
Phylodynamic and Evolution of the Hemagglutinin (HA) and Neuraminidase (NA) Genes of Influenza A(H1N1) pdm09 Viruses Circulating in the 2009 and 2023 Seasons in Italy

 ,
, 
 ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
3. Results
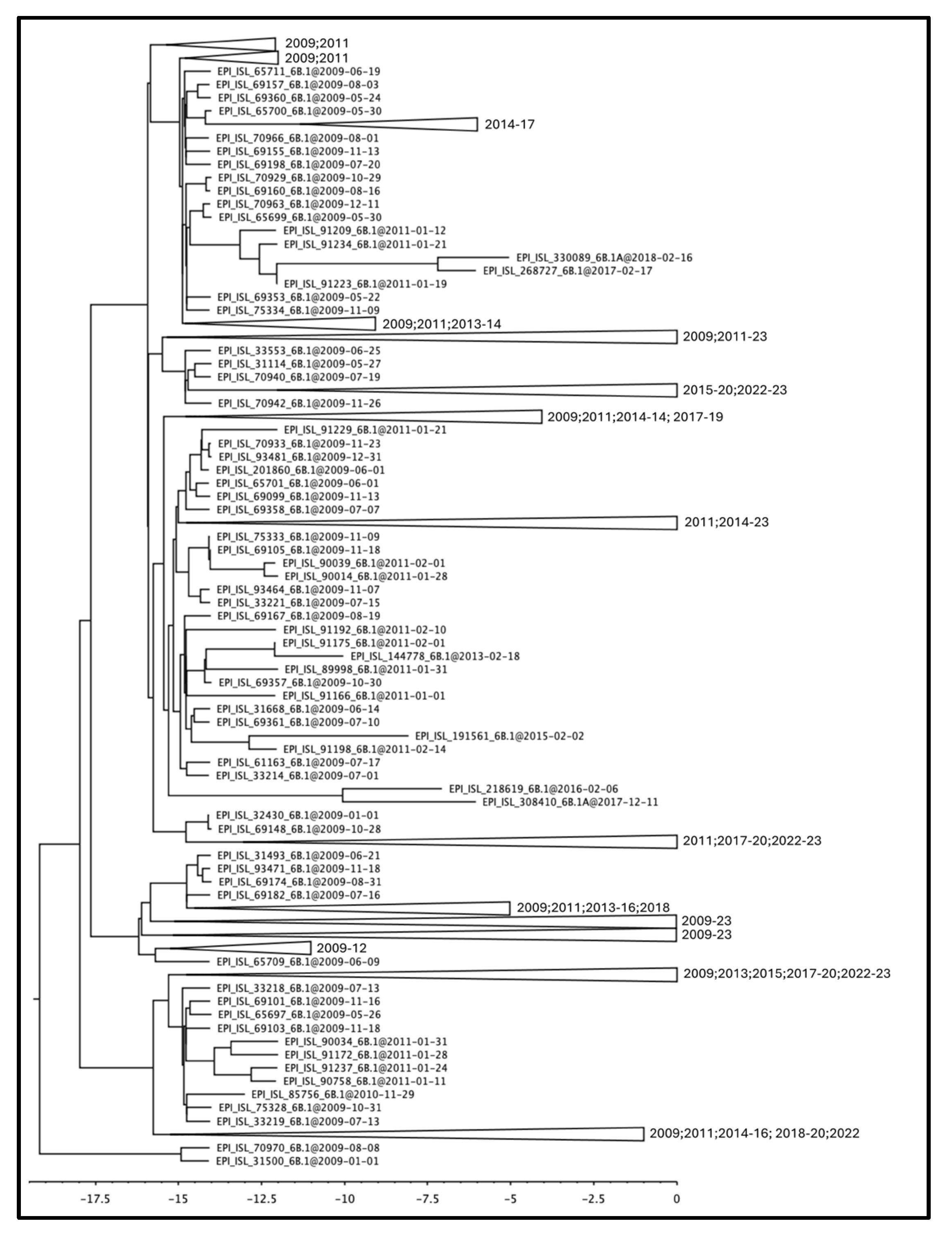
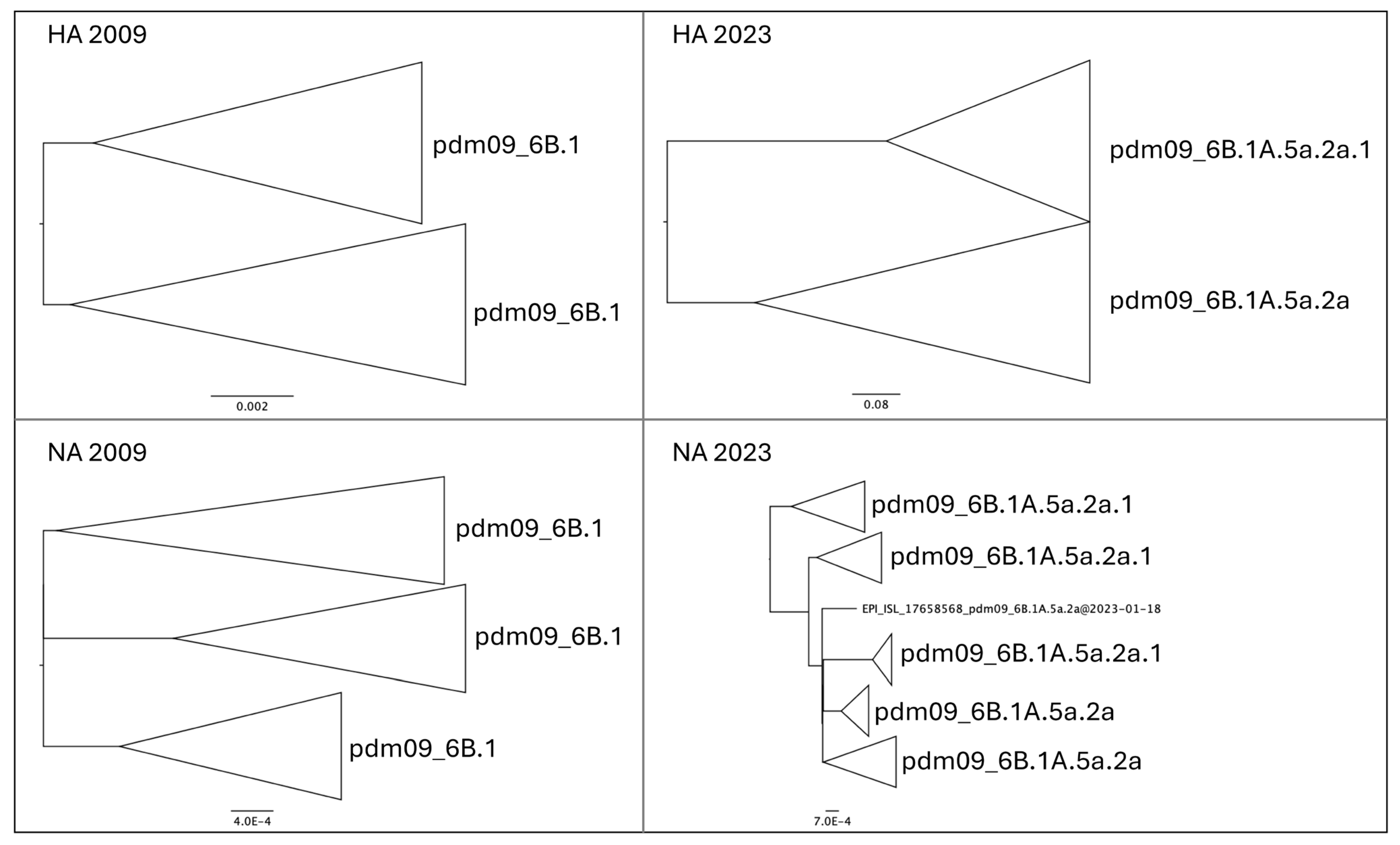
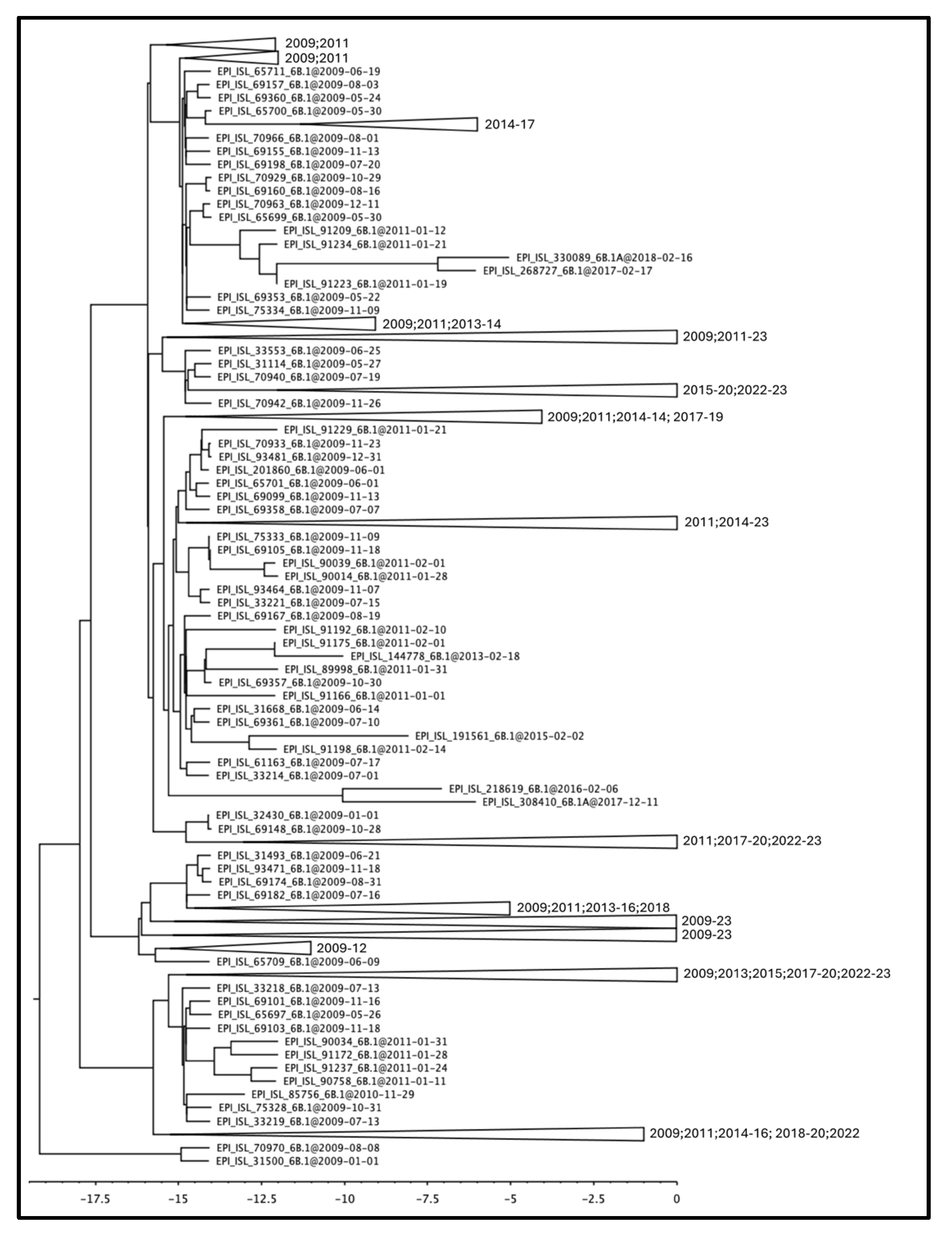
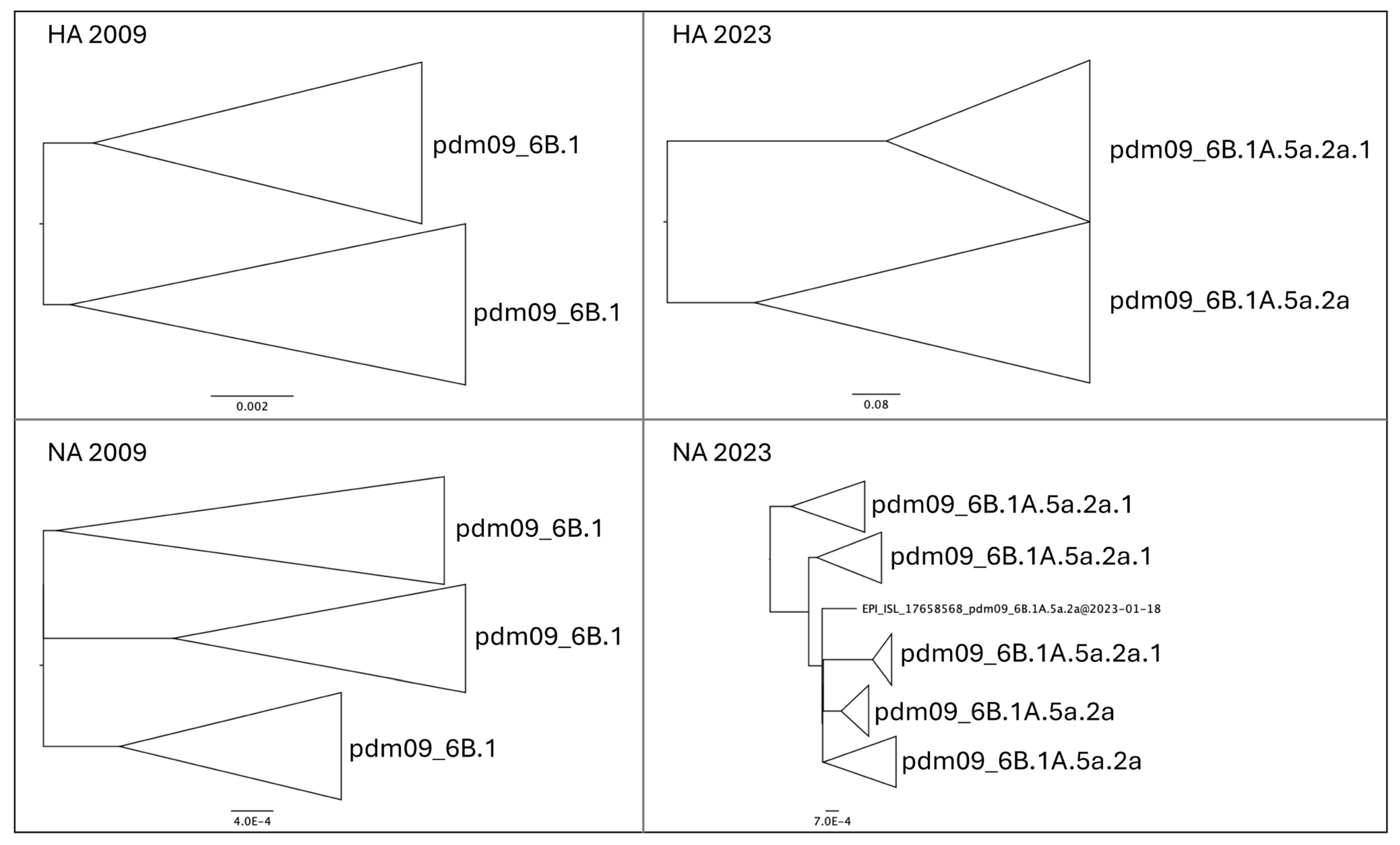
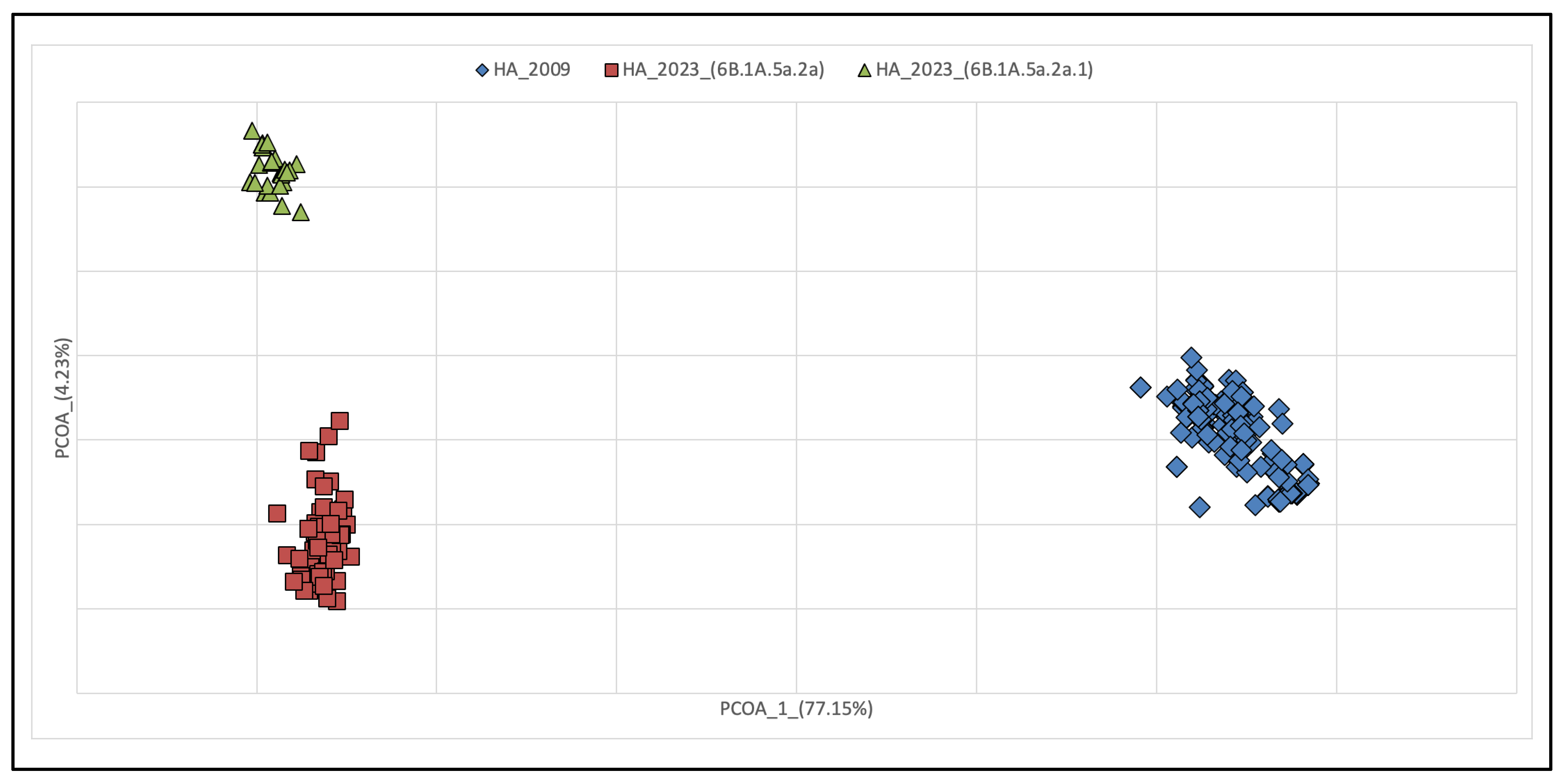
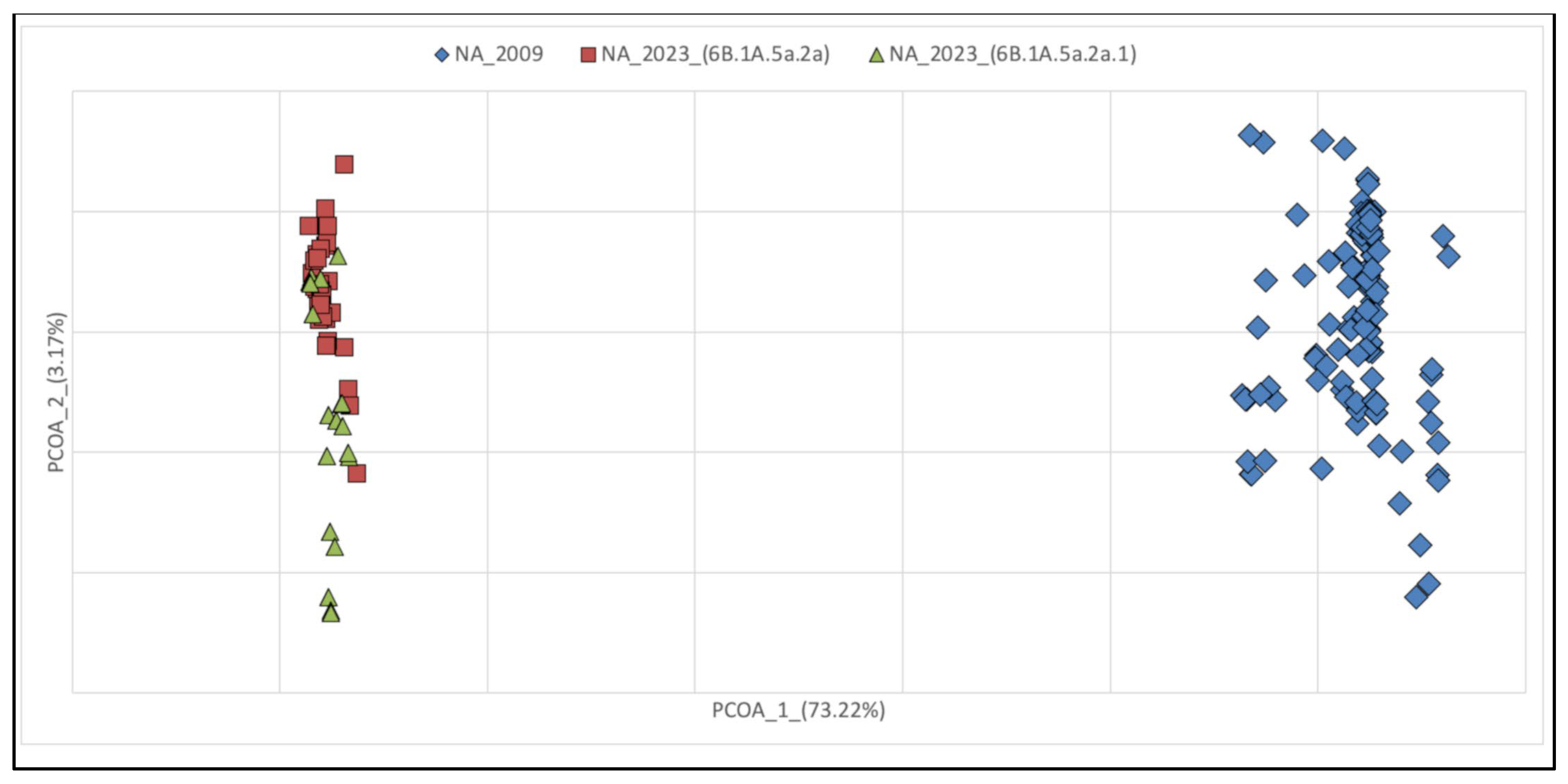
3.1. Phylogenetic Reconstruction of Global and Italian A(H1N1) pdm09 Epidemics
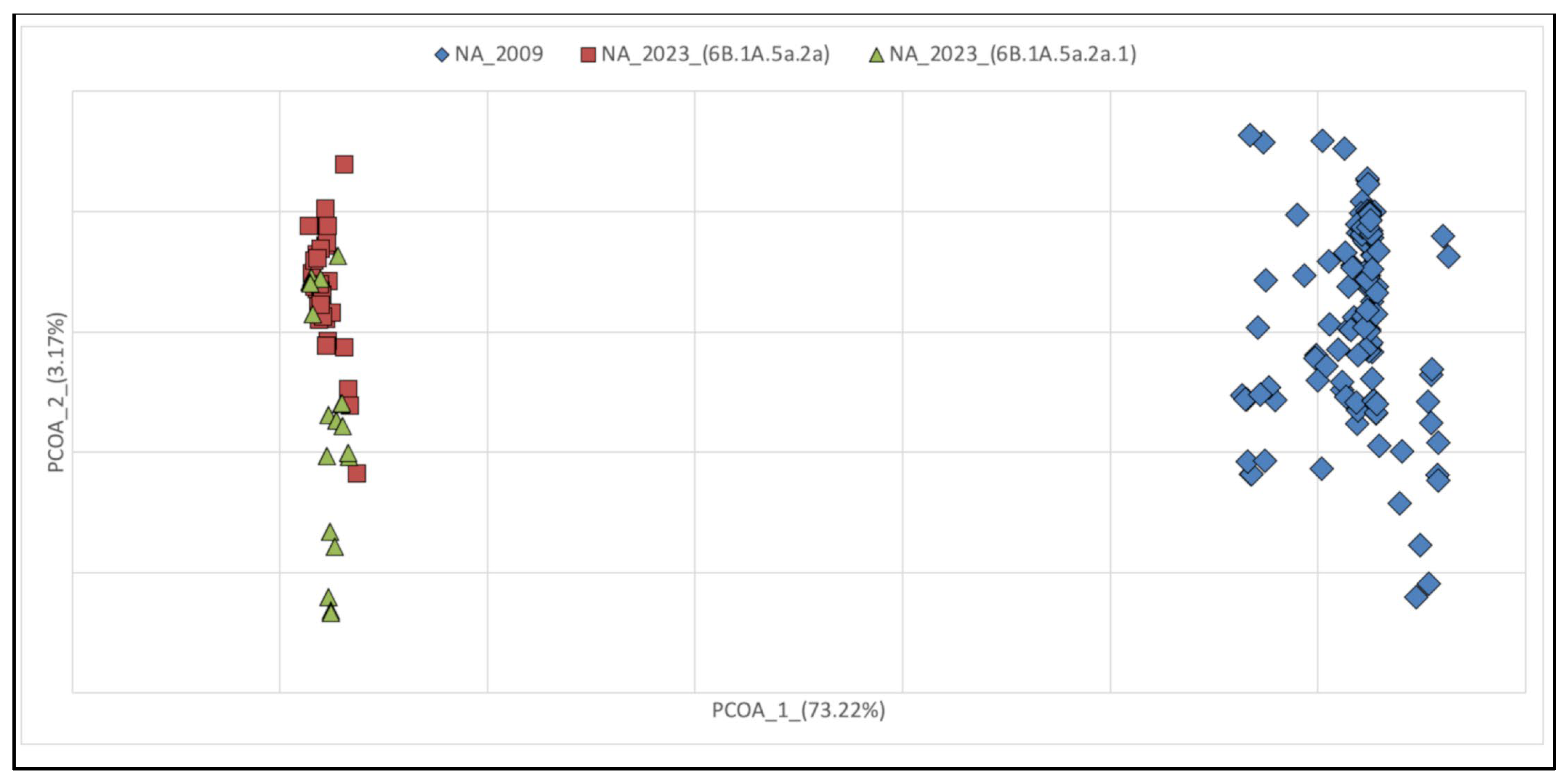
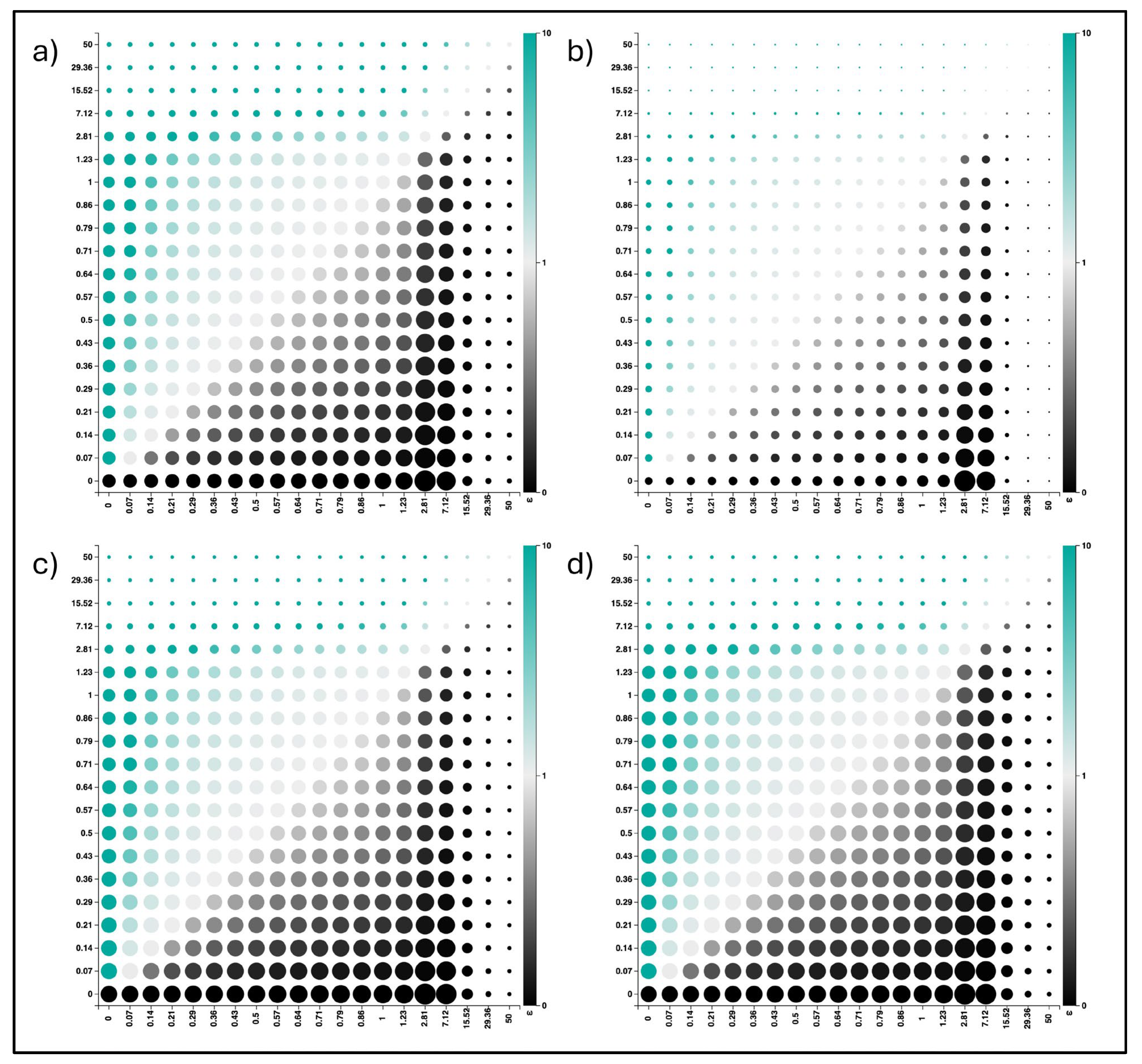
3.2. Genetic Variations of A(H1N1) pdm09 Virus Isolates from 2009 and 2023
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petrova, V.N.; Russell, C.A. The evolution of seasonal influenza viruses. Nat. Rev. Microbiol. 2018, 16, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–959. [Google Scholar] [CrossRef] [PubMed]
- WHO. Pandemic Influenza. Available online: https://www.who.int/emergencies/situations/influenza-a-(h1n1)-outbreak (accessed on 1 February 2024).
- Zehender, G.; Pariani, E.; Piralla, A.; Lai, A.; Gabanelli, E.; Ranghiero, A.; Ebranati, E.; Amendola, A.; Campanini, G.; Rovida, F.; et al. Reconstruction of the Evolutionary Dynamics of the A(H1N1)pdm09 Influenza Virus in Italy during the Pandemic and Post-Pandemic Phases. PLoS ONE 2012, 7, e47517. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Gramer, M.R.; Vincent, A.L.; Holmes, E.C. Global transmission of influenza viruses from humans to swine. J. Gen. Virol. 2012, 93, 2195–2203. [Google Scholar] [CrossRef]
- Vincent, A.; Awada, L.; Brown, I.; Chen, H.; Claes, F.; Dauphin, G.; Donis, R.; Culhane, M.; Hamilton, K.; Lewis, N.; et al. Review of influenza a virus in swine worldwide: A call for increased surveillance and research. Zoonoses Public Health 2013, 61, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Ciacci-Zanella, J.R.; Schaefer, R.; Gava, D.; Haach, V.; Cantão, M.E.; Coldebella, A. Influenza a virus infection in Brazilian swine herds following the introduction of pandemic 2009 H1N1. Vet. Microbiol. 2015, 180, 118–122. [Google Scholar] [CrossRef]
- Chastagner, A.; Hervé, S.; Bonin, E.; Quéguiner, S.; Hirchaud, E.; Henritzi, D.; Béven, V.; Gorin, S.; Barbier, N.; Blanchard, Y.; et al. Spatiotemporal distribution and evolution of the a/H1N1 2009 pandemic influenza virus in pigs in France from 2009 to 2017: Identification of a potential swine-specific lineage. J. Virol. 2018, 92, e00988-18. [Google Scholar] [CrossRef] [PubMed]
- Rajão, D.S.; Walia, R.R.; Campbell, B.; Gauger, P.C.; Janas-Martindale, A.; Killian, M.L.; Vincent, A.L. Reassortment between swine H3N2 and 2009 pandemic H1N1 in the United States resulted in influenza a viruses with diverse genetic constellations with variable virulence in pigs. J. Virol. 2017, 91, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Russell, C.A.; Langat, P.; Anderson, T.K.; Berger, K.; Bielejec, F.; Burke, D.F.; Dudas, G.; Fonville, J.M.; Fouchier, R.A.; et al. The global antigenic diversity of swine influenza A viruses. eLife 2016, 5, e12217. [Google Scholar] [CrossRef]
- Radovanov, J.; Ristic, M.; Medic, S.; Kovacevic, G.; Dopud, N.; Nikolic, N.; Patic, A.; Cvjetkovic, I.H.; Petrovic, V. Genetic variability of the neuraminidase gene of influenza A(H1N1)pdm09 viruses circulating from the 2012/2013 to 2017/2018 season in Vojvodina Province, Serbia. Mol. Cell. Probes 2020, 52, 101557. [Google Scholar] [CrossRef]
- Junqueira, D.M.; Tochetto, C.; Anderson, T.K.; Gava, D.; Haach, V.; Cantão, M.E.; Baker, A.L.V.; Schaefer, R. Human-to-swine introductions and onward transmission of 2009 H1N1 pandemic influenza viruses in Brazil. Front. Microbiol. 2023, 14, 1243567. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, Y.; Liu, H.; Pang, Z.; Cui, X.; Zhao, R.; Liu, Y.; Qu, X.; Huang, M.; Ke, C.; et al. The genetic diversity, replication, and transmission of 2009 pandemic H1N1 viruses in China. Front. Microbiol. 2023, 14, 1110100. [Google Scholar] [CrossRef] [PubMed]
- Kolosova, N.P.; Boldyrev, N.D.; Svyatchenko, S.V.; Danilenko, A.V.; Goncharova, N.I.; Shadrinova, K.N.; Danilenko, E.I.; Onkhonova, G.S.; Kosenko, M.N.; Antonets, M.E.; et al. An Investigation of Severe Influenza Cases in Russia during the 2022-2023 Epidemic Season and an Analysis of HA-D222G/N Polymorphism in Newly Emerged and Dominant Clade 6B.1A.5a.2a A(H1N1)pdm09 Viruses. Pathogens 2023, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Zeller, M.A.; Ma, J.; Wong, F.Y.; Tum, S.; Hidano, A.; Holt, H.; Chhay, T.; Sorn, S.; Koeut, D.; Seng, B.; et al. The genomic landscape of swine influenza A viruses in Southeast Asia. PNAS 2023, 120, e2301926120. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M. UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, e214. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarisation in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Pond, S.L.K.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Sanna, D.; Giovanetti, M.; Pascarella, S.; Casu, M.; Ciccozzi, M. Avian influenza A H5N1: Are we really sure it is a spillover? Pathog. Glob. Health 2023, 117, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin-Yang Proteins Coevolving to Thwart Immunity. Viruses 2019, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Holmes, E.C. The phylogenomics of evolving virus virulence. Nat. Rev. Genet. 2018, 19, 756–769. [Google Scholar] [CrossRef] [PubMed]
- LaTourrette, K.; Garcia-Ruiz, H. Determinants of Virus Variation, Evolution, and Host Adaptation. Pathogens 2022, 11, 1039. [Google Scholar] [CrossRef] [PubMed]
- McCrone, J.T.; Lauring, A.S. Genetic bottlenecks in intraspecies virus transmission. Curr. Opin. Virol. 2018, 28, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Kosik, I.; Ince, W.L.; Gentles, L.E.; Oler, A.J.; Kosikova, M.; Angel, M.; Magadán, J.G.; Xie, H.; Brooke, C.B.; Yewdell, J.W. Influenza A virus hemagglutinin glycosylation compensates for antibody escape fitness costs. PLoS Pathog. 2018, 14, e1006796. [Google Scholar] [CrossRef]
- Bao, D.; Xue, R.; Zhang, M.; Lu, C.; Ma, T.; Ren, C.; Zhang, T.; Yang, J.; Teng, Q.; Li, X.; et al. N-Linked Glycosylation Plays an Important Role in Budding of Neuraminidase Protein and Virulence of Influenza Viruses. J. Virol. 2021, 95, e02042-20. [Google Scholar] [CrossRef] [PubMed]
- Al Khatib, H.A.; Al Thani, A.A.; Yassine, H.M. Evolution and dynamics of the pandemic H1N1 influenza hemagglutinin protein from 2009 to 2017. Arch. Virol. 2018, 163, 3035–3049. [Google Scholar] [CrossRef]
- Wei, C.J.; Boyington, J.C.; Dai, K.; Houser, K.V.; Pearce, M.B.; Kong, W.P.; Yang, Z.Y.; Tumpey, T.M.; Nabel, G.J. Cross-neutralization of 1918 and 2009 influenza viruses: Role of glycans in viral evolution and vaccine design. Sci. Transl. Med. 2010, 2, 24ra21. [Google Scholar] [CrossRef]
- Takahashi, T.; Song, J.; Suzuki, T.; Kawaoka, Y. Mutations in NA that induced low pH-stability and enhanced the replication of pandemic (H1N1) 2009 influenza A virus at an early stage of the pandemic. PLoS ONE 2013, 8, e64439. [Google Scholar] [CrossRef] [PubMed]
- Furuse, Y.; Shimabukuro, K.; Odagiri, T.; Sawayama, R.; Okada, T.; Khandaker, I.; Suzuki, A.; Oshitani, H. Comparison of selection pressures on the HA gene of pandemic (2009) and seasonal human and swine influenza A H1 subtype viruses. Virology 2010, 405, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Joseph, U.; Vijaykrishna, D.; Smith, G.J.D.; Su, Y.C.F. Adaptive evolution during the establishment of European avian-like H1N1 influenza A virus in swine. Evol. Appl. 2017, 11, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, U.; Chakrabarti, A.K.; Kanungo, S.; Dutta, S. Evolutionary dynamics of influenza A/H1N1 virus circulating in India from 2011 to 2021. Infect. Genet. Evol. 2023, 110, 105424. [Google Scholar] [CrossRef]
- Xu, D.; Chen, L.; Ji, L.; Yan, W. Genetic characterization of influenza A(H1N1) pdm09 virus in 2023 in Huzhou, China. J. Clin. Virol. Plus 2024, 4, 100178. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | nt Position | α | β | Prob [α > β] | Prob [α < β] | BF [α < β] | β-α | |
|---|---|---|---|---|---|---|---|---|
| HA_2009 | 177 | 529–531 | 0.895 | 6.484 | 0.025 | 0.945 | 24.681 | 5.589 |
| 41 | 121–123 | 1.077 | 9.591 | 0.030 | 0.942 | 23.428 | 8.514 | |
| HA_2023 | 123 | 367–369 | 0.880 | 6.570 | 0.049 | 0.918 | 19.964 | 5.690 |
| 22 | 64–66 | 0.889 | 5.870 | 0.058 | 0.906 | 17.231 | 4.981 | |
| 513 | 1537–1539 | 0.926 | 5.960 | 0.059 | 0.903 | 16.787 | 5.034 | |
| NA_2009 | 286 | 856–858 | 0.942 | 10.977 | 0.010 | 0.970 | 47.275 | 10.035 |
| 248 | 742–744 | 0.957 | 10.843 | 0.011 | 0.969 | 45.162 | 9.886 | |
| 455 | 1363–1365 | 5.037 | 28.782 | 0.048 | 0.906 | 14.059 | 23.745 | |
| NA_2023 | 339 | 1015–1017 | 1.021 | 13.242 | 0.049 | 0.925 | 18.000 | 12.221 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpa, F.; Sernicola, L.; Farcomeni, S.; Ciccozzi, A.; Sanna, D.; Casu, M.; Vitale, M.; Cicenia, A.; Giovanetti, M.; Romano, C.; et al. Phylodynamic and Evolution of the Hemagglutinin (HA) and Neuraminidase (NA) Genes of Influenza A(H1N1) pdm09 Viruses Circulating in the 2009 and 2023 Seasons in Italy. Pathogens 2024, 13, 334. https://doi.org/10.3390/pathogens13040334
Scarpa F, Sernicola L, Farcomeni S, Ciccozzi A, Sanna D, Casu M, Vitale M, Cicenia A, Giovanetti M, Romano C, et al. Phylodynamic and Evolution of the Hemagglutinin (HA) and Neuraminidase (NA) Genes of Influenza A(H1N1) pdm09 Viruses Circulating in the 2009 and 2023 Seasons in Italy. Pathogens. 2024; 13(4):334. https://doi.org/10.3390/pathogens13040334
Chicago/Turabian StyleScarpa, Fabio, Leonardo Sernicola, Stefania Farcomeni, Alessandra Ciccozzi, Daria Sanna, Marco Casu, Marco Vitale, Alessia Cicenia, Marta Giovanetti, Chiara Romano, and et al. 2024. "Phylodynamic and Evolution of the Hemagglutinin (HA) and Neuraminidase (NA) Genes of Influenza A(H1N1) pdm09 Viruses Circulating in the 2009 and 2023 Seasons in Italy" Pathogens 13, no. 4: 334. https://doi.org/10.3390/pathogens13040334
APA StyleScarpa, F., Sernicola, L., Farcomeni, S., Ciccozzi, A., Sanna, D., Casu, M., Vitale, M., Cicenia, A., Giovanetti, M., Romano, C., Branda, F., Ciccozzi, M., & Borsetti, A. (2024). Phylodynamic and Evolution of the Hemagglutinin (HA) and Neuraminidase (NA) Genes of Influenza A(H1N1) pdm09 Viruses Circulating in the 2009 and 2023 Seasons in Italy. Pathogens, 13(4), 334. https://doi.org/10.3390/pathogens13040334