

Equivocating and Deliberating on the Probability of COVID-19 Infection Serving as a Risk Factor for Lung Cancer and Common Molecular Pathways Serving as a Link
 , , , ,
, , , , {kind=link}
Abstract
:1. Introduction
2. Persistent Symptoms and Long-Term Respiratory Sequelae in COVID-19 Survivors
3. Lung Cancer
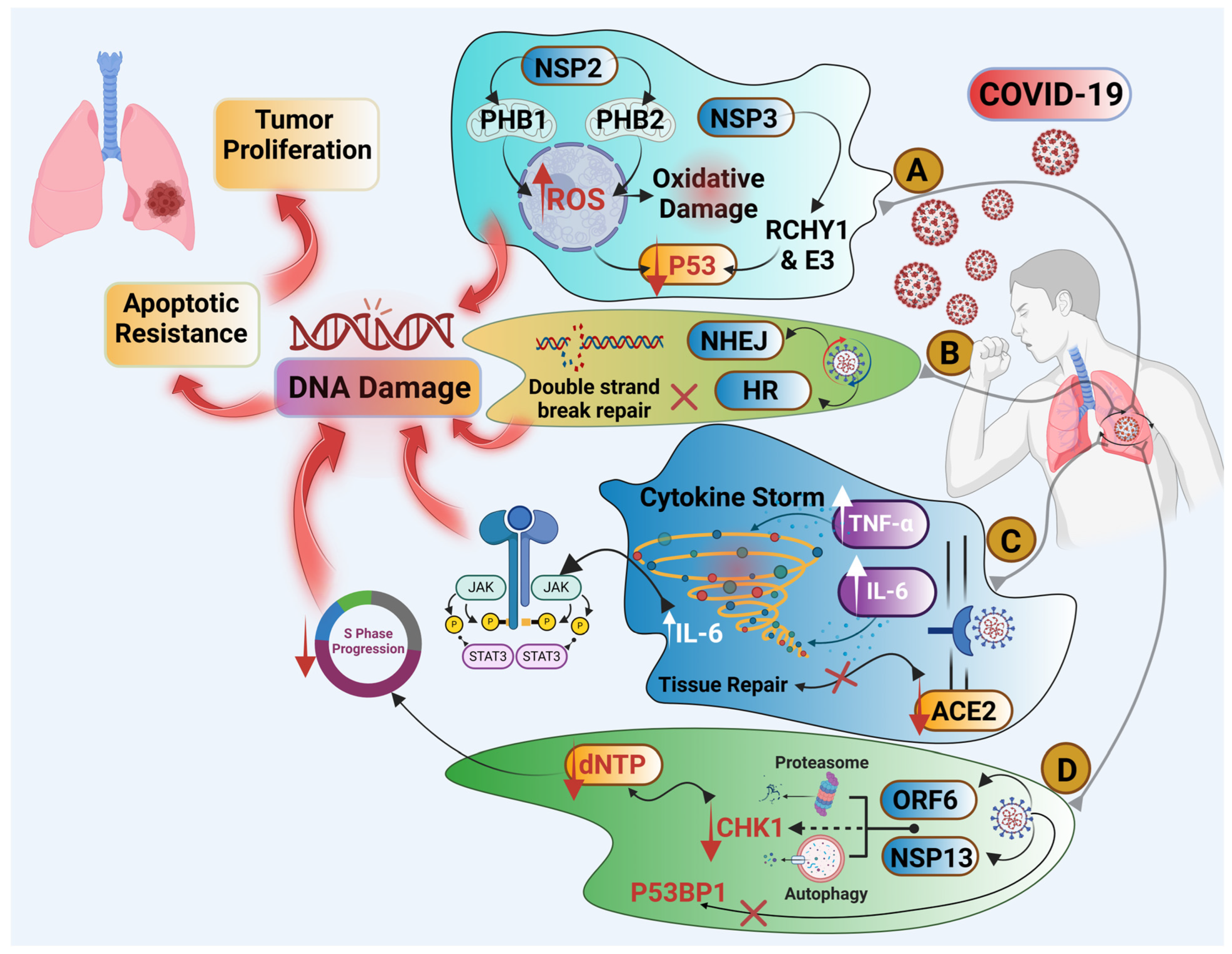
4. Is There a Link Between COVID-19 Infection and Lung Cancer?
4.1. Cytokine Storm and Chronic Inflammation
4.2. SARS-CoV-2 Could Be an Oncogenic Virus
4.3. COVID-19 Infection Is a Cause of DNA Damage and Impaired DNA Repair
4.4. Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response (UPR)
4.5. COVID-19 and ACE2 Receptor
5. SLC22A18, ZEB2, and ZEB1 as Molecular Markers of Lung Cancer That Could Be Overexpressed by COVID-19 Infection
6. Challenges in Proving Causality Between COVID-19 and Lung Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Coronavirus Disease (COVID-19): Symptoms. Available online: https://www.who.int/health-topics/coronavirus#tab=tab_3 (accessed on 1 October 2024).
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Ahmad Farouk, I.; Lal, S.K. COVID-19: A Review on the Novel Coronavirus Disease Evolution, Transmission, Detection, Control and Prevention. Viruses 2021, 13, 202. [Google Scholar] [CrossRef]
- COVID19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University. Available online: https://www.arcgis.com/apps/dashboards/bda7594740fd40299423467b48e9ecf6 (accessed on 1 September 2024).
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Population Health and Public Health Practice; Board on Health Sciences Policy; Committee on Evidence-Based Practices for Public Health Emergency Preparedness and Response. Evidence-Based Practice for Public Health Emergency Preparedness and Response; Downey, A., Brown, L., Calonge, N., Eds.; National Academies Press (US): Washington, DC, USA, 2020. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Nicola, M.; Alsafi, Z.; Sohrabi, C.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, M.; Agha, R. The socio-economic implications of the coronavirus pandemic (COVID-19): A review. Int. J. Surg. 2020, 78, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19); StatPearls: Treasure Island, FL, USA, 2024. [Google Scholar]
- Huang, L.; Yao, Q.; Gu, X.; Wang, Q.; Ren, L.; Wang, Y.; Hu, P.; Guo, L.; Liu, M.; Xu, J.; et al. 1-year outcomes in hospital survivors with COVID-19: A longitudinal cohort study. Lancet 2021, 398, 747–758. [Google Scholar] [CrossRef]
- Alrajhi, N.N. Post-COVID-19 pulmonary fibrosis: An ongoing concern. Ann. Thorac. Med. 2023, 18, 173–181. [Google Scholar] [CrossRef]
- Parotto, M.; Gyongyosi, M.; Howe, K.; Myatra, S.N.; Ranzani, O.; Shankar-Hari, M.; Herridge, M.S. Post-acute sequelae of COVID-19: Understanding and addressing the burden of multisystem manifestations. Lancet Respir. Med. 2023, 11, 739–754. [Google Scholar] [CrossRef]
- Wilson, G.N. A Clinical Qualification Protocol Highlights Overlapping Genomic Influences and Neuro-Autonomic Mechanisms in Ehlers-Danlos and Long COVID-19 Syndromes. Curr. Issues Mol. Biol. 2023, 45, 6003–6023. [Google Scholar] [CrossRef]
- Johnston, J.; Dorrian, D.; Linden, D.; Stanel, S.C.; Rivera-Ortega, P.; Chaudhuri, N. Pulmonary Sequelae of COVID-19: Focus on Interstitial Lung Disease. Cells 2023, 12, 2238. [Google Scholar] [CrossRef]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Ledda, R.E.; Milanese, G.; Milone, F.; Leo, L.; Balbi, M.; Silva, M.; Sverzellati, N. Interstitial lung abnormalities: New insights between theory and clinical practice. Insights Imaging 2022, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Palmucci, S.; Roccasalva, F.; Puglisi, S.; Torrisi, S.E.; Vindigni, V.; Mauro, L.A.; Ettorre, G.C.; Piccoli, M.; Vancheri, C. Clinical and radiological features of idiopathic interstitial pneumonias (IIPs): A pictorial review. Insights Imaging 2014, 5, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Salehi, S.; Reddy, S.; Gholamrezanezhad, A. Long-term Pulmonary Consequences of Coronavirus Disease 2019 (COVID-19): What We Know and What to Expect. J. Thorac. Imaging 2020, 35, W87–W89. [Google Scholar] [CrossRef] [PubMed]
- Parimon, T.; Espindola, M.; Marchevsky, A.; Rampolla, R.; Chen, P.; Hogaboam, C.M. Potential mechanisms for lung fibrosis associated with COVID-19 infection. QJM 2023, 116, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Y.; Pinsky, P.F.; Caporaso, N.E.; Chatterjee, N.; Baumgarten, M.; Langenberg, P.; Furuno, J.P.; Lan, Q.; Engels, E.A. Lung cancer risk following detection of pulmonary scarring by chest radiography in the prostate, lung, colorectal, and ovarian cancer screening trial. Arch. Intern. Med. 2008, 168, 2326–2332; discussion 2332. [Google Scholar] [CrossRef]
- Dugerdil, A.; Semenzato, L.; Weill, A.; Zureik, M.; Flahault, A. Severe SARS-CoV-2 infection as a marker of undiagnosed cancer: A population-based study. Sci. Rep. 2023, 13, 8729. [Google Scholar] [CrossRef]
- Sadigov, A.; Akhundov, S.; Agayeva, A. Risk Factors for Lung Cancer in Individuals with COVID-19 Without Cancer History. Respir. Care 2021, 66, 3566906. [Google Scholar]
- WHO. Lung Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/lung-cancer (accessed on 1 September 2024).
- Schabath, M.B.; Cote, M.L. Cancer Progress and Priorities: Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1563–1579. [Google Scholar] [CrossRef]
- Rami-Porta, R.; Bolejack, V.; Giroux, D.J.; Chansky, K.; Crowley, J.; Asamura, H.; Goldstraw, P.; International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee, Advisory Board Members; Participating Institutions. The IASLC lung cancer staging project: The new database to inform the eighth edition of the TNM classification of lung cancer. J. Thorac. Oncol. 2014, 9, 1618–1624. [Google Scholar] [CrossRef]
- Parris, B.A.; O’Farrell, H.E.; Fong, K.M.; Yang, I.A. Chronic obstructive pulmonary disease (COPD) and lung cancer: Common pathways for pathogenesis. J. Thorac. Dis. 2019, 11, S2155–S2172. [Google Scholar] [CrossRef]
- Xing, P.Y.; Zhu, Y.X.; Wang, L.; Hui, Z.G.; Liu, S.M.; Ren, J.S.; Zhang, Y.; Song, Y.; Liu, C.C.; Huang, Y.C.; et al. What are the clinical symptoms and physical signs for non-small cell lung cancer before diagnosis is made? A nation-wide multicenter 10-year retrospective study in China. Cancer Med. 2019, 8, 4055–4069. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Valyi-Nagy, T.; Fredericks, B.; Wilson, J.; Shukla, S.D.; Setty, S.; Slavin, K.V.; Valyi-Nagy, K. Detection of SARS-CoV-2 RNA by In Situ Hybridization in Lung-Cancer Cells Metastatic to Brain and in Adjacent Brain Parenchyma. Pathogens 2023, 12, 772. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol. 2011, 2, 98. [Google Scholar] [CrossRef] [PubMed]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Montazersaheb, S.; Hosseiniyan Khatibi, S.M.; Hejazi, M.S.; Tarhriz, V.; Farjami, A.; Ghasemian Sorbeni, F.; Farahzadi, R.; Ghasemnejad, T. COVID-19 infection: An overview on cytokine storm and related interventions. Virol. J. 2022, 19, 92. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Longo, D.L.; June, C.H. Cytokine Storm. Ne. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Yasmeen, N.; Selvaraj, H.; Lakhawat, S.S.; Datta, M.; Sharma, P.K.; Jain, A.; Khanna, R.; Srinivasan, J.; Kumar, V. Possibility of averting cytokine storm in SARS-COV 2 patients using specialized pro-resolving lipid mediators. Biochem. Pharmacol. 2023, 209, 115437. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef]
- Megha, K.B.; Joseph, X.; Akhil, V.; Mohanan, P.V. Cascade of immune mechanism and consequences of inflammatory disorders. Phytomedicine 2021, 91, 153712. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Wei, Y.; Ren, J.; Zhang, L.; Zhou, F. The intersection of COVID-19 and cancer: Signaling pathways and treatment implications. Mol. Cancer 2021, 20, 76. [Google Scholar] [CrossRef]
- Dethlefsen, C.; Hojfeldt, G.; Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 2013, 138, 657–664. [Google Scholar] [CrossRef]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour. Biol. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Vargas, A.J.; Harris, C.C. Biomarker development in the precision medicine era: Lung cancer as a case study. Nat. Rev. Cancer 2016, 16, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Hsiao, C.F.; Yeh, Y.M.; Chang, G.C.; Tsai, Y.H.; Chen, Y.M.; Huang, M.S.; Chen, H.L.; Li, Y.J.; Yang, P.C.; et al. Circulating interleukin-6 level is a prognostic marker for survival in advanced nonsmall cell lung cancer patients treated with chemotherapy. Int. J. Cancer 2013, 132, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Chang, Y.F. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 2003, 83, 222–226. [Google Scholar] [CrossRef]
- Culig, Z.; Puhr, M. Interleukin-6: A multifunctional targetable cytokine in human prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 52–58. [Google Scholar] [CrossRef]
- Miura, T.; Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Takahashi, H.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; et al. Characterization of patients with advanced pancreatic cancer and high serum interleukin-6 levels. Pancreas 2015, 44, 756–763. [Google Scholar] [CrossRef]
- Song, L.; Rawal, B.; Nemeth, J.A.; Haura, E.B. JAK1 activates STAT3 activity in non-small-cell lung cancer cells and IL-6 neutralizing antibodies can suppress JAK1-STAT3 signaling. Mol. Cancer Ther. 2011, 10, 481–494. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Akira, S.; Taga, T. Interleukin-6 and its receptor: A paradigm for cytokines. Science 1992, 258, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002, 3, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Yeh, H.H.; Lai, W.W.; Chen, H.H.W.; Liu, H.S.; Su, W.C. Autocrine IL-6-induced Stat3 activation contributes to the pathogenesis of lung adenocarcinoma and malignant pleural effusion. Oncogene 2006, 25, 4300–4309. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Munger, K. Viruses associated with human cancer. Biochim. Biophys Acta 2008, 1782, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, C.M.; Hainaut, P. Viral strategies for circumventing p53: The case of severe acute respiratory syndrome coronavirus. Curr. Opin. Oncol. 2021, 33, 149–158. [Google Scholar] [CrossRef]
- Fusaro, G.; Dasgupta, P.; Rastogi, S.; Joshi, B.; Chellappan, S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J. Biol. Chem. 2003, 278, 47853–47861. [Google Scholar] [CrossRef]
- Ma-Lauer, Y.; Carbajo-Lozoya, J.; Hein, M.Y.; Muller, M.A.; Deng, W.; Lei, J.; Meyer, B.; Kusov, Y.; von Brunn, B.; Bairad, D.R.; et al. p53 down-regulates SARS coronavirus replication and is targeted by the SARS-unique domain and PLpro via E3 ubiquitin ligase RCHY1. Proc. Natl. Acad. Sci. USA 2016, 113, E5192–E5201. [Google Scholar] [CrossRef]
- Stingi, A.; Cirillo, L. SARS-CoV-2 infection and cancer: Evidence for and against a role of SARS-CoV-2 in cancer onset. Bioessays 2021, 43, e2000289. [Google Scholar] [CrossRef]
- Bhardwaj, K.; Liu, P.; Leibowitz, J.L.; Kao, C.C. The coronavirus endoribonuclease Nsp15 interacts with retinoblastoma tumor suppressor protein. J. Virol. 2012, 86, 4294–4304. [Google Scholar] [CrossRef]
- Gomez-Carballa, A.; Martinon-Torres, F.; Salas, A. Is SARS-CoV-2 an oncogenic virus? J. Infect. 2022, 85, 573–607. [Google Scholar] [CrossRef] [PubMed]
- Grand, R.J. SARS-CoV-2 and the DNA damage response. J. Gen. Virol. 2023, 104, 001918. [Google Scholar] [CrossRef] [PubMed]
- Panico, P.; Ostrosky-Wegman, P.; Salazar, A.M. The potential role of COVID-19 in the induction of DNA damage. Mutat. Res. Rev. Mutat. Res. 2022, 789, 108411. [Google Scholar] [CrossRef] [PubMed]
- Gioia, U.; Tavella, S.; Martínez-Orellana, P.; Cicio, G.; Colliva, A.; Ceccon, M.; Cabrini, M.; Henriques, A.C.; Fumagalli, V.; Paldino, A.; et al. SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nat. Cell Biol. 2023, 25, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.H.; Huang, M.; Fang, S.G.; Liu, D.X. Coronavirus Infection Induces DNA Replication Stress Partly Through Interaction of Its Nonstructural Protein 13 with the p125 Subunit of DNA Polymerase δ. J. Biol. Chem. 2011, 286, 39546–39559. [Google Scholar] [CrossRef]
- Berti, M.; Cortez, D.; Lopes, M. The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat. Rev. Mol. Cell Biol. 2020, 21, 633–651. [Google Scholar] [CrossRef]
- Gordenin, D.A.; Koussa, N.C.; Smith, D.J. Limiting DNA polymerase delta alters replication dynamics and leads to a dependence on checkpoint activation and recombination-mediated DNA repair. PLoS Genet. 2021, 17, e1009322. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef]
- Caracciolo, D.; Montesano, M.; Tagliaferri, P.; Tassone, P. Alternative non-homologous end joining repair: A master regulator of genomic instability in cancer. Precis. Cancer Med. 2019, 2, 8. [Google Scholar] [CrossRef]
- SARS-CoV-2 causes DNA damage, cellular senescence and inflammation. Nat. Cell Biol. 2023, 25, 526–527. [CrossRef]
- Garcia, G.; Sharma, A.; Ramaiah, A.; Sen, C.; Purkayastha, A.; Kohn, D.B.; Parcells, M.S.; Beck, S.; Kim, H.; Bakowski, M.A.; et al. Antiviral drug screen identifies DNA-damage response inhibitor as potent blocker of SARS-CoV-2 replication. Cell Rep. 2021, 35, 108940. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.C.; Renner, D.M.; Silva, D.; Yang, D.; Parenti, N.A.; Medina, K.M.; Nicolaescu, V.; Gula, H.; Drayman, N.; Valdespino, A.; et al. SARS-CoV-2 Diverges from Other Betacoronaviruses in Only Partially Activating the IRE1alpha/XBP1 Endoplasmic Reticulum Stress Pathway in Human Lung-Derived Cells. mBio 2022, 13, e0241522. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K.; Murakami, S. Role of the unfolded protein response in cell death. Apoptosis 2006, 11, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Bhat, T.A.; Chaudhary, A.K.; Kumar, S.; O’Malley, J.; Inigo, J.R.; Kumar, R.; Yadav, N.; Chandra, D. Endoplasmic reticulum-mediated unfolded protein response and mitochondrial apoptosis in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 58–66. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gökçen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-κB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef]
- Morganstein, T.; Haidar, Z.; Trivlidis, J.; Azuelos, I.; Huang, M.J.; Eidelman, D.H.; Baglole, C.J. Involvement of the ACE2/Ang-(1-7)/MasR Axis in Pulmonary Fibrosis: Implications for COVID-19. Int. J. Mol. Sci. 2021, 22, 12955. [Google Scholar] [CrossRef] [PubMed]
- Bridi, G.D.P.; Tanni, S.E.; Baldi, B.G. Current Understanding of Post-COVID Pulmonary Fibrosis: Where Are We? Arch. Bronconeumol. 2023, 59, 69–70. [Google Scholar] [CrossRef]
- Tao, S.-L.; Wang, X.-m.; Feng, Y.-g.; Kang, P.-m.; Li, Q.-y.; Sun, T.-y.; Tan, Q.-y.; Deng, B. Is the presence of lung injury in COVID-19 an independent risk factor for secondary lung cancer? Med. Hypotheses 2020, 143, 110074. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Yang, S.; Thangaraju, M.; Ganapathy, V. SLC transporters as a novel class of tumour suppressors: Identity, function and molecular mechanisms. Biochem. J. 2016, 473, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Cheng, Q.; Zhao, Y.; Liu, T.; Wang, X.; Deng, Y.; Yang, J.; Zhang, Z. Expression and its clinical significance of SLC22A18 in non-small cell lung cancer. Zhongguo Fei Ai Za Zhi 2012, 15, 17–20. [Google Scholar] [CrossRef]
- Dutta, R.K.; Chinnapaiyan, S.; Unwalla, H. Aberrant MicroRNAomics in Pulmonary Complications: Implications in Lung Health and Diseases. Mol. Ther. Nucleic. Acids 2019, 18, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, T.; Wu, T.; Wang, Z.; Rao, Z.; Gao, J. microRNA-137 functions as a tumor suppressor in human non-small cell lung cancer by targeting SLC22A18. Int. J. Biol. Macromol. 2015, 74, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Ucles, J.F.; Boyero, L.; Salinas, A.; Cordero Varela, J.A.; Benedetti, J.C.; Bernabe-Caro, R.; Sanchez-Gastaldo, A.; Alonso, M.; Paz-Ares, L.; Molina-Pinelo, S. The Roles of Imprinted SLC22A18 and SLC22A18AS Gene Overexpression Caused by Promoter CpG Island Hypomethylation as Diagnostic and Prognostic Biomarkers for Non-Small Cell Lung Cancer Patients. Cancers 2020, 12, 2075. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Ramkumar, K.; Cargill, K.R.; Cardnell, R.J.; Nilsson, M.B.; Heeke, S.; Park, E.M.; Kundu, S.T.; Diao, L.; et al. SARS-CoV-2 infection induces EMT-like molecular changes, including ZEB1-mediated repression of the viral receptor ACE2, in lung cancer models. bioRxiv 2021. [Google Scholar] [CrossRef]
- Jaiswal, A.; Shrivastav, S.; Kushwaha, H.R.; Chaturvedi, R.; Singh, R.P. Oncogenic potential of SARS-CoV-2-targeting hallmarks of cancer pathways. Cell Commun. Signal 2024, 22, 447. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The roles of ZEB1 in tumorigenic progression and epigenetic modifications. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Nathan, V.; Osborne, J.K.; Farrow, R.K.; Deb, D.; Sullivan, J.P.; Dospoy, P.D.; Augustyn, A.; Hight, S.K.; Sato, M.; et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J. Clin. Investig. 2016, 126, 3219–3235. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Bi, H.; Bai, S.; Zhao, L.; Zhang, J.; Qi, C. Targeting ZEB2 by microRNA-129 in Non-Small Cell Lung Cancer Suppresses Cell Proliferation, Invasion and Migration via Regulating Wnt/β-Catenin Signaling Pathway and Epithelial—Mesenchymal Transition. OncoTargets Ther. 2019, 12, 9165–9175. [Google Scholar] [CrossRef]
- Hossain, M.A.; Rahman, M.Z.; Bhuiyan, T.; Moni, M.A. Identification of Biomarkers and Molecular Pathways Implicated in Smoking and COVID-19 Associated Lung Cancer Using Bioinformatics and Machine Learning Approaches. Int. J. Environ. Res. Public Health 2024, 21, 1392. [Google Scholar] [CrossRef]
- Shirbhate, E.; Pandey, J.; Patel, V.K.; Kamal, M.; Jawaid, T.; Gorain, B.; Kesharwani, P.; Rajak, H. Understanding the role of ACE-2 receptor in pathogenesis of COVID-19 disease: A potential approach for therapeutic intervention. Pharmacol. Rep. 2021, 73, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amara, A.; Trabelsi, S.; Hai, A.; Zaidi, S.H.H.; Siddiqui, F.; Alsaeed, S. Equivocating and Deliberating on the Probability of COVID-19 Infection Serving as a Risk Factor for Lung Cancer and Common Molecular Pathways Serving as a Link. Pathogens 2024, 13, 1070. https://doi.org/10.3390/pathogens13121070
Amara A, Trabelsi S, Hai A, Zaidi SHH, Siddiqui F, Alsaeed S. Equivocating and Deliberating on the Probability of COVID-19 Infection Serving as a Risk Factor for Lung Cancer and Common Molecular Pathways Serving as a Link. Pathogens. 2024; 13(12):1070. https://doi.org/10.3390/pathogens13121070
Chicago/Turabian StyleAmara, Abdelbasset, Saoussen Trabelsi, Abdul Hai, Syeda Huma H. Zaidi, Farah Siddiqui, and Sami Alsaeed. 2024. "Equivocating and Deliberating on the Probability of COVID-19 Infection Serving as a Risk Factor for Lung Cancer and Common Molecular Pathways Serving as a Link" Pathogens 13, no. 12: 1070. https://doi.org/10.3390/pathogens13121070
APA StyleAmara, A., Trabelsi, S., Hai, A., Zaidi, S. H. H., Siddiqui, F., & Alsaeed, S. (2024). Equivocating and Deliberating on the Probability of COVID-19 Infection Serving as a Risk Factor for Lung Cancer and Common Molecular Pathways Serving as a Link. Pathogens, 13(12), 1070. https://doi.org/10.3390/pathogens13121070