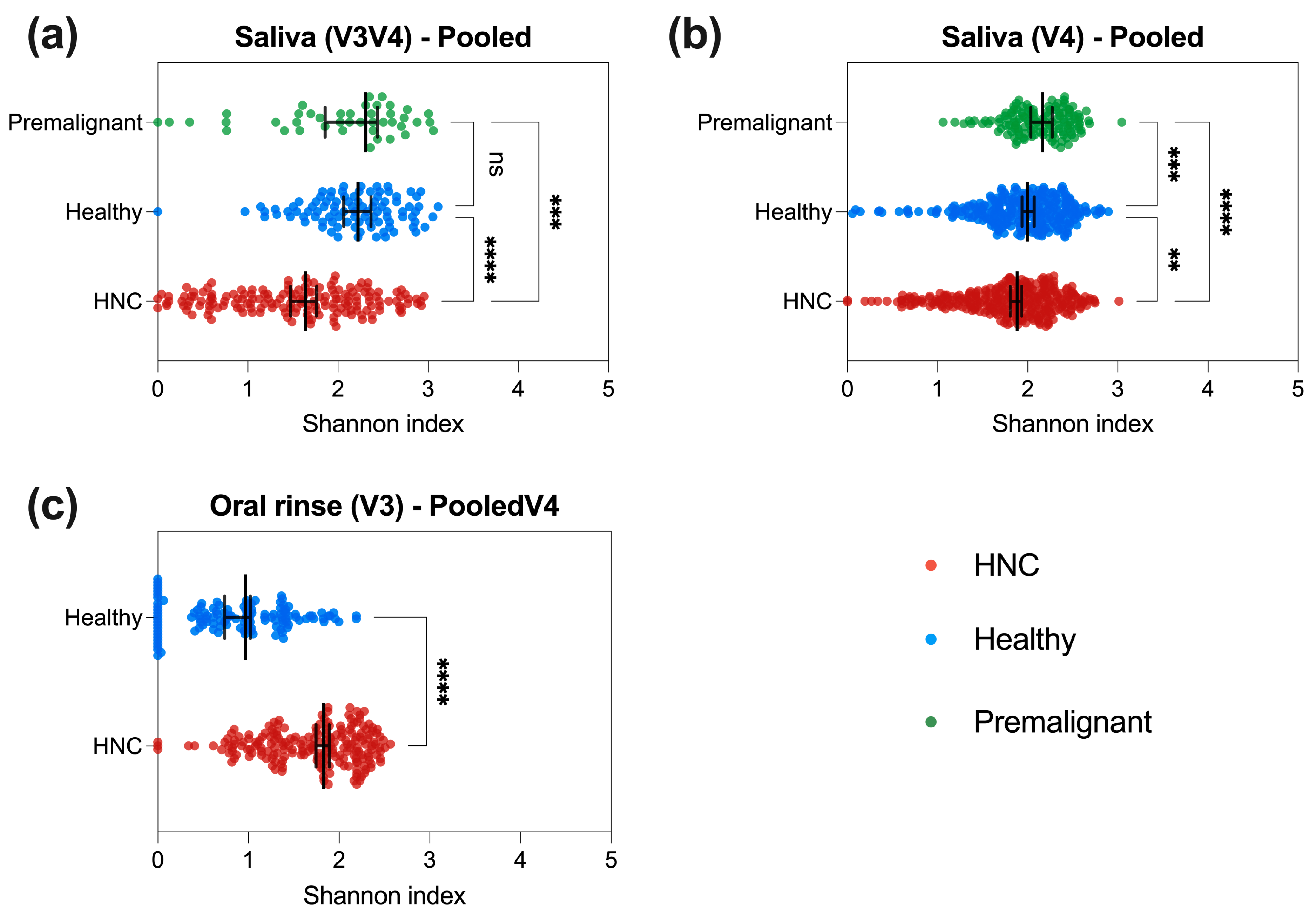
3.3. HNC Alpha Diversity Differs between Saliva and Oral Rinse
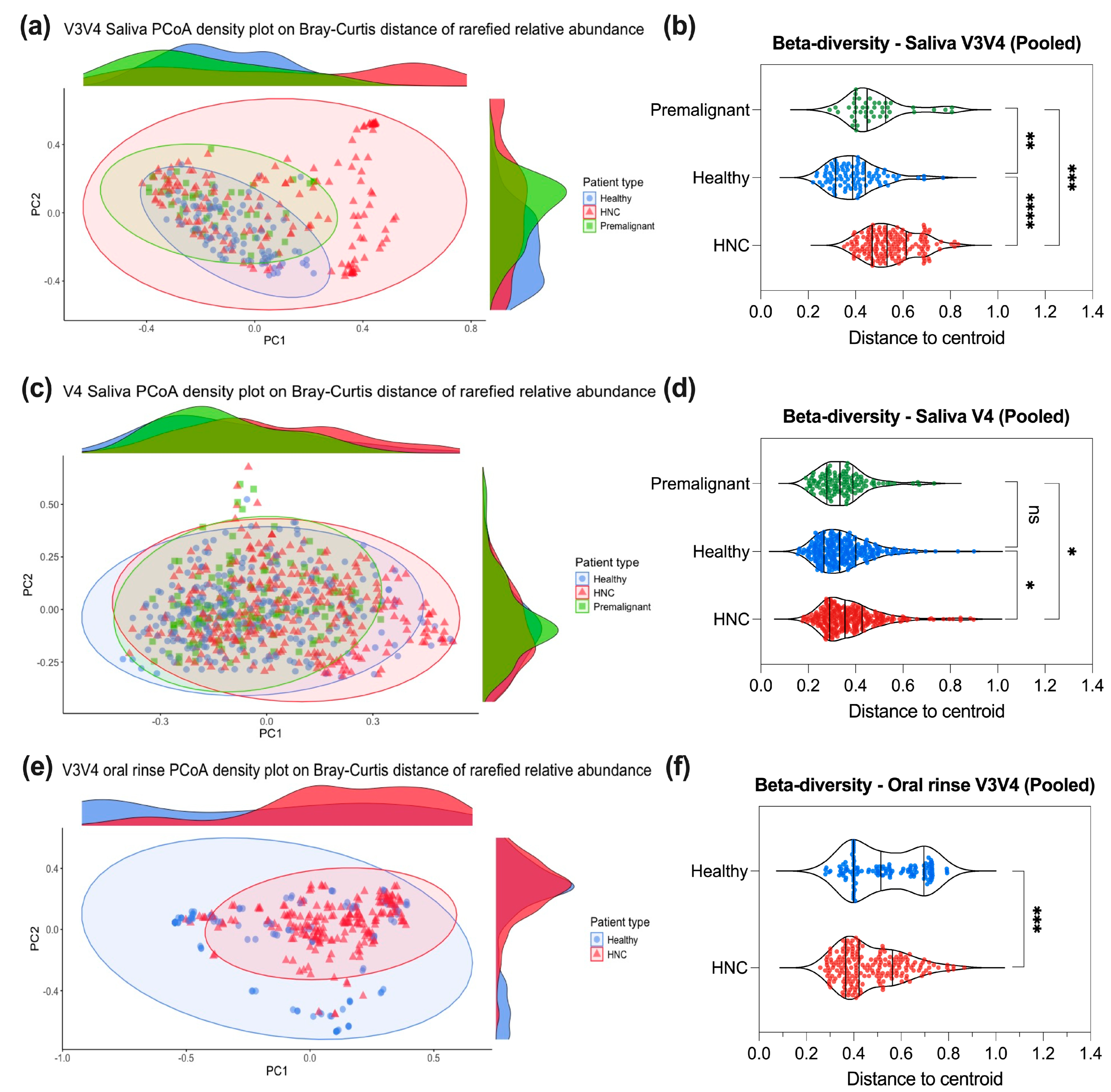
3.4. Saliva or Oral Rinse from HNC, Premalignant and Healthy Donors Have Similar Beta Diversities at the Genus Level
3.5. Differentially Abundant Genera between HNC and Healthy Oral Samples
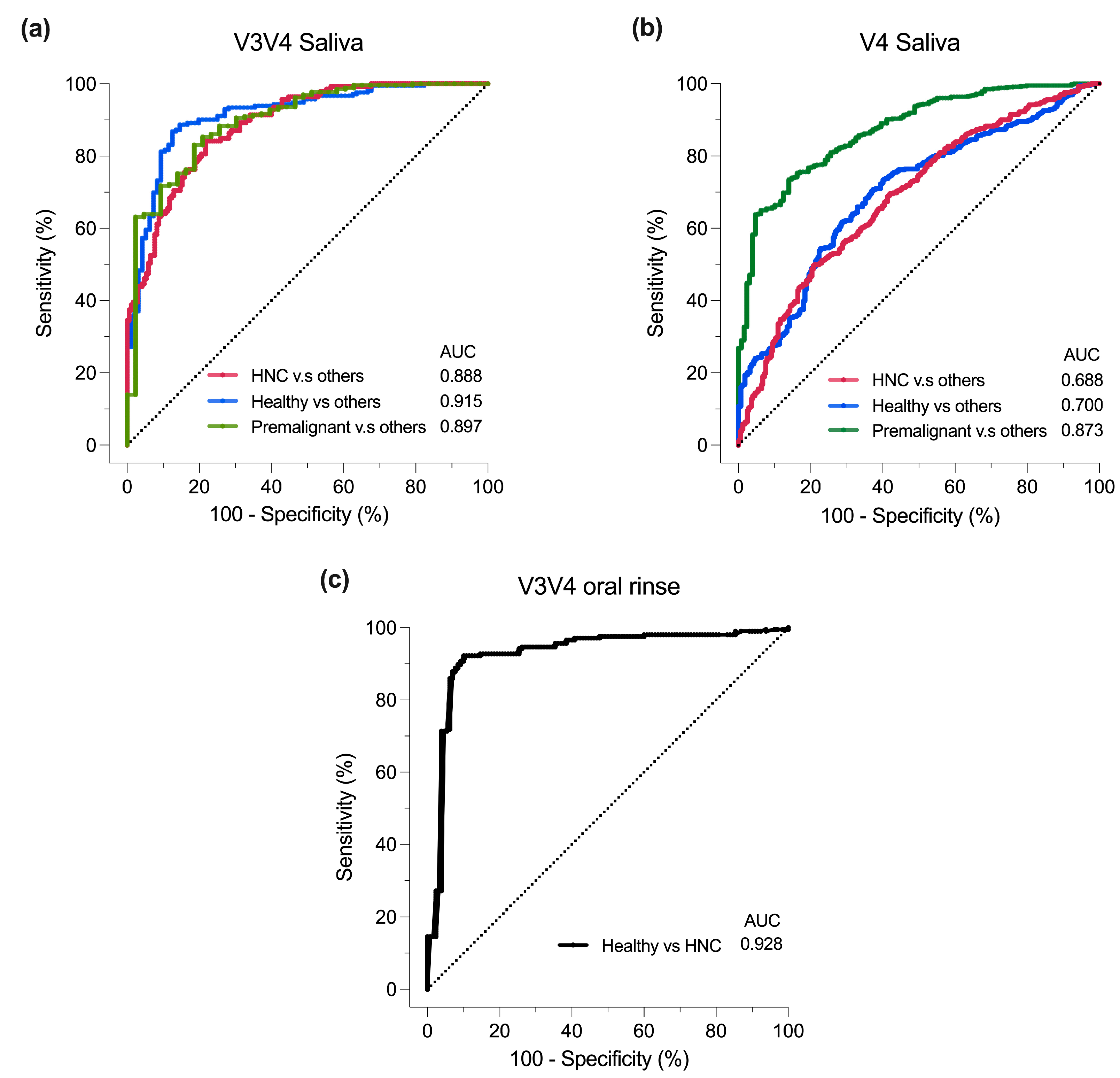
3.6. Classification of Oral Samples Based on sPLS-DA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peng, X.; Cheng, L.; You, Y.; Tang, C.; Ren, B.; Li, Y.; Xu, X.; Zhou, X. Oral microbiota in human systematic diseases. Int. J. Oral Sci. 2022, 14, 14. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Irfan, M.; Delgado, R.Z.R.; Frias-Lopez, J. The oral microbiome and cancer. Front. Immunol. 2020, 11, 591088. [Google Scholar] [CrossRef] [PubMed]
- Gormley, M.; Creaney, G.; Schache, A.; Ingarfield, K.; Conway, D.I. Reviewing the epidemiology of head and neck cancer: Definitions, trends and risk factors. Br. Dent. J. 2022, 233, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Sykes, E.A.; Weisbrod, N.; Rival, E.; Haque, A.; Fu, R.; Eskander, A. Methods, Detection Rates, and Survival Outcomes of Screening for Head and Neck Cancers: A Systematic Review. JAMA Otolaryngol.–Head Neck Surg. 2023, 149, 1047–1056. [Google Scholar] [CrossRef]
- Li, Q.; Tie, Y.; Alu, A.; Ma, X.; Shi, H. Targeted therapy for head and neck cancer: Signaling pathways and clinical studies. Signal Transduct. Target. Ther. 2023, 8, 31. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef]
- Ting, H.S.L.; Chen, Z.; Chan, J.Y.K. Systematic review on oral microbial dysbiosis and its clinical associations with head and neck squamous cell carcinoma. Head Neck 2023, 45, 2120–2135. [Google Scholar] [CrossRef]
- Wolf, A.; Moissl-Eichinger, C.; Perras, A.; Koskinen, K.; Tomazic, P.V.; Thurnher, D. The salivary microbiome as an indicator of carcinogenesis in patients with oropharyngeal squamous cell carcinoma: A pilot study. Sci. Rep. 2017, 7, 5867. [Google Scholar] [CrossRef]
- Kumpitsch, C.; Moissl-Eichinger, C.; Pock, J.; Thurnher, D.; Wolf, A. Preliminary insights into the impact of primary radiochemotherapy on the salivary microbiome in head and neck squamous cell carcinoma. Sci. Rep. 2020, 10, 16582. [Google Scholar] [CrossRef]
- Du, Y.; Feng, R.; Chang, E.T.; Debelius, J.W.; Yin, L.; Xu, M.; Huang, T.; Zhou, X.; Xiao, X.; Li, Y. Influence of pre-treatment saliva microbial diversity and composition on nasopharyngeal carcinoma prognosis. Front. Cell. Infect. Microbiol. 2022, 12, 831409. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Chen, J.W.; Wu, L.W.; Huang, K.C.; Chen, J.Y.; Wu, W.S.; Chiang, W.F.; Shih, C.J.; Tsai, K.N.; Hsieh, W.T.; et al. Carcinogenesis of Male Oral Submucous Fibrosis Alters Salivary Microbiomes. J. Dent. Res. 2021, 100, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.; Wu, J.-H.; Chiang, W.-F.; Chen, Y.-L.; Wu, W.-S.; Wu, L.-W. Taxonomic and functional dysregulation in salivary microbiomes during oral carcinogenesis. Front. Cell. Infect. Microbiol. 2021, 11, 663068. [Google Scholar] [CrossRef]
- Lee, W.H.; Chen, H.M.; Yang, S.F.; Liang, C.; Peng, C.Y.; Lin, F.M.; Tsai, L.L.; Wu, B.C.; Hsin, C.H.; Chuang, C.Y.; et al. Bacterial alterations in salivary microbiota and their association in oral cancer. Sci. Rep. 2017, 7, 16540. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Hao, Y.; Peng, X.; Li, B.; Han, Q.; Ren, B.; Li, M.; Li, L.; Li, Y.; Cheng, G.; et al. The Clinical Potential of Oral Microbiota as a Screening Tool for Oral Squamous Cell Carcinomas. Front. Cell Infect. Microbiol. 2021, 11, 728933. [Google Scholar] [CrossRef]
- Vesty, A.; Gear, K.; Biswas, K.; Radcliff, F.J.; Taylor, M.W.; Douglas, R.G. Microbial and inflammatory-based salivary biomarkers of head and neck squamous cell carcinoma. Clin. Exp. Dent. Res. 2018, 4, 255–262. [Google Scholar] [CrossRef]
- Oyeyemi, B.F.; Kaur, U.S.; Paramraj, A.; Tandon, R.; Kumar, A.; Bhavesh, N.S. Microbiome analysis of saliva from oral squamous cell carcinoma (OSCC) patients and tobacco abusers with potential biomarkers for oral cancer screening. Heliyon 2023, 9, e21773. [Google Scholar] [CrossRef]
- Torralba, M.G.; Aleti, G.; Li, W.; Moncera, K.J.; Lin, Y.H.; Yu, Y.; Masternak, M.M.; Golusinski, W.; Golusinski, P.; Lamperska, K.; et al. Oral Microbial Species and Virulence Factors Associated with Oral Squamous Cell Carcinoma. Microb. Ecol. 2021, 82, 1030–1046. [Google Scholar] [CrossRef]
- Zakrzewski, M.; Gannon, O.M.; Panizza, B.J.; Saunders, N.A.; Antonsson, A. Human papillomavirus infection and tumor microenvironment are associated with the microbiota in patients with oropharyngeal cancers-pilot study. Head Neck 2021, 43, 3324–3330. [Google Scholar] [CrossRef]
- Granato, D.C.; Neves, L.X.; Trino, L.D.; Carnielli, C.M.; Lopes, A.F.; Yokoo, S.; Pauletti, B.A.; Domingues, R.R.; Sa, J.O.; Persinoti, G. Meta-omics analysis indicates the saliva microbiome and its proteins associated with the prognosis of oral cancer patients. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2021, 1869, 140659. [Google Scholar] [CrossRef]
- Hao, Y.; Zeng, Z.; Peng, X.; Ai, P.; Han, Q.; Ren, B.; Li, M.; Wang, H.; Zhou, X.; Zhou, X. The human oral–nasopharynx microbiome as a risk screening tool for nasopharyngeal carcinoma. Front. Cell. Infect. Microbiol. 2022, 12, 1013920. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Tan, J.; Guo, G.; Li, Z.; Yang, L.; Lao, X.; Wang, D.; Ma, J.; Zhang, S.; Liao, G. The oral cancer microbiome contains tumor space–specific and clinicopathology-specific bacteria. Front. Cell. Infect. Microbiol. 2022, 12, 942328. [Google Scholar] [CrossRef]
- Nie, F.; Wang, L.; Huang, Y.; Yang, P.; Gong, P.; Feng, Q.; Yang, C. Characteristics of microbial distribution in different oral niches of oral squamous cell carcinoma. Front. Cell. Infect. Microbiol. 2022, 12, 905653. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.; Szczesniak, M.; Maclean, J.; Yim, H.C.H.; Zhang, F.; Graham, P.; El-Omar, E.M.; Wu, P. Dysbiosis in head and neck cancer: Determining optimal sampling site for oral microbiome collection. Pathogens 2022, 11, 1550. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, M.C.d.; The, S.; Bellile, E.; Russo, N.; Schmitd, L.; Danella, E.; Singh, P.; Banerjee, R.; Bassis, C.; Murphy, G.R., III. Salivary microbiome changes distinguish response to chemoradiotherapy in patients with oral cancer. Microbiome 2023, 11, 268. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, A.I.; Pappalardo, V.Y.; Buijs, M.J.; Brandt, B.W.; Mäkitie, A.A.; Meurman, J.H.; Zaura, E. Salivary microbiome profiles of oral cancer patients analyzed before and after treatment. Microbiome 2023, 11, 171. [Google Scholar] [CrossRef]
- Chen, Z.; Wong, P.Y.; Ng, C.W.K.; Lan, L.; Fung, S.; Li, J.W.; Cai, L.; Lei, P.; Mou, Q.; Wong, S.H.; et al. The Intersection between Oral Microbiota, Host Gene Methylation and Patient Outcomes in Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 3425. [Google Scholar] [CrossRef]
- Chan, J.Y.K.; Ng, C.W.K.; Lan, L.; Fung, S.; Li, J.W.; Cai, L.; Lei, P.; Mou, Q.; Meehan, K.; Lau, E.H.L.; et al. Restoration of the Oral Microbiota after Surgery for Head and Neck Squamous Cell Carcinoma Is Associated with Patient Outcomes. Front. Oncol. 2021, 11, 737843. [Google Scholar] [CrossRef]
- Sawant, S.; Dugad, J.; Parikh, D.; Srinivasan, S.; Singh, H. Identification & correlation of bacterial diversity in oral cancer and long-term tobacco chewers—A case-control pilot study. J. Med. Microbiol. 2021, 70, 001417. [Google Scholar] [CrossRef]
- Zhu, H.; Yip, H.C.; Cheung, M.K.; Chan, H.C.; Ng, C.; Lau, E.H.; Yeung, Z.W.; Wong, E.W.; Leung, L.; Qu, X. Convergent dysbiosis of upper aerodigestive microbiota between patients with esophageal and oral cavity squamous cell carcinoma. Int. J. Cancer 2023, 152, 1903–1915. [Google Scholar] [CrossRef]
- Benjamin, W.; Wang, K.; Zarins, K.; Bellile, E.; Blostein, F.; Argirion, I. Oral microbiome community composition in head and neck squamous cell carcinoma. Cancers 2023, 15, 2549. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Yeh, Y.-M.; Yu, H.-Y.; Chin, C.-Y.; Hsu, C.-W.; Liu, H.; Huang, P.-J.; Hu, S.-N.; Liao, C.-T.; Chang, K.-P. Oral microbiota community dynamics associated with oral squamous cell carcinoma staging. Front. Microbiol. 2018, 9, 862. [Google Scholar] [CrossRef]
- Lim, Y.; Fukuma, N.; Totsika, M.; Kenny, L.; Morrison, M.; Punyadeera, C. The performance of an oral microbiome biomarker panel in predicting oral cavity and oropharyngeal cancers. Front. Cell. Infect. Microbiol. 2018, 8, 267. [Google Scholar] [CrossRef]
- Srivastava, A.; Mishra, S.; Garg, P.K.; Dubey, A.K.; Deo, S.; Verma, D. Comparative and analytical characterization of the oral bacteriome of smokeless tobacco users with oral squamous cell carcinoma. Appl. Microbiol. Biotechnol. 2022, 106, 4115–4128. [Google Scholar] [CrossRef] [PubMed]
- Shitozawa, Y.; Haro, K.; Ogawa, M.; Miyawaki, A.; Saito, M.; Fukuda, K. Differences in the microbiota of oral rinse, lesion, and normal site samples from patients with mucosal abnormalities on the tongue. Sci. Rep. 2022, 12, 16839. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shi, Y.; Yuan, R.; Chen, Z.; Dong, Q.; Han, L.; Wang, L.; Zhou, J. Microbial dysbiosis in oral squamous cell carcinoma: A systematic review and meta-analysis. Heliyon 2023, 9, e13198. [Google Scholar] [CrossRef]
- Gopinath, D.; Menon, R.K.; Banerjee, M.; Yuxiong, R.S.; Botelho, M.G.; Johnson, N.W. Culture-independent studies on bacterial dysbiosis in oral and oropharyngeal squamous cell carcinoma: A systematic review. Crit. Rev. Oncol./Hematol. 2019, 139, 31–40. [Google Scholar] [CrossRef]
- Bronzato, J.D.; Bomfim, R.A.; Edwards, D.H.; Crouch, D.; Hector, M.P.; Gomes, B.P. Detection of Fusobacterium in oral and head and neck cancer samples: A systematic review and meta-analysis. Arch. Oral Biol. 2020, 112, 104669. [Google Scholar] [CrossRef]
- Baker, J.L.; Mark Welch, J.L.; Kauffman, K.M.; McLean, J.S.; He, X. The oral microbiome: Diversity, biogeography and human health. Nat. Rev. Microbiol. 2024, 22, 89–104. [Google Scholar] [CrossRef]
- Caselli, E.; Fabbri, C.; D’Accolti, M.; Soffritti, I.; Bassi, C.; Mazzacane, S.; Franchi, M. Defining the oral microbiome by whole-genome sequencing and resistome analysis: The complexity of the healthy picture. BMC Microbiol. 2020, 20, 120. [Google Scholar] [CrossRef]
- Ruan, X.; Luo, J.; Zhang, P.; Howell, K. The salivary microbiome shows a high prevalence of core bacterial members yet variability across human populations. NPJ Biofilms Microbiomes 2022, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Park, J.; Hosomi, K.; Yamada, T.; Kobayashi, A.; Yamaguchi, Y.; Iketani, S.; Kunisawa, J.; Mizuguchi, K.; Maeda, N. Analysis of oral microbiota in Japanese oral cancer patients using 16S rRNA sequencing. J. Oral Biosci. 2019, 61, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Yeo, K.; Li, R.; Wu, F.; Bouras, G.; Mai, L.T.H.; Smith, E.; Wormald, P.J.; Valentine, R.; Psaltis, A.J.; Vreugde, S.; et al. Identification of consensus head and neck cancer-associated microbiota signatures: A systematic review and meta-analysis of 16S rRNA and the Cancer Microbiome Atlas datasets. J. Med. Microbiol. 2024, 73, 001799. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.K.; Cheung, M.K.; Lan, L.; Ng, C.; Lau, E.H.L.; Yeung, Z.W.C.; Wong, E.W.Y.; Leung, L.; Qu, X.; Cai, L.; et al. Characterization of oral microbiota in HPV and non-HPV head and neck squamous cell carcinoma and its association with patient outcomes. Oral Oncol. 2022, 135, 106245. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Marchais, M.; Vacher, S.; Hilmi, M.; Schnitzler, A.; Meseure, D.; Leclere, R.; Lecerf, C.; Dubot, C.; Jeannot, E.; et al. Prognostic value of intratumoral Fusobacterium nucleatum and association with immune-related gene expression in oral squamous cell carcinoma patients. Sci. Rep. 2021, 11, 7870. [Google Scholar] [CrossRef]
- Hayes, R.B.; Ahn, J.; Fan, X.; Peters, B.A.; Ma, Y.; Yang, L.; Agalliu, I.; Burk, R.D.; Ganly, I.; Purdue, M.P. Association of Oral Microbiome with Risk for Incident Head and Neck Squamous Cell Cancer. JAMA Oncol. 2018, 4, 358–365. [Google Scholar] [CrossRef]
- Shay, E.; Sangwan, N.; Padmanabhan, R.; Lundy, S.; Burkey, B.; Eng, C. Bacteriome and mycobiome and bacteriome-mycobiome interactions in head and neck squamous cell carcinoma. Oncotarget 2020, 11, 2375. [Google Scholar] [CrossRef]
- Heng, W.; Wang, W.; Dai, T.; Jiang, P.; Lu, Y.; Li, R.; Zhang, M.; Xie, R.; Zhou, Y.; Zhao, M. Oral bacteriome and mycobiome across stages of oral carcinogenesis. Microbiol. Spectr. 2022, 10, e0273722. [Google Scholar] [CrossRef]
- Li, Y.; Tan, X.; Zhao, X.; Xu, Z.; Dai, W.; Duan, W.; Huang, S.; Zhang, E.; Liu, J.; Zhang, S. Composition and function of oral microbiota between gingival squamous cell carcinoma and periodontitis. Oral. Oncol. 2020, 107, 104710. [Google Scholar] [CrossRef]
- Zuo, H.J.; Fu, M.R.; Zhao, H.L.; Du, X.W.; Hu, Z.Y.; Zhao, X.Y.; Ji, X.Q.; Feng, X.Q.; Zhumajiang, W.; Zhou, T.H.; et al. Study on the Salivary Microbial Alteration of Men with Head and Neck Cancer and Its Relationship With Symptoms in Southwest China. Front. Cell. Infect. Microbiol. 2020, 10, 514943. [Google Scholar] [CrossRef]
- Peter, T.K.; Withanage, M.H.H.; Comnick, C.L.; Pendleton, C.; Dabdoub, S.; Ganesan, S.; Drake, D.; Banas, J.; Xie, X.J.; Zeng, E. Systematic review and meta-analysis of oral squamous cell carcinoma associated oral microbiome. Front. Microbiol. 2022, 13, 968304. [Google Scholar] [CrossRef]
- Metsäniitty, M.; Hasnat, S.; Salo, T.; Salem, A. Oral Microbiota-A New Frontier in the Pathogenesis and Management of Head and Neck Cancers. Cancers 2021, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Su Mun, L.; Wye Lum, S.; Kong Yuiin Sze, G.; Hock Yoong, C.; Ching Yung, K.; Kah Lok, L.; Gopinath, D. Association of Microbiome with Oral Squamous Cell Carcinoma: A Systematic Review of the Metagenomic Studies. Int. J. Environ. Res. Public Health 2021, 18, 7224. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.T.; Sodré, C.S.; de Sousa Rodrigues, P.M.G.R.; da Silva, A.M.P.; Fuly, M.S.; Dos Santos, H.F.; Gonçalves, L.S.; de Carvalho Ferreira, D.; Ribeiro, M.G. High-throughput nucleotide sequencing for bacteriome studies in oral squamous cell carcinoma: A systematic review. Oral Maxillofac. Surg. 2020, 24, 387–401. [Google Scholar] [CrossRef]
- Delaney, C.; Veena, C.L.R.; Butcher, M.C.; McLean, W.; Shaban, S.M.A.; Nile, C.J.; Ramage, G. Limitations of using 16S rRNA microbiome sequencing to predict oral squamous cell carcinoma. APMIS 2023, 131, 262–276. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [PubMed]
- Nearing, J.T.; Comeau, A.M.; Langille, M.G.I. Identifying biases and their potential solutions in human microbiome studies. Microbiome 2021, 9, 113. [Google Scholar] [CrossRef]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef]
- Wang, S.; Song, F.; Gu, H.; Wei, X.; Zhang, K.; Zhou, Y.; Luo, H. Comparative evaluation of the salivary and buccal mucosal microbiota by 16S rRNA sequencing for forensic investigations. Front. Microbiol. 2022, 13, 777882. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. Rev. Esp. Cardiol. (Engl. Ed.) 2021, 74, 790–799. [Google Scholar] [CrossRef]
- Sterne, J.A.; Savović, J.; Page, M.J.; Elbers, R.G.; Blencowe, N.S.; Boutron, I.; Cates, C.J.; Cheng, H.-Y.; Corbett, M.S.; Eldridge, S.M. RoB 2: A revised tool for assessing risk of bias in randomised trials. BMJ 2019, 366, l4898. [Google Scholar] [CrossRef] [PubMed]
- Team STD. SRA Toolkit. 2020. Available online: https://hpc.nih.gov/apps/sratoolkit.html (accessed on 20 September 2024).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Wang, Y.; Lê Cao, K.-A. PLSDA-batch: A multivariate framework to correct for batch effects in microbiome data. Brief. Bioinform. 2023, 24, bbac622. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Ma, S.; Shungin, D.; Mallick, H.; Schirmer, M.; Nguyen, L.H.; Kolde, R.; Franzosa, E.; Vlamakis, H.; Xavier, R.; Huttenhower, C. Population structure discovery in meta-analyzed microbial communities and inflammatory bowel disease using MMUPHin. Genome Biol. 2022, 23, 208. [Google Scholar] [CrossRef]
- Curry, K.D.; Wang, Q.; Nute, M.G.; Tyshaieva, A.; Reeves, E.; Soriano, S.; Wu, Q.; Graeber, E.; Finzer, P.; Mendling, W.; et al. Emu: Species-level microbial community profiling of full-length 16S rRNA Oxford Nanopore sequencing data. Nat. Methods 2022, 19, 845–853. [Google Scholar] [CrossRef]
- Kodikara, S.; Ellul, S.; Lê Cao, K.-A. Statistical challenges in longitudinal microbiome data analysis. Brief. Bioinform. 2022, 23, bbac273. [Google Scholar] [CrossRef]
- Susin, A.; Wang, Y.; Lê Cao, K.-A.; Calle, M.L. Variable selection in microbiome compositional data analysis. NAR Genom. Bioinform. 2020, 2, lqaa029. [Google Scholar] [CrossRef]
- Yeo, K.; Connell, J.; Bouras, G.; Smith, E.; Murphy, W.; Hodge, J.C.; Krishnan, S.; Wormald, P.J.; Valentine, R.; Psaltis, A.J.; et al. A comparison between full-length 16S rRNA Oxford nanopore sequencing and Illumina V3-V4 16S rRNA sequencing in head and neck cancer tissues. Arch. Microbiol. 2024, 206, 248. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community Ecol. Package Version 2013, 2, 1–295. [Google Scholar]
- Zhou, H.; He, K.; Chen, J.; Zhang, X. LinDA: Linear models for differential abundance analysis of microbiome compositional data. Genome Biol. 2022, 23, 95. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. Msphere 2021, 6, e01202-20. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, S.M.; Duvallet, C.; Alm, E.J. Correcting for batch effects in case-control microbiome studies. PLoS Comput. Biol. 2018, 14, e1006102. [Google Scholar] [CrossRef]
- Topçuoğlu, B.D.; Lapp, Z.; Sovacool, K.L.; Snitkin, E.; Wiens, J.; Schloss, P.D. mikropml: User-friendly R package for supervised machine learning pipelines. J. Open Source Softw. 2021, 6, 3073. [Google Scholar] [CrossRef]
- Sharma, A.K.; DeBusk, W.T.; Stepanov, I.; Gomez, A.; Khariwala, S.S. Oral Microbiome Profiling in Smokers with and without Head and Neck Cancer Reveals Variations between Health and Disease. Cancer Prev. Res. 2020, 13, 463–474. [Google Scholar] [CrossRef]
- Guerrero-Preston, R.; Godoy-Vitorino, F.; Jedlicka, A.; Rodríguez-Hilario, A.; González, H.; Bondy, J.; Lawson, F.; Folawiyo, O.; Michailidi, C.; Dziedzic, A.; et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget 2016, 7, 51320–51334. [Google Scholar] [CrossRef]
- Frank, D.N.; Qiu, Y.; Cao, Y.; Zhang, S.; Lu, L.; Kofonow, J.M.; Robertson, C.E.; Liu, Y.; Wang, H.; Levens, C.L.; et al. A dysbiotic microbiome promotes head and neck squamous cell carcinoma. Oncogene 2022, 41, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Shen, W.; Wang, J.; Xu, Y.; Zhai, R.; Zhang, J.; Wang, M.; Wang, M.; Liu, L. Capnocytophaga gingivalis is a potential tumor promotor in oral cancer. Oral Dis. 2024, 30, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Y.; Liao, Y.; Wu, Y.X.; Diao, H.; Du, Y.; Chen, Y.W.; Xie, J.R.; Xue, W.Q.; He, Y.Q.; Wang, T.M.; et al. The Oral Microbiome as Mediator between Oral Hygiene and Its Impact on Nasopharyngeal Carcinoma. Microorganisms 2023, 11, 719. [Google Scholar] [CrossRef]
- Banavar, G.; Ogundijo, O.; Toma, R.; Rajagopal, S.; Lim, Y.K.; Tang, K.; Camacho, F.; Torres, P.J.; Gline, S.; Parks, M.; et al. The salivary metatranscriptome as an accurate diagnostic indicator of oral cancer. NPJ Genom. Med. 2021, 6, 105. [Google Scholar] [CrossRef] [PubMed]
- Omori, M.; Kato-Kogoe, N.; Sakaguchi, S.; Fukui, N.; Yamamoto, K.; Nakajima, Y.; Inoue, K.; Nakano, H.; Motooka, D.; Nakano, T. Comparative evaluation of microbial profiles of oral samples obtained at different collection time points and using different methods. Clin. Oral Investig. 2021, 25, 2779–2789. [Google Scholar] [CrossRef]
- Zaura, E.; Keijser, B.J.F.; Huse, S.M.; Crielaard, W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009, 9, 259. [Google Scholar] [CrossRef]
- Dhakal, A.; Upadhyay, R.; Wheeler, C.; Hoyd, R.; Karivedu, V.; Gamez, M.E.; Valentin, S.; Vanputten, M.; Bhateja, P.; Bonomi, M.; et al. Association between Tumor Microbiome and Hypoxia across Anatomic Subsites of Head and Neck Cancers. Int. J. Mol. Sci. 2022, 23, 15531. [Google Scholar] [CrossRef]
- Yano, Y.; Hua, X.; Wan, Y.; Suman, S.; Zhu, B.; Dagnall, C.L.; Hutchinson, A.; Jones, K.; Hicks, B.D.; Shi, J.; et al. Comparison of Oral Microbiota Collected Using Multiple Methods and Recommendations for New Epidemiologic Studies. Msystems 2020, 5, e00156-20. [Google Scholar] [CrossRef]
- Mougeot, J.C.; Beckman, M.F.; Langdon, H.C.; Lalla, R.V.; Brennan, M.T.; Bahrani Mougeot, F.K. Haemophilus pittmaniae and Leptotrichia spp. Constitute a Multi-Marker Signature in a Cohort of Human Papillomavirus-Positive Head and Neck Cancer Patients. Front. Microbiol. 2021, 12, 794546. [Google Scholar] [CrossRef]
- Kers, J.G.; Saccenti, E. The Power of Microbiome Studies: Some Considerations on Which Alpha and Beta Metrics to Use and How to Report Results. Front. Microbiol. 2021, 12, 796025. [Google Scholar] [CrossRef]
- Unlu, O.; Demirci, M.; Paksoy, T.; Eden, A.B.; Tansuker, H.D.; Dalmizrak, A.; Aktan, C.; Senel, F.; Sunter, A.V.; Yigit, O.; et al. Oral microbial dysbiosis in patients with oral cavity cancers. Clin. Oral Investig. 2024, 28, 377. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, B.; Hu, X.; Li, J.; Wu, M.; Yan, C.; Yang, Y.; Li, Y. Neisseria sicca and Corynebacterium matruchotii inhibited oral squamous cell carcinomas by regulating genome stability. Bioengineered 2022, 13, 14094–14106. [Google Scholar] [CrossRef] [PubMed]
- Baraniya, D.; Jain, V.; Lucarelli, R.; Tam, V.; Vanderveer, L.; Puri, S.; Yang, M.; Al-Hebshi, N.N. Screening of Health-Associated Oral Bacteria for Anticancer Properties in vitro. Front. Cell. Infect. Microbiol. 2020, 10, 575656. [Google Scholar] [CrossRef]
- Connell, J.T.; Yeo, K.; Bouras, G.; Bassiouni, A.; Fenix, K.; Cooksley, C.; Vreugde, S.; Wormald, P.J.; Psaltis, A.J. Enhanced phylogenetic insights into the microbiome of chronic rhinosinusitis through the novel application of long read 16S rRNA gene amplicon sequencing. Rhinology 2024, 62, 152–162. [Google Scholar] [CrossRef]
- Kumar, B.; Lorusso, E.; Fosso, B.; Pesole, G. A comprehensive overview of microbiome data in the light of machine learning applications: Categorization, accessibility, and future directions. Front. Microbiol. 2024, 15, 1343572. [Google Scholar] [CrossRef]
- Microbiome test spots oral cancers. Nat. Biotechnol. 2021, 39, 650. [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Moher, D.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phylum | Saliva Relative Abundance (Mean ± SD) | Oral Rinse Relative Abundance (Mean ± SD) |
|---|---|---|
| Firmicutes | V3V4 = 32.4 ± 23.4% V4 = 49.7 ± 19.0% V4V5 = 43.2 ± 14.5% | V3V4 = 20.9 ± 20.4% |
| Bacteroidota | V3V4 = 32.2 ± 27.2% V4 = 19.1 ± 14.3% V4V5 = 33.0 ± 14.8% | V3V4 = 46.5 ± 29.5% |
| Fusobacteriota | V3V4 = 12.2 ± 21.0% V4 = 8.00 ± 9.01% V4V5 = 2.93 ± 2.82% | V3V4 = 7.06 ± 11.3% |
| Proteobacteria | V3V4 = 12.0 ± 16.0% V4 = 12.4 ± 11.5% V4V5 = 16.2 ± 13.8% | V3V4 = 20.3 ± 24.2% |
| Actinobacteriota | V3V4 = 4.15 ± 7.78% V4 = 8.32 ± 7.64% V4V5 = 3.29 ± 3.46% | V3V4 = 2.90 ± 5.79% |
| Genera | Saliva Relative Abundance (Mean ± SD) | Oral Rinse Relative Abundance (Mean ± SD) |
| Streptococcus | V3V4 = 13.4 ± 17.4% V4 = 31.1 ± 18.1% V4V5 = 29.3 ± 16.2% | V3V4 = 10.3 ± 15.4% |
| Neisseria | V3V4 = 8.38 ± 13.8% V4 = 10.5 ± 10.7% V4V5 = 9.17 ± 7.65% | V3V4 = 12.7 ± 19.4% |
| Prevotella | V3V4 = 19.6 ± 21.9% V4 = 13.0 ± 12.5% V4V5 = 13.1 ± 9.35% | V3V4 = 25.8 ± 28.1% |
| Porphyromonas | V3V4 = 6.05 ± 13.4% V4 = 4.40 ± 7.04% V4V5 = 12.9 ± 9.89% | V3V4 = 12.5 ± 21.6% |
| Veillonella | V3V4 = 8.78 ± 10.8% V4 = 11.6 ± 11.5% V4V5 = 4.84 ± 4.93% | V3V4 = 4.1 ± 9.2% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeo, K.; Wu, F.; Li, R.; Smith, E.; Wormald, P.-J.; Valentine, R.; Psaltis, A.J.; Vreugde, S.; Fenix, K. Is Short-Read 16S rRNA Sequencing of Oral Microbiome Sampling a Suitable Diagnostic Tool for Head and Neck Cancer? Pathogens 2024, 13, 826. https://doi.org/10.3390/pathogens13100826
Yeo K, Wu F, Li R, Smith E, Wormald P-J, Valentine R, Psaltis AJ, Vreugde S, Fenix K. Is Short-Read 16S rRNA Sequencing of Oral Microbiome Sampling a Suitable Diagnostic Tool for Head and Neck Cancer? Pathogens. 2024; 13(10):826. https://doi.org/10.3390/pathogens13100826
Chicago/Turabian StyleYeo, Kenny, Fangmeinuo Wu, Runhao Li, Eric Smith, Peter-John Wormald, Rowan Valentine, Alkis James Psaltis, Sarah Vreugde, and Kevin Fenix. 2024. "Is Short-Read 16S rRNA Sequencing of Oral Microbiome Sampling a Suitable Diagnostic Tool for Head and Neck Cancer?" Pathogens 13, no. 10: 826. https://doi.org/10.3390/pathogens13100826
APA StyleYeo, K., Wu, F., Li, R., Smith, E., Wormald, P.-J., Valentine, R., Psaltis, A. J., Vreugde, S., & Fenix, K. (2024). Is Short-Read 16S rRNA Sequencing of Oral Microbiome Sampling a Suitable Diagnostic Tool for Head and Neck Cancer? Pathogens, 13(10), 826. https://doi.org/10.3390/pathogens13100826