


Genetic Diversity of Salmonella enterica subsp. enterica Serovar Enteritidis from Human and Non-Human Sources in Portugal

 , , , , and
, , , , and 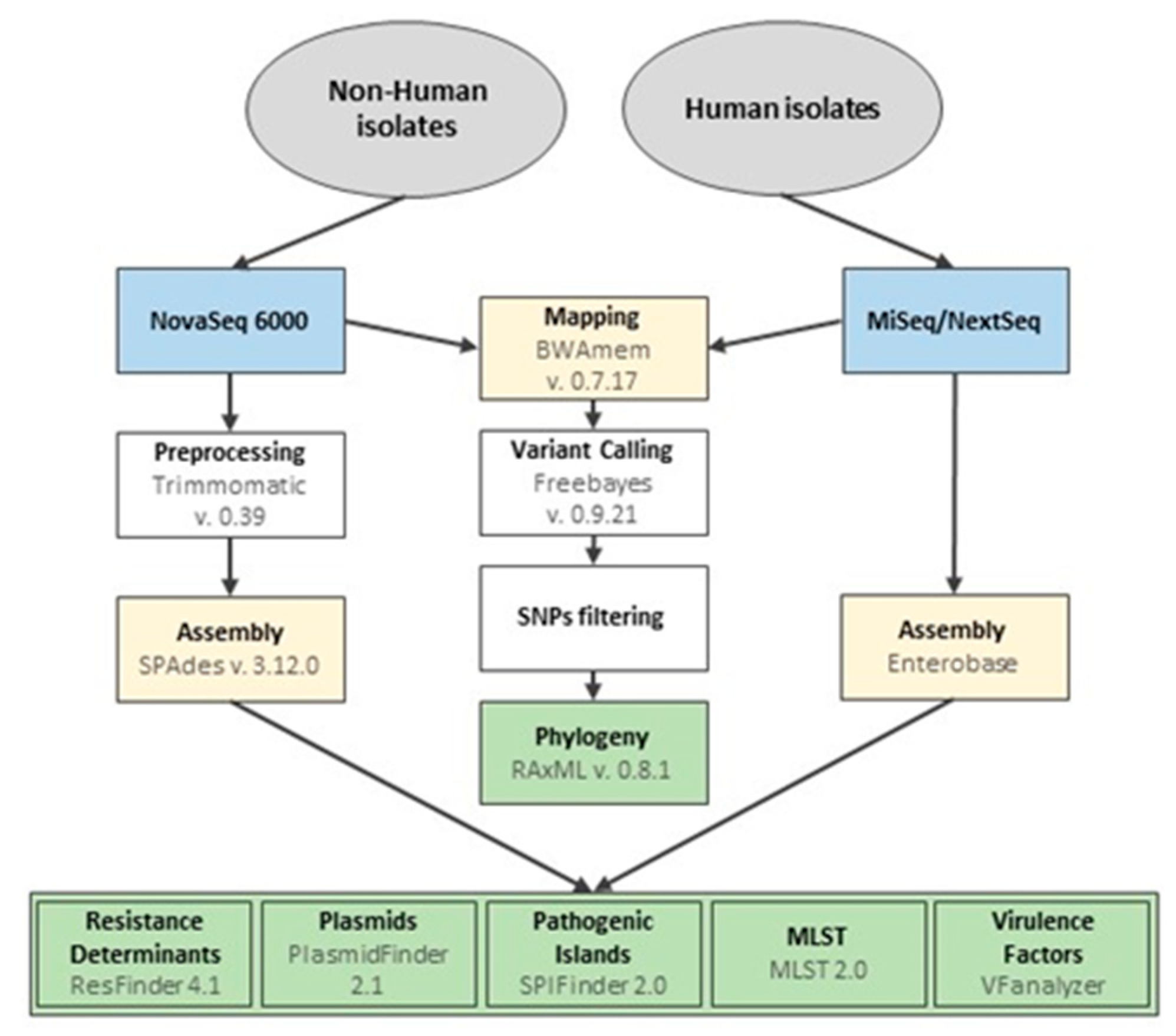{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Isolates
2.2. Serotyping of Salmonella Isolates
2.3. Antimicrobial Susceptibility Testing
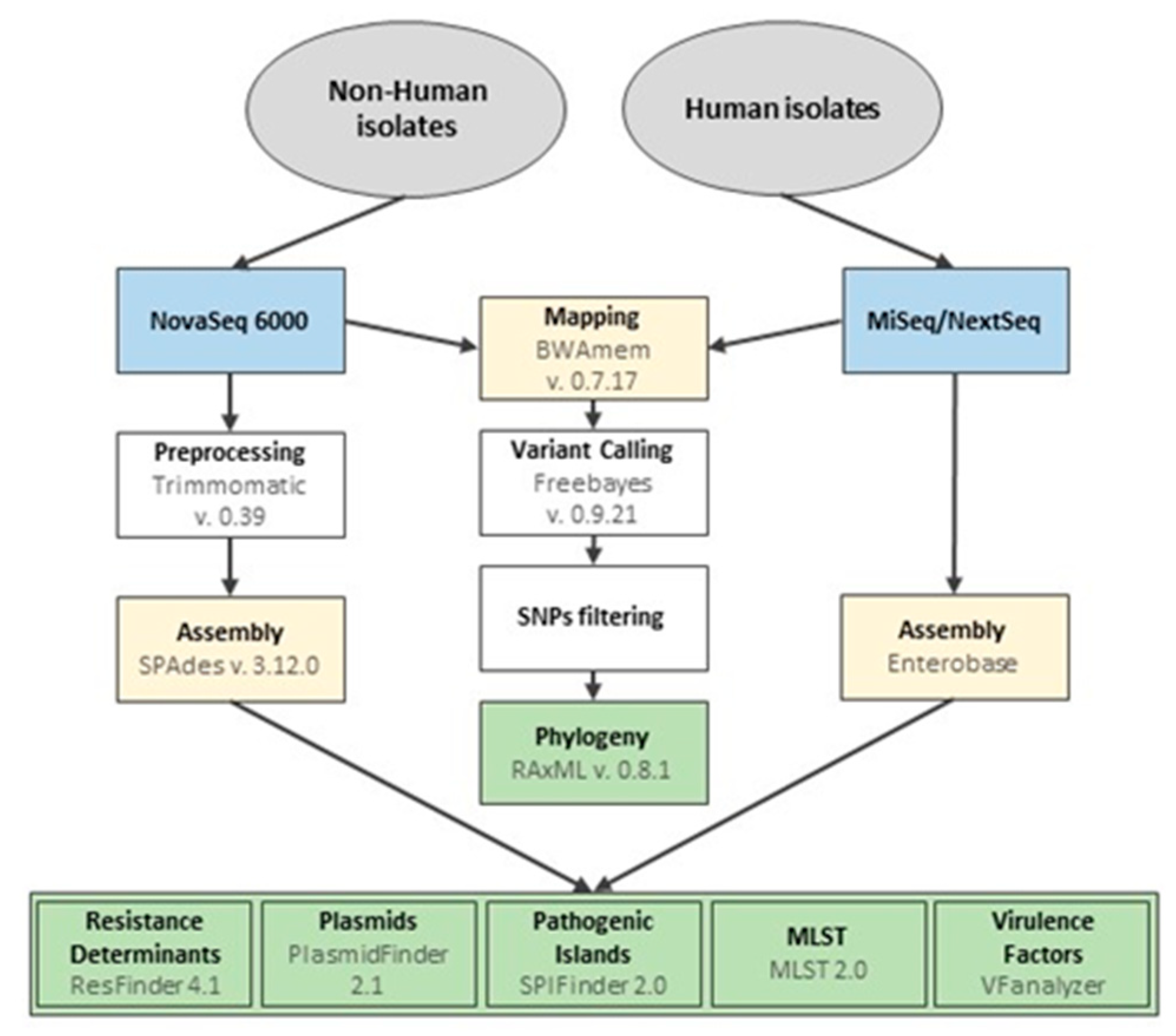
2.4. Whole Genome Sequencing and Bioinformatics Analysis
3. Results
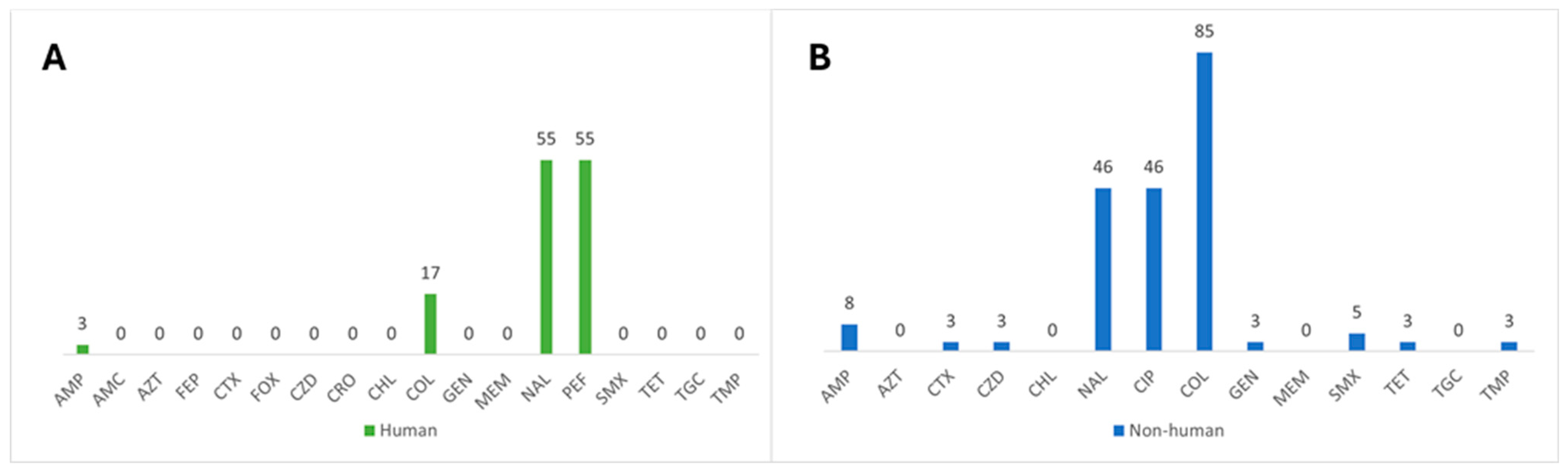
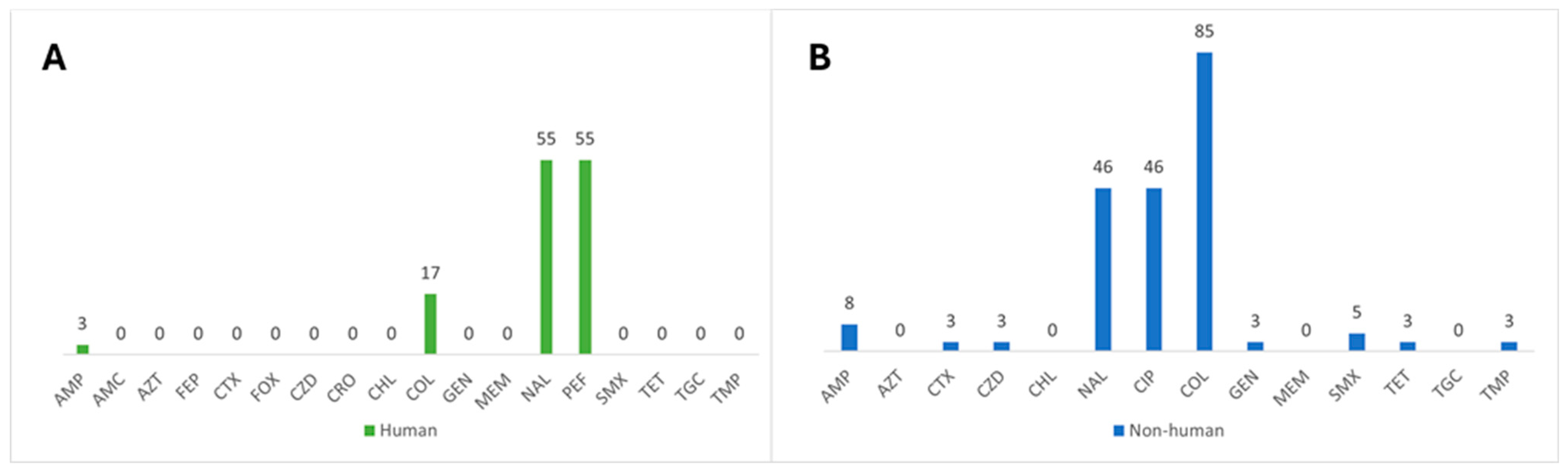
3.1. Characterization of Antimicrobial Resistance
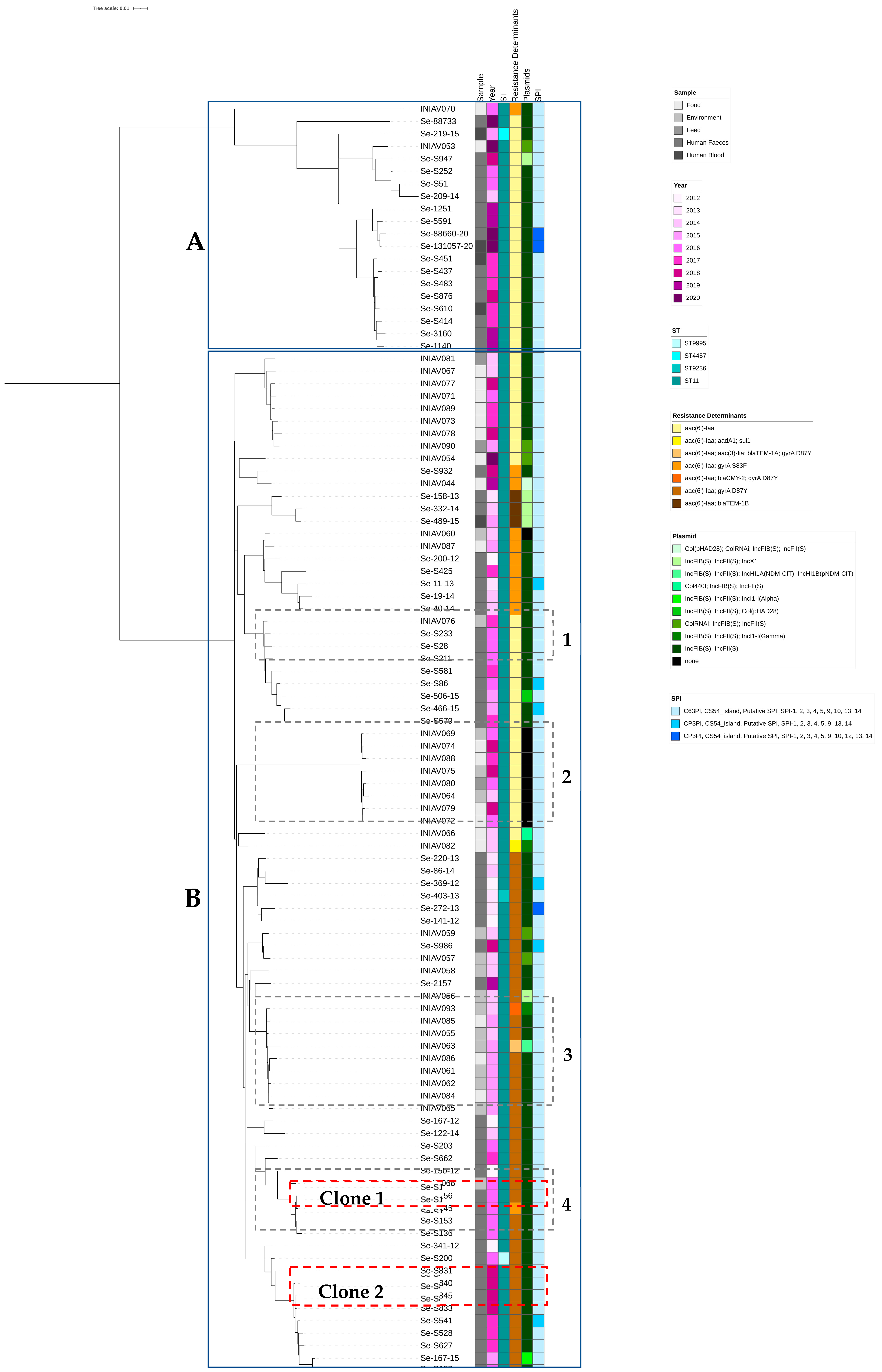
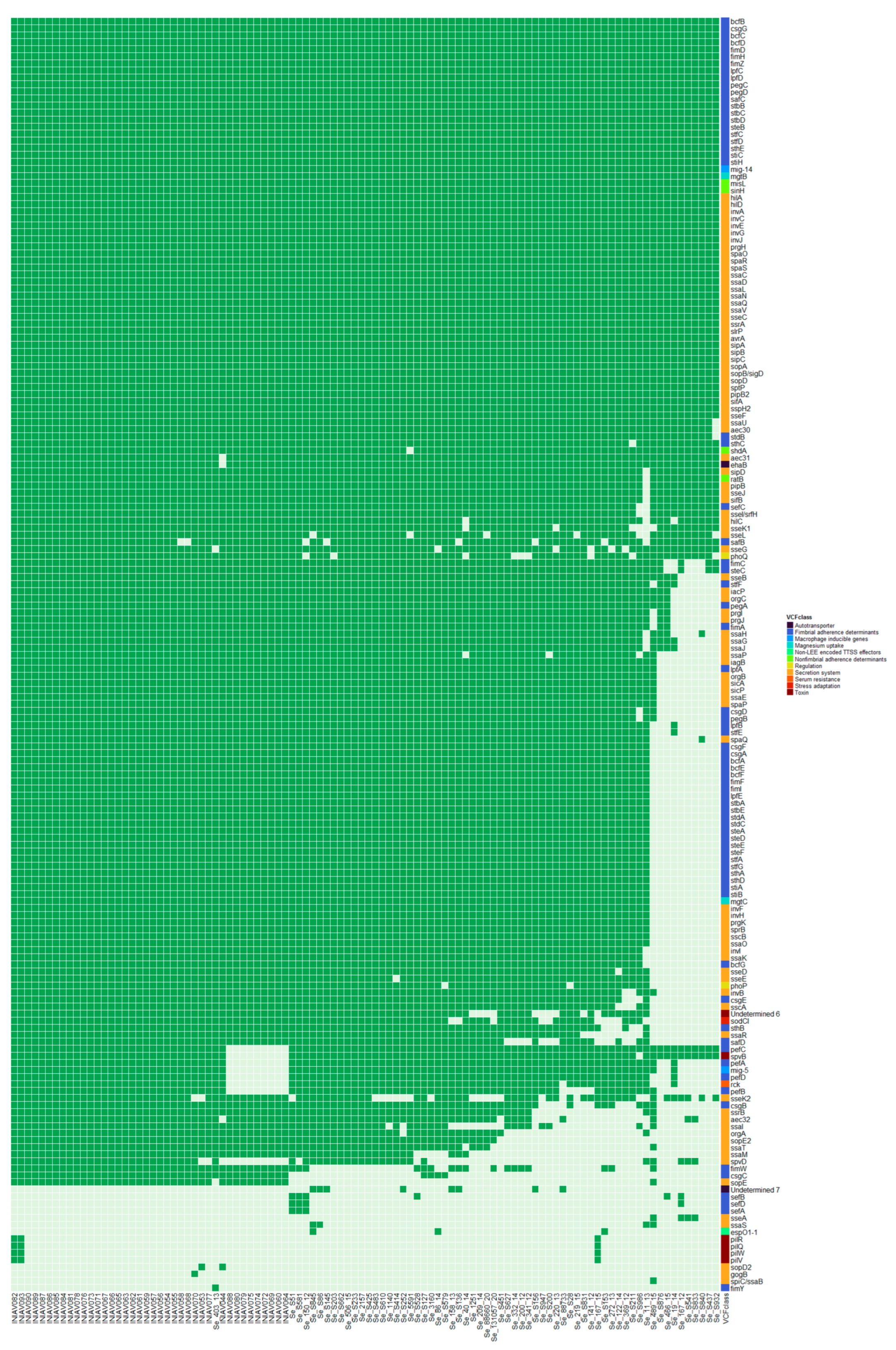
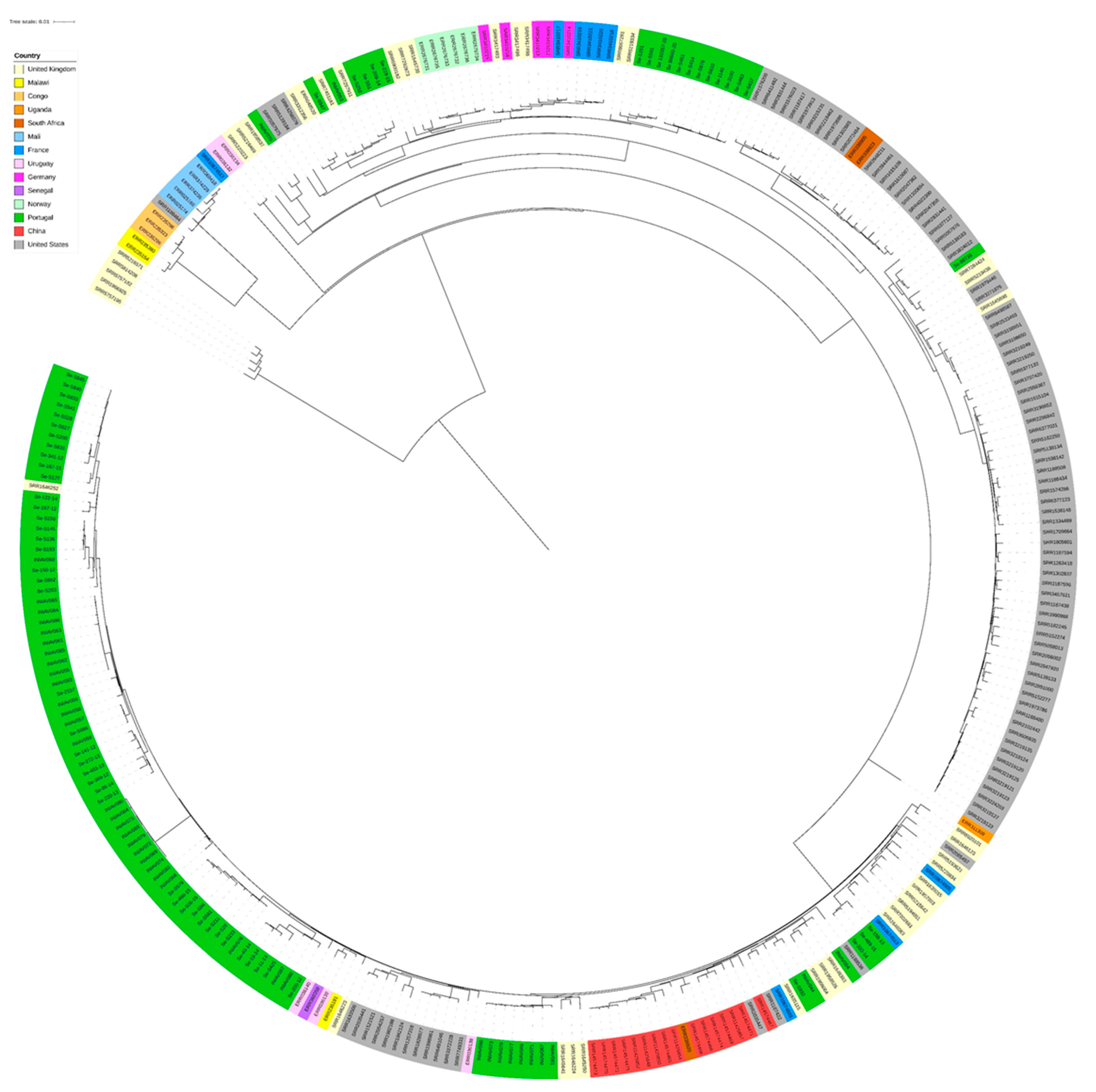
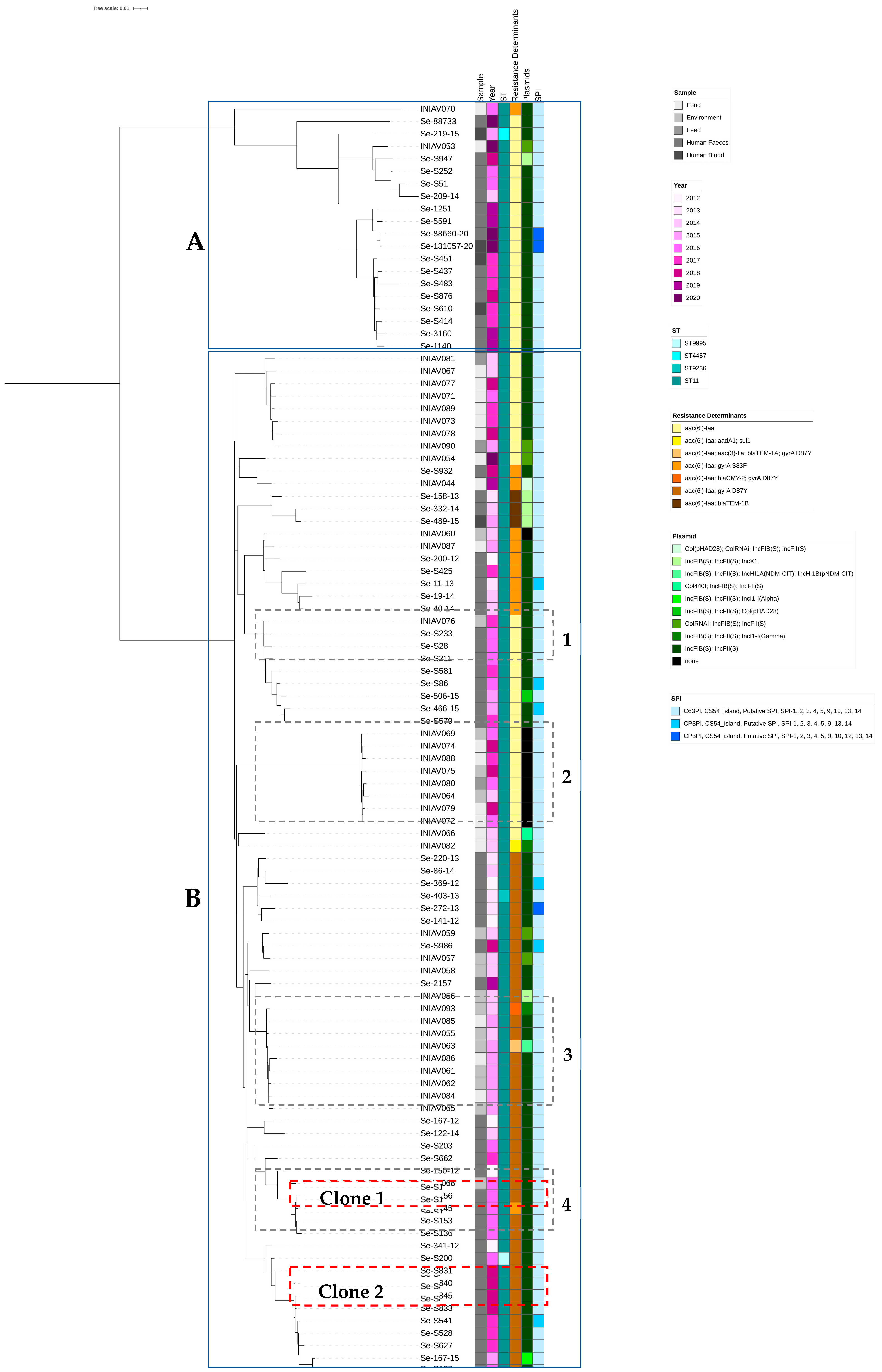
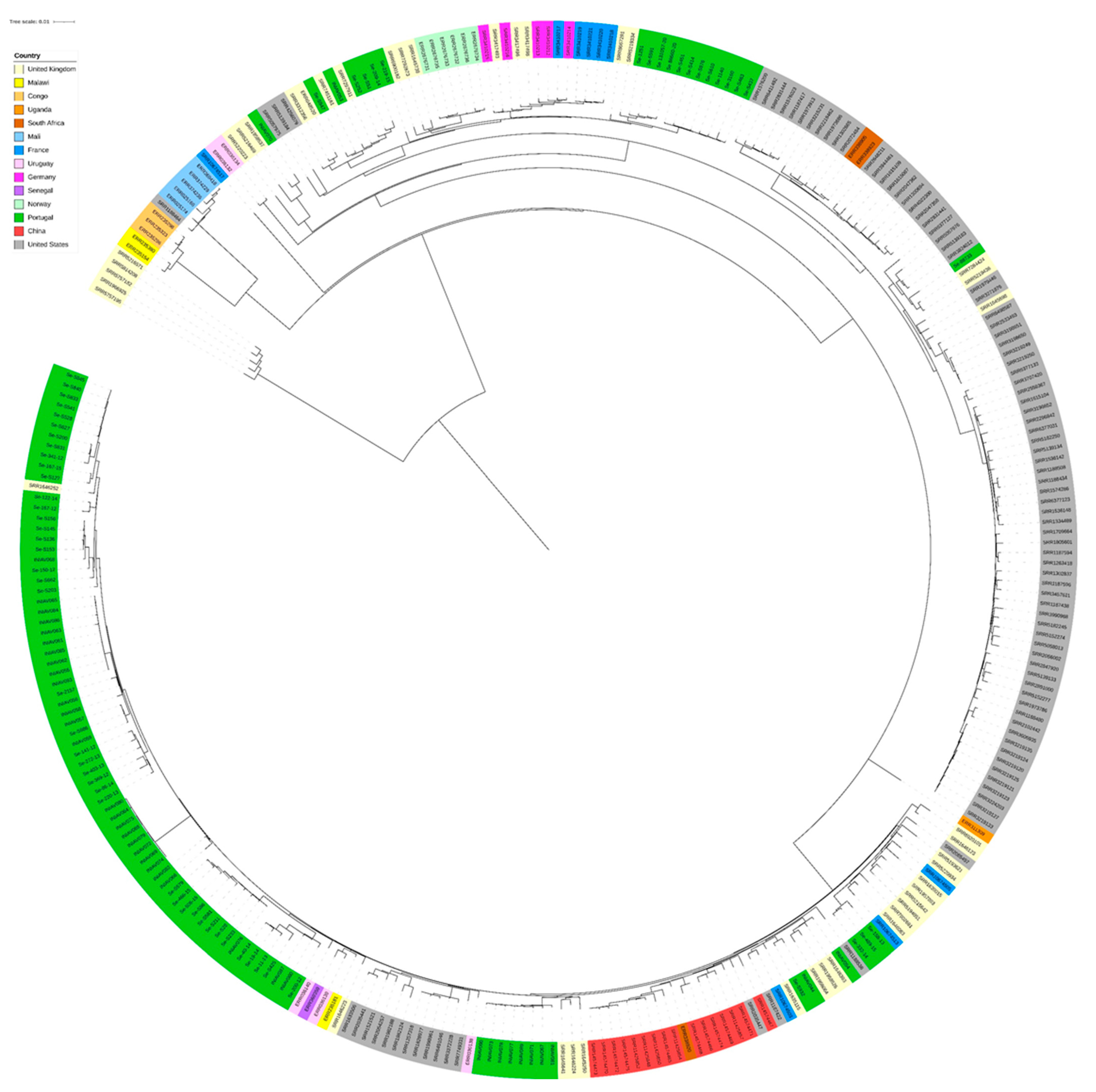
3.2. Genome Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Centre for Disease Prevention and Control. Salmonellosis: Annual Epidemiological Report for 2021; European Centre for Disease Prevention and Control: Solna, Sweden, 2022. [Google Scholar]
- European Food Safety Authority; European Centre for Disease Prevention and Control. The European Union One Health 2021 Zoonoses Report. Eur. Food Saf. Auth. J. 2022, 20, e07666. [Google Scholar] [CrossRef]
- Chanamé Pinedo, L.; Mughini-Gras, L.; Franz, E.; Hald, T.; Pires, S.M. Sources and Trends of Human Salmonellosis in Europe, 2015–2019: An Analysis of Outbreak Data. Int. J. Food Microbiol. 2022, 379, 2015–2019. [Google Scholar] [CrossRef]
- Mughini-Gras, L.; Chanamé Pinedo, L.; Pijnacker, R.; Van Den Beld, M.; Wit, B.; Veldman, K.; Bosh, T.; Franz, E. Impact of the COVID-19 Pandemic on Human Salmonellosis in the Netherlands. Epidemiol. Infect. 2021, 149, 1–5. [Google Scholar] [CrossRef]
- Uelze, L.; Becker, N.; Borowiak, M.; Busch, U.; Dangel, A.; Deneke, C.; Fischer, J.; Flieger, A.; Hepner, S.; Huber, I.; et al. Toward an Integrated Genome-Based Surveillance of Salmonella enterica in Germany. Front. Microbiol. 2021, 12, 626941. [Google Scholar] [CrossRef]
- Ashton, P.M.; Nair, S.; Peters, T.M.; Bale, J.A.; Powell, D.G.; Painset, A.; Tewolde, R.; Schaefer, U.; Jenkins, C.; Dallman, T.J.; et al. Identification of Salmonella for Public Health Surveillance Using Whole Genome Sequencing. PeerJ 2016, 4, e1752. [Google Scholar] [CrossRef] [PubMed]
- Alikhan, N.-F.F.; Zhou, Z.; Sergeant, M.J.; Achtman, M. A Genomic Overview of the Population Structure of Salmonella. PLoS Genet. 2018, 14, e1007261. [Google Scholar] [CrossRef]
- Grimont, P.A.D.; Weill, F.-X. WHO Collaborating Centre for Reference and Research on Salmonella. In Antigenic Formulae of The Salmonella Serovars, 9th ed.; Grimont, P.A.D., Weill, F.X., Eds.; Institut Pasteur: Paris, France, 2007; pp. 1–166. [Google Scholar]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters, Version 7.1; 2017. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_7.1_Breakpoint_Tables.pdf (accessed on 30 January 2017).
- CLSI Clinical and Laboratory Standards Institute. M07 Methods Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approval CDM-A; CLSI Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing Breakpoint Tables for Interpretation of MICs and Zone Diameters Version 10.0. 2020. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_10.0_Breakpoint_Tables.pdf (accessed on 30 January 2020).
- Agersø, Y.; Torpdahl, M.; Zachariasen, C.; Seyfarth, A.; Hammerum, A.M.; Nielsen, E.M. Tentative Colistin Epidemiological Cut-off Value for Salmonella spp. Foodborne Pathog. Dis. 2012, 9, 367–369. [Google Scholar] [CrossRef]
- Zhou, Z.; Alikhan, N.-F.; Mohamed, K.; Fan, Y.; Achtman, M. The EnteroBase User’s Guide, with Case Studies on Salmonella Transmissions, Yersinia pestis Phylogeny, and Escherichia Core Genomic Diversity. Genome Res. 2020, 30, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Achtman, M.; Zhou, Z.; Alikhan, N.F.; Tyne, W.; Parkhill, J.; Cormican, M.; Chiou, C.S.; Torpdahl, M.; Litrup, E.; Prendergast, D.M.; et al. Genomic Diversity of Salmonella enterica—The UoWUCC 10K Genomes Project. Wellcome Open Res. 2021, 5, 223. [Google Scholar] [CrossRef] [PubMed]
- Leão, C.; Clemente, L.; Cara d’Anjo, M.; Albuquerque, T.; Amaro, A. Emergence of Cfr-Mediated Linezolid Resistance among Livestock-Associated Methicillin-Resistant Staphylococcus aureus (LA-MRSA) from Healthy Pigs in Portugal. Antibiotics 2022, 11, 1439. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A Novel Web Tool for WGS-Based Detection of Antimicrobial Resistance Associated with Chromosomal Point Mutations in Bacterial Pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Clausen, P.T.L.C.; Aarestrup, F.M.; Lund, O. Rapid and Precise Alignment of Raw Reads against Redundant Databases with KMA. BMC Bioinform. 2018, 19, 307. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Voldby Larsen, M.; Lund, O.; Villa, L.; Møller Aarestrup, F.; Hasman, H. In Silico Detection and Typing of Plasmids Using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Ponten, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yin, Y.; Jones, M.B.; Zhang, Z.; Deatherage Kaiser, B.L.; Dinsmore, B.A.; Fitzgerald, C.; Fields, P.I.; Deng, X.; Kaiser, B.L.D.; et al. Salmonella Serotype Determination Utilizing High-Throughput Genome Sequencing Data. J. Clin. Microbiol. 2015, 53, 1685–1692. [Google Scholar] [CrossRef]
- Roer, L.; Hendriksen, R.S.; Leekitcharoenphon, P.; Lukjancenko, O.; Kaas, R.S.; Hasman, H.; Aarestrup, F.M. Is the Evolution of Salmonella enterica subsp. Enterica Linked to Restriction-Modification Systems? mSystems 2016, 1, e00009-16. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A General Classification Scheme for Bacterial Virulence Factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Gu, Z. Complex Heatmap Visualization. iMeta 2022, 1, e43. [Google Scholar] [CrossRef]
- Cao, G.; Zhao, S.; Kuang, D.; Hsu, C.H.; Yin, L.; Luo, Y.; Chen, Z.; Xu, X.; Strain, E.; McDermott, P.; et al. Geography Shapes the Genomics and Antimicrobial Resistance of Salmonella enterica Serovar Enteritidis Isolated from Humans. Sci. Rep. 2023, 13, 1331. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ciccarelli, F.D.; Doerks, T.; Von Mering, C.; Creevey, C.J.; Snel, B.; Bork, P. Toward Automatic Reconstruction of a Highly Resolved Tree of Life. Science 2006, 311, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Payne, M.; Kaur, S.; Hu, D.; Cheney, L.; Octavia, S.; Wang, Q.; Tanaka, M.M.; Sintchenko, V.; Lan, R. Elucidation of Global and National Genomic Epidemiology of Salmonella enterica Serovar Enteritidis through Multilevel Genome Typing. Microb. Genom. 2021, 7, 000605. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority) and ECDC (European Centre for Disease Prevention and Control). The European Union Summary Report on Antimicrobial Resistance in Zoonotic and Indicator Bacteria from Humans, Animals and Food in 2020/2021. Eur. Food Saf. Auth. J. 2023, 21, e07867. [Google Scholar] [CrossRef]
- Clemente, L.; Manageiro, V.; Jones-Dias, D.; Correia, I.; Themudo, P.; Albuquerque, T.; Geraldes, M.; Matos, F.; Almendra, C.; Ferreira, E.; et al. Antimicrobial Susceptibility and Oxymino-β-Lactam Resistance Mechanisms in Salmonella enterica and Escherichia coli Isolates from Different Animal Sources. Res. Microbiol. 2015, 166, 574–583. [Google Scholar] [CrossRef]
- Clemente, L.; Correia, I.; Themudo, P.; Neto, I.; Caniça, M.; Bernardo, F. Antimicrobial Susceptibility of Salmonella enterica Isolates from Healthy Breeder and Broiler Flocks in Portugal. Vet. J. 2014, 200, 276–281. [Google Scholar] [CrossRef]
- Clemente, L.; Manageiro, V.; Ferreira, E.; Jones-Dias, D.; Correia, I.; Themudo, P.; Albuquerque, T.; Caniça, M. Occurrence of Extended-Spectrum β-Lactamases among Isolates of Salmonella enterica subsp. Enterica from Food-Producing Animals and Food Products, in Portugal. Int. J. Food Microbiol. 2013, 167, 221–228. [Google Scholar] [CrossRef]
- European Medicines Agency. European Medicines Agency. European Surveillance of Veterinary Antimicrobial Consumption. In Sales of Veterinary Antimicrobial Agents in 31 European Countries in 2021; European Medicines Agency: Amsterdam, The Netherlands, 2022; ISSN 2315-1455. [Google Scholar]
- Koutsoumanis, K.; Allende, A.; Alvarez-Ordóñez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; De Cesare, A.; Herman, L.; Hilbert, F.; Lindqvist, R.; et al. Salmonella Control in Poultry Flocks and Its Public Health Impact. Eur. Food Saf. Auth. J. 2019, 17, e05596. [Google Scholar] [CrossRef]
- Schmerold, I.; van Geijlswijk, I.; Gehring, R. European Regulations on the Use of Antibiotics in Veterinary Medicine. Eur. J. Pharm. Sci. 2023, 189, 106473. [Google Scholar] [CrossRef]
- Clemente, L.; Manageiro, V.; Correia, I.; Amaro, A.; Albuquerque, T.; Themudo, P.; Ferreira, E.; Caniça, M. Revealing Mcr-1-Positive ESBL-Producing Escherichia coli Strains among Enterobacteriaceae from Food-Producing Animals (Bovine, Swine and Poultry) and Meat (Bovine and Swine), Portugal, 2010–2015. Int. J. Food Microbiol. 2019, 296, 37–42. [Google Scholar] [CrossRef]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial Activity, Susceptibility Testing, and Resistance Mechanisms Encoded by Plasmids or Chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef]
- Ricci, V.; Zhang, D.; Teale, C.; Piddock, L.J.V. The O-Antigen Epitope Governs Susceptibility to Colistin in Salmonella enterica. MBio 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Silveira, L.; Nunes, A.; Pista, A.; Isidro, J.; Belo Correia, C.; Saraiva, M.; Batista, R.; Castanheira, I.; MacHado, J.; Gomes, J.P. Characterization of Multidrug-Resistant Isolates of Salmonella enterica Serovars Heidelberg and Minnesota from Fresh Poultry Meat Imported to Portugal. Microb. Drug Resist. 2021, 27, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Magnet, S.; Courvalin, P.; Lambert, T. Activation of the Cryptic Aac(6′)-Iy Aminoglycoside Resistance Gene of Salmonella by a Chromosomal Deletion Generating a Transcriptional Fusion. J. Bacteriol. 1999, 181, 6650–6655. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, R.R.; Dissel, S.; Rapallini, M.L.; van Der Weijden, C.C.; Wit, B.; Heymans, R. Characterization and Whole Genome Sequencing of Closely Related Multidrug-Resistant Salmonella enterica Serovar Heidelberg Isolates from Imported Poultry Meat In The Netherlands. PLoS ONE 2019, 14, e0219795. [Google Scholar] [CrossRef]
- Abdelhamid, A.G.; Yousef, A.E. Egg-Associated Salmonella enterica Serovar Enteritidis: Comparative Genomics Unveils Phylogenetic Links, Virulence Potential, and Antimicrobial Resistance Traits. Front. Microbiol. 2023, 14, 1278821. [Google Scholar] [CrossRef] [PubMed]
- Feasey, N.A.; Hadfield, J.; Keddy, K.H.; Dallman, T.J.; Jacobs, J.; Deng, X.; Wigley, P.; Barquist, L.; Langridge, G.C.; Feltwell, T.; et al. Distinct Salmonella Enteritidis Lineages Associated with Enterocolitis in High-Income Settings and Invasive Disease in Low-Income Settings. Nat. Genet. 2016, 48, 1211–1217. [Google Scholar] [CrossRef]
- Shen, X.; Yin, L.; Zhang, A.; Zhao, R.; Yin, D.; Wang, J.; Dai, Y.; Hou, H.; Pan, X.; Hu, X.; et al. Prevalence and Characterization of Salmonella Isolated from Chickens in Anhui, China. Pathogens 2023, 12, 465. [Google Scholar] [CrossRef] [PubMed]
- Ksibi, B.; Ktari, S.; Othman, H.; Ghedira, K.; Maalej, S.; Mnif, B.; Abbassi, M.s.; Fabre, L.; Rhimi, F.; Le Hello, S.; et al. Comparison of Conventional Molecular and Whole-Genome Sequencing Methods for Subtyping Salmonella enterica Serovar Enteritidis Strains from Tunisia. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Fandiño, L.C.; García, N.V. A Common Salmonella Enteritidis Sequence Type from Poultry and Human Gastroenteritis in Ibagué, Colombia. Biomedica 2019, 39, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Marcus, S.L.; Brumell, J.H.; Pfeifer, C.G.; Finlay, B.B. Salmonella Pathogenicity Islands: Big Virulence in Small Packages. Microbes Infect. 2000, 2, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Kombade, S.; Kaur, N. Pathogenicity Island in Salmonella. In Salmonella spp.—A Global Challenge; IntechOpen: London, UK, 2021; Volume 11, p. 13. ISBN 0000957720. [Google Scholar]
- Nikiema, M.E.M.; Kakou-ngazoa, S.; Ky/Ba, A.; Sylla, A.; Bako, E.; Addablah, A.Y.A.; Ouoba, J.B.; Sampo, E.; Gnada, K.; Zongo, O.; et al. Characterization of Virulence Factors of Salmonella Isolated from Human Stools and Street Food in Urban Areas of Burkina Faso. BMC Microbiol. 2021, 21, 338. [Google Scholar] [CrossRef] [PubMed]
- Diab, M.S.; Thabet, A.S.; Elsalam, M.A.; Ewida, R.M.; Sotohy, S.A. Detection of Virulence and β-Lactamase Resistance Genes of Non-Typhoidal Salmonella Isolates from Human and Animal Origin in Egypt “One Health Concern”. Gut Pathog. 2023, 15, 16. [Google Scholar] [CrossRef]
- Cao, G.; Meng, J.; Strain, E.; Stones, R.; Pettengill, J.; Zhao, S.; McDermott, P.; Brown, E.; Allard, M. Phylogenetics and Differentiation of Salmonella Newport Lineages by Whole Genome Sequencing. PLoS ONE 2013, 8, e55687. [Google Scholar] [CrossRef]
- Lozano-Villegas, K.J.; Herrera-Sánchez, M.P.; Beltrán-Martínez, M.A.; Cárdenas-Moscoso, S.; Rondón-Barragán, I.S. Molecular Detection of Virulence Factors in Salmonella Serovars Isolated from Poultry and Human Samples. Vet. Med. Int. 2023, 2023, 1875253. [Google Scholar] [CrossRef]
- Romero-Calle, D.X.; Pedrosa-Silva, F.; Tomé, L.M.R.; Sousa, T.J.; de Oliveira Santos, L.T.S.; de Carvalho Azevedo, V.A.; Brenig, B.; Benevides, R.G.; Venancio, T.M.; Billington, C.; et al. Hybrid Genomic Analysis of Salmonella enterica Serovar Enteritidis SE3 Isolated from Polluted Soil in Brazil. Microorganisms 2023, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Andesfha, E.; Indrawati, A.; Mayasari, N.L.P.I.; Rahayuningtyas, I.; Jusa, I. Detection of Salmonella Pathogenicity Island and Salmonella Plasmid Virulence Genes in Salmonella Enteritidis Originated from Layer and Broiler Farms in Java Island. J. Adv. Vet. Anim. Res. 2019, 6, 384–393. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leão, C.; Silveira, L.; Usié, A.; Gião, J.; Clemente, L.; Themudo, P.; Amaro, A.; Pista, A. Genetic Diversity of Salmonella enterica subsp. enterica Serovar Enteritidis from Human and Non-Human Sources in Portugal. Pathogens 2024, 13, 112. https://doi.org/10.3390/pathogens13020112
Leão C, Silveira L, Usié A, Gião J, Clemente L, Themudo P, Amaro A, Pista A. Genetic Diversity of Salmonella enterica subsp. enterica Serovar Enteritidis from Human and Non-Human Sources in Portugal. Pathogens. 2024; 13(2):112. https://doi.org/10.3390/pathogens13020112
Chicago/Turabian StyleLeão, Célia, Leonor Silveira, Ana Usié, Joana Gião, Lurdes Clemente, Patricia Themudo, Ana Amaro, and Angela Pista. 2024. "Genetic Diversity of Salmonella enterica subsp. enterica Serovar Enteritidis from Human and Non-Human Sources in Portugal" Pathogens 13, no. 2: 112. https://doi.org/10.3390/pathogens13020112
APA StyleLeão, C., Silveira, L., Usié, A., Gião, J., Clemente, L., Themudo, P., Amaro, A., & Pista, A. (2024). Genetic Diversity of Salmonella enterica subsp. enterica Serovar Enteritidis from Human and Non-Human Sources in Portugal. Pathogens, 13(2), 112. https://doi.org/10.3390/pathogens13020112