

Hepatitis Delta Virus and Hepatocellular Carcinoma

{kind=link}
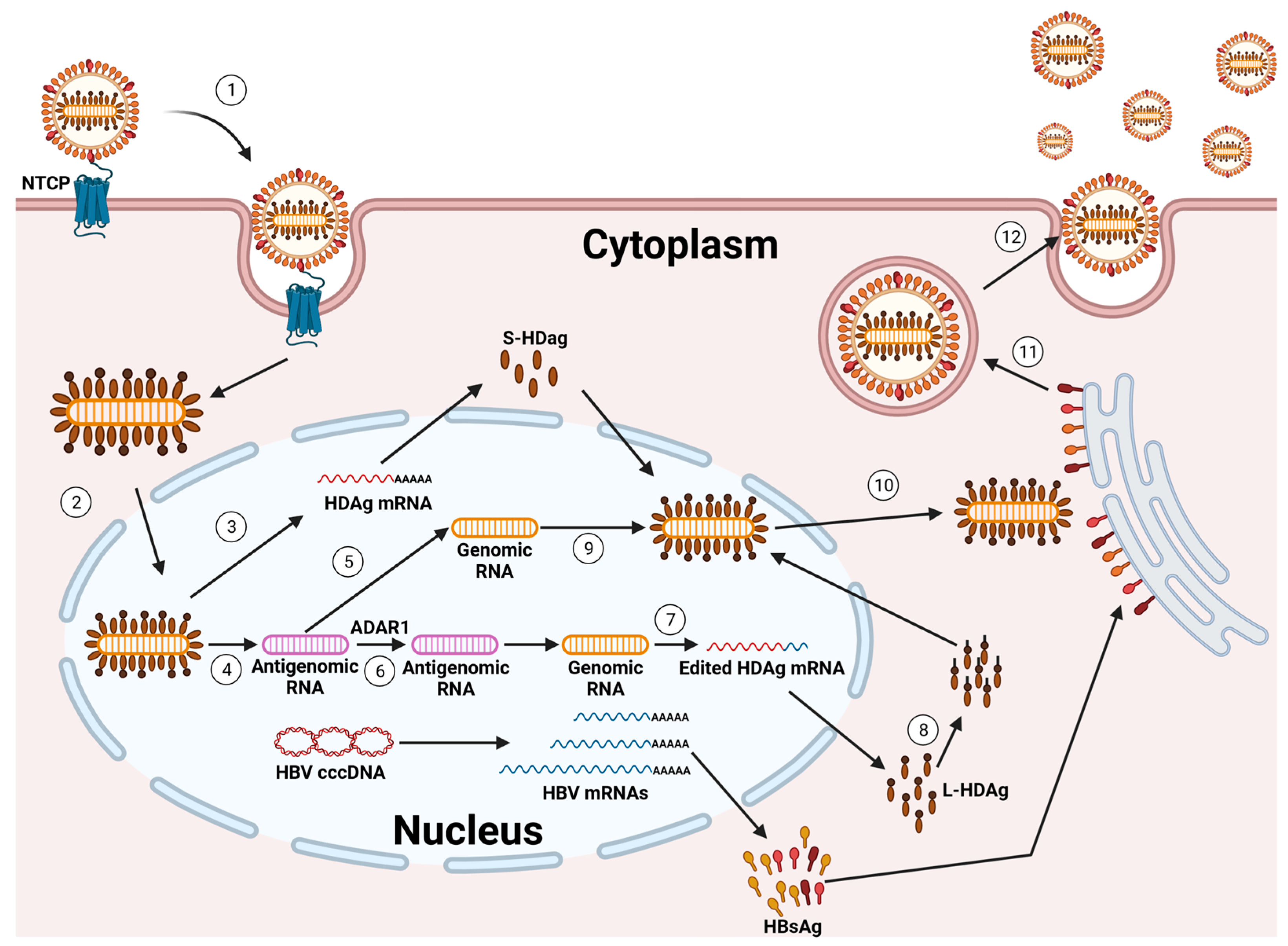
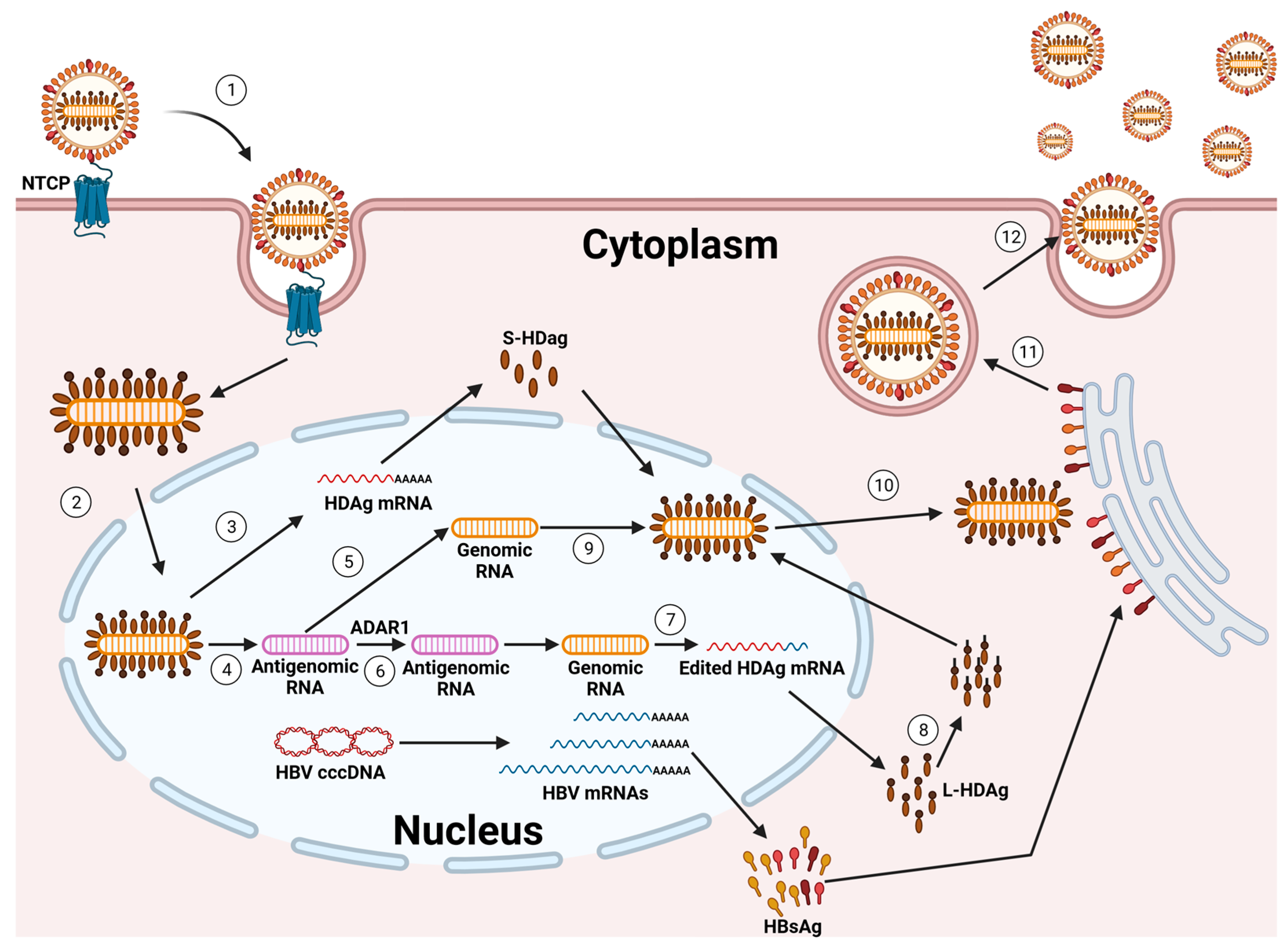{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Virological Aspects
3. Epidemiology
4. Clinical Outcomes of HDV Infection
5. HDV Pathogenesis
Immunological Aspects
6. HDV and HCC Development: Potential Oncogenic Mechanisms
7. Clinical Features of HDV Infection and HCC Development
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brunetto, M.R.; Ricco, G.; Negro, F.; Wedemeyer, H.; Yurdaydin, C.; Asselah, T.; Papatheodoridis, G.; Gheorghe, L.; Agarwal, K.; Farci, P.; et al. EASL Clinical Practice Guidelines on Hepatitis Delta Virus. J. Hepatol. 2023, 79, 433–460. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; Niro, G.A.; Zamboni, F.; Diaz, G. Hepatitis D Virus and Hepatocellular Carcinoma. Viruses 2021, 13, 830. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M. Hepatitis D: Thirty Years After. J. Hepatol. 2009, 50, 1043–1050. [Google Scholar] [CrossRef]
- Sureau, C.; Negro, F. The Hepatitis Delta Virus: Replication and Pathogenesis. J. Hepatol. 2016, 64, S102–S116. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Shen, D.-T.; Ji, D.-Z.; Han, P.-C.; Zhang, W.-M.; Ma, J.-F.; Chen, W.-S.; Goyal, H.; Pan, S.; Xu, H.-G. Prevalence and Burden of Hepatitis D Virus Infection in the Global Population: A Systematic Review and Meta-Analysis. Gut 2019, 68, 512–521. [Google Scholar] [CrossRef]
- Cross, T.J.S.; Rizzi, P.; Horner, M.; Jolly, A.; Hussain, M.J.; Smith, H.M.; Vergani, D.; Harrison, P.M. The Increasing Prevalence of Hepatitis Delta Virus (HDV) Infection in South London. J. Med. Virol. 2008, 80, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Peck, M.; Gacic-Dobo, M.; Diallo, M.S.; Nedelec, Y.; Sodha, S.S.; Wallace, A.S. Global Routine Vaccination Coverage, 2018; CDC: Atlanta, GA, USA, 2019; Volume 68. [Google Scholar]
- World Health Organization. Hepatitis D. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-d (accessed on 4 March 2024).
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; De Martel, C.; Hutin, Y.; Geretti, A.M. The Global Prevalence of Hepatitis D Virus Infection: Systematic Review and Meta-Analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef]
- Miao, Z.; Zhang, S.; Ou, X.; Li, S.; Ma, Z.; Wang, W.; Peppelenbosch, M.P.; Liu, J.; Pan, Q. Estimating the Global Prevalence, Disease Progression, and Clinical Outcome of Hepatitis Delta Virus Infection. J. Infect. Dis. 2020, 221, 1677–1687. [Google Scholar] [CrossRef]
- Giannini, E.G.; Pasta, A.; Pieri, G.; Plaz Torres, M.C.; Marseglia, M.; Pelizzaro, F.; Sangiovanni, A.; Cabibbo, G.; Ghittoni, G.; Di Marco, M.; et al. Characteristics and Outcome of Anti-Hepatitis D Virus Positive Patients with Hepatocellular Carcinoma. Liver Int. 2024, 1–12. [Google Scholar] [CrossRef]
- Alfaiate, D.; Clément, S.; Gomes, D.; Goossens, N.; Negro, F. Chronic Hepatitis D and Hepatocellular Carcinoma: A Systematic Review and Meta-Analysis of Observational Studies. J. Hepatol. 2020, 73, 533–539. [Google Scholar] [CrossRef]
- Rizzetto, M.; Hamid, S. The Medical Impact of Hepatitis D Virus Infection in Asia and Africa; Time for a Reappraisal. Liver Int. 2021, 41, 16–19. [Google Scholar] [CrossRef]
- Da, B.L.; Rahman, F.; Lai, W.C.; Kleiner, D.E.; Heller, T.; Koh, C. Risk Factors for Delta Hepatitis in a North American Cohort: Who Should Be Screened? Am. J. Gastroenterol. 2021, 116, 206–209. [Google Scholar] [CrossRef]
- Osiowy, C.; Swidinsky, K.; Haylock-Jacobs, S.; Sadler, M.D.; Fung, S.; Wong, D.; Minuk, G.Y.; Doucette, K.E.; Wong, P.; Tam, E.; et al. Molecular Epidemiology and Clinical Characteristics of Hepatitis D Virus Infection in Canada. JHEP Rep. 2022, 4, 100461. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef]
- Global Burden of Disease Liver Cancer Collaboration; Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005.e1. [Google Scholar] [CrossRef]
- Raimondo, G.; Craxi, A.; Longo, G.; Giannuoli, G.; Caltagirone, M.; Aragona, M.; Pecoraro, G.; Squadrito, G.; Pagliaro, L. Delta Infection in Hepatocellular Carcinoma Positive for Hepatitis B Surface Antigen. Ann. Intern. Med. 1984, 101, 343–344. [Google Scholar] [CrossRef]
- Puigvehí, M.; Moctezuma-Velázquez, C.; Villanueva, A.; Llovet, J.M. The Oncogenic Role of Hepatitis Delta Virus in Hepatocellular Carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef]
- Tseligka, E.D.; Clément, S.; Negro, F. HDV Pathogenesis: Unravelling Ariadne’s Thread. Viruses 2021, 13, 778. [Google Scholar] [CrossRef]
- Rizzetto, M.; Canese, M.G.; Arico, S.; Trepo, C.; Bonino, F.; Verme, G. Immunofluorescence Detection of New Antigen- Antibody System (S/Anti-5) Associated to Hepatitis B Virus in Liver and in Serum of HBsAg Carriers. Gut 1977, 18, 997–1003. [Google Scholar] [CrossRef]
- Rizzetto, M.; Gerin, J.L. A Agent: Association of a Antigen with Hepatitis B Surface Antigen and RNA in Serum of B-Infected Chimpanzees. Proc. Natl. Acad. Sci. USA 1980, 77, 6124–6128. [Google Scholar] [CrossRef]
- Gerin, J.L. The Taxonomy of Hepatitis Delta Virus. In Proceedings of the Viral Hepatitis and Liver Disease, Tokyo, Japan, 10–14 May 1993; Nishioka, K., Suzuki, H., Mishiro, S., Oda, T., Eds.; Springer: Tokyo, Japan, 1994; pp. 63–64. [Google Scholar]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to Virus Taxonomy and to the International Code of Virus Classification and Nomenclature Ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, F.; Brichler, S.; Drugan, T.; Alloui, C.; Roulot, D.; Pawlotsky, J.; Dény, P.; Gordien, E. Genetic Diversity and Worldwide Distribution of the Deltavirus Genus: A Study of 2,152 Clinical Strains. Hepatology 2017, 66, 1826–1841. [Google Scholar] [CrossRef]
- Rizzetto, M. The Adventure of Delta. Liver Int. 2016, 36, 135–140. [Google Scholar] [CrossRef]
- Lempp, F.A.; Ni, Y.; Urban, S. Hepatitis Delta Virus: Insights into a Peculiar Pathogen and Novel Treatment Options. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 580–589. [Google Scholar] [CrossRef]
- Zhang, Z.; Urban, S. Interplay between Hepatitis D Virus and the Interferon Response. Viruses 2020, 12, 1334. [Google Scholar] [CrossRef]
- Wong, S.K.; Lazinski, D.W. Replicating Hepatitis Delta Virus RNA Is Edited in the Nucleus by the Small Form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef]
- Casey, J.L. RNA Editing in Hepatitis Delta Virus. In Hepatitis Delta Virus; Casey, J.L., Ed.; Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 67–89. ISBN 978-3-540-29802-1. [Google Scholar]
- Chen, P.-J.; Kalpana, G.; Goldberg, J.; Mason, W.; WERNERt, B.; GERINt, J.; Taylor, J. Structure and Replication of the Genome of the Hepatitis 6 Virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef]
- Kos, A.; Dijkema, R.; Arnberg, A.C.; van der Meide, P.H.; Schellekens, H. The Hepatitis Delta (δ) Virus Possesses a Circular RNA. Nature 1986, 323, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Zuccola, H.J.; Rozzelle, J.E.; Lemon, S.M.; Erickson, B.W.; Hogle, J.M. Structural Basis of the Oligomerization of Hepatitis Delta Antigen. Structure 1998, 6, 821–830. [Google Scholar] [CrossRef]
- Asselah, T.; Rizzetto, M. Hepatitis D Virus Infection. N. Engl. J. Med. 2023, 389, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.-C.; Hsieh, T.-Y.; Sheu, G.-T.; Lai, M.M.C. Hepatitis Delta Antigen Mediates the Nuclear Import of Hepatitis Delta Virus RNA. J. Virol. 1998, 72, 3684–3690. [Google Scholar] [CrossRef] [PubMed]
- Tavanez, J.P.; Cunha, C.; Silva, M.C.A.; David, E.; Monjardino, J.; Carmo-Fonseca, M. Hepatitis Delta Virus Ribonucleoproteins Shuttle between the Nucleus and the Cytoplasm. RNA 2002, 8, 637–646. [Google Scholar] [CrossRef]
- Filipovska, J.; Konarska, M.M. Specific HDV RNA-Templated Transcription by Pol II in Vitro. RNA 2000, 6, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Modahl, L.E.; Macnaughton, T.B.; Zhu, N.; Johnson, D.L.; Lai, M.M.C. RNA-Dependent Replication and Transcription of Hepatitis Delta Virus RNA Involve Distinct Cellular RNA Polymerases. Mol. Cell. Biol. 2000, 20, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Urban, S. Virus Entry and Its Inhibition to Prevent and Treat Hepatitis B and Hepatitis D Virus Infections. Curr. Opin. Virol. 2018, 30, 68–79. [Google Scholar] [CrossRef] [PubMed]
- FDA Rejects Bulevirtide for Hepatitis D, Compensated Liver Disease. Available online: https://www.ajmc.com/view/fda-rejects-bulevirtide-for-hepatitis-d-compensated-liver-disease (accessed on 1 March 2024).
- Wedemeyer, H.; Aleman, S.; Brunetto, M.R.; Blank, A.; Andreone, P.; Bogomolov, P.; Chulanov, V.; Mamonova, N.; Geyvandova, N.; Morozov, V.; et al. A Phase 3, Randomized Trial of Bulevirtide in Chronic Hepatitis D. N. Engl. J. Med. 2023, 389, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.; Canini, L.; Dahari, H.; Zhao, X.; Uprichard, S.L.; Haynes-Williams, V.; Winters, M.A.; Subramanya, G.; Cooper, S.L.; Pinto, P.; et al. Oral Prenylation Inhibition with Lonafarnib in Chronic Hepatitis D Infection: A Proof-of-Concept Randomised, Double-Blind, Placebo-Controlled Phase 2A Trial. Lancet Infect. Dis. 2015, 15, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Yurdaydin, C.; Keskin, O.; Kalkan, Ç.; Karakaya, F.; Çalişkan, A.; Karatayli, E.; Karatayli, S.; Bozdayi, A.M.; Koh, C.; Heller, T.; et al. Optimizing Lonafarnib Treatment for the Management of Chronic Delta Hepatitis: The LOWR HDV-1 Study. Hepatology 2018, 67, 1224–1236. [Google Scholar] [CrossRef]
- Week 48 Results of the Phase 3 D-LIVR Study, a Randomized Double-Blind, Placebo-Controlled Trial Evaluating the Safety and Efficacy of Lonafarnib-Boosted with Ritonavir with or without Peginterferon Alfa in Patients with Chronic Hepatitis Delta. Available online: https://www.natap.org/2023/EASL/EASL_34.htm (accessed on 1 March 2024).
- Rizzetto, M.; Chiaberge, E.; Negro, F.; Giacomo, C.D.; Cortesini, R.; Doglia, M.; Macagno, S.; Verme, G.; Marinucci, G.; Alfani, D.; et al. Liver Transplantation in Hepatitis Delta Virus Disease. Lancet 1987, 330, 469–471. [Google Scholar] [CrossRef]
- Ottobrelli, A.; Marzano, A.; Smedile, A.; Recchia, S.; Salizzoni, M.; Cornu, C.; Lamy, M.E.; Otte, J.B.; De Hemptinne, B.; Geubel, A.; et al. Patterns of Hepatitis Delta Virus Reinfection and Disease in Liver Transplantation. Gastroenterology 1991, 101, 1649–1655. [Google Scholar] [CrossRef] [PubMed]
- Samuel, D.; Zignego, A.L.; Reynes, M.; Feray, C.; Arulnaden, J.L.; David, M.F.; Gigou, M.; Bismuth, A.; Mathieu, D.; Gentilini, P. Long-Term Clinical and Virological Outcome after Liver Transplantation for Cirrhosis Caused by Chronic Delta Hepatitis. Hepatology 1995, 21, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Filmann, N.; Yurdaydin, C.; Bremer, B.; Puls, F.; Zacher, B.J.; Heidrich, B.; Tillmann, H.L.; Rosenau, J.; Bock, C.-T.; et al. Rapid Early HDV RNA Decline in the Peripheral Blood but Prolonged Intrahepatic Hepatitis Delta Antigen Persistence after Liver Transplantation. J. Hepatol. 2012, 56, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Helbig, M.; Volz, T.; Allweiss, L.; Mancke, L.V.; Lohse, A.W.; Polywka, S.; Pollok, J.M.; Petersen, J.; Taylor, J.; et al. Persistent Hepatitis D Virus Mono-Infection in Humanized Mice Is Efficiently Converted by Hepatitis B Virus to a Productive Co-Infection. J. Hepatol. 2014, 60, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Hamid, S.; Negro, F. The Changing Context of Hepatitis D. J. Hepatol. 2021, 74, 1200–1211. [Google Scholar] [CrossRef]
- Razavi-Shearer, D.; Child, H.; Razavi-Shearer, K.; Voeller, A.; Razavi, H.; Buti, M.; Tacke, F.; Terrault, N.; Zeuzem, S.; Abbas, Z.; et al. Adjusted Estimate of the Prevalence of Hepatitis Delta Virus in 25 Countries and Territories. J. Hepatol. 2024, 80, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Lago, B.V.; Mello, F.C.A.; Barros, T.M.; Mello, V.M.; Villar, L.M.; Lewis-Ximenez, L.L.; Pardini, M.I.M.C.; Lampe, E.; on behalf of The Brazilian Hepatitis B Research Group. Hepatitis D Infection in Brazil: Prevalence and Geographical Distribution of Anti-Delta Antibody. J. Med. Virol. 2018, 90, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Demirel, A.; Uraz, S.; Deniz, Z.; Daglilar, E.; Basar, O.; Tahan, V.; Ozaras, R. Epidemiology of Hepatitis D Virus Infection in Europe: Is It Vanishing? J. Viral Hepat. 2024, 31, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oidovsambuu, O.; Liu, P.; Grosely, R.; Elazar, M.; Winn, V.D.; Fram, B.; Boa, Z.; Dai, H.; Dashtseren, B.; et al. A Novel Quantitative Microarray Antibody Capture Assay Identifies an Extremely High Hepatitis Delta Virus Prevalence among Hepatitis B Virus–Infected Mongolians. Hepatology 2017, 66, 1739–1749. [Google Scholar] [CrossRef]
- Sarin, S.K.; Kumar, M.; Lau, G.K.; Abbas, Z.; Chan, H.L.Y.; Chen, C.J.; Chen, D.S.; Chen, H.L.; Chen, P.J.; Chien, R.N.; et al. Asian-Pacific Clinical Practice Guidelines on the Management of Hepatitis B: A 2015 Update. Hepatol. Int. 2016, 10, 1–98. [Google Scholar] [CrossRef]
- Lampertico, P.; Agarwal, K.; Berg, T.; Buti, M.; Janssen, H.L.A.; Papatheodoridis, G.; Zoulim, F.; Tacke, F. EASL 2017 Clinical Practice Guidelines on the Management of Hepatitis B Virus Infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Lok, A.S.F.; McMahon, B.J.; Chang, K.; Hwang, J.P.; Jonas, M.M.; Brown, R.S.; Bzowej, N.H.; Wong, J.B. Update on Prevention, Diagnosis, and Treatment of Chronic Hepatitis B: AASLD 2018 Hepatitis B Guidance. Hepatology 2018, 67, 1560–1599. [Google Scholar] [CrossRef] [PubMed]
- Lampertico, P.; Degasperi, E.; Sandmann, L.; Wedemeyer, H.; Yurdaydin, C.; Roulot, D.; Zoulim, F.; Caruntu, F.A.; Wedemeyer, H.; Kefalakes, H.; et al. Hepatitis D Virus Infection: Pathophysiology, Epidemiology and Treatment. Report from the First International Delta Cure Meeting 2022. JHEP Rep. 2023, 5, 100818. [Google Scholar] [CrossRef] [PubMed]
- Caviglia, G.P.; Ciancio, A.; Rizzetto, M. A Review of HDV Infection. Viruses 2022, 14, 1749. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Lok, A.S. Hepatitis D: A Review. JAMA 2023, 330, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Yurdaydın, C.; Idilman, R.; Bozkaya, H.; Bozdayi, A.M. Natural History and Treatment of Chronic Delta Hepatitis: Chronic Delta Hepatitis. J. Viral Hepat. 2010, 17, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Smedile, A.; Casey, J.L.; Cote, P.J.; Durazzo, M.; Lavezzo, B.; Purcell, R.H.; Rizzetto, M.; Gerin, J.L. Hepatitis D Viremia Following Orthotopic Liver Transplantation Involves a Typical HDV Virion with a Hepatitis B Surface Antigen Envelope. Hepatology 1998, 27, 1723–1729. [Google Scholar] [CrossRef] [PubMed]
- Smedile, A.; Verme, G.; Cargnel, A.; Dentico, P.; Opolon, P.; Vergani, D.; Farci, P.; Caredda, F.; Caporaso, N.; Trepo, C.; et al. Influence of delta infection on severity of hepatitis B. Lancet 1982, 320, 945–947. [Google Scholar] [CrossRef] [PubMed]
- Moestrup, T.; Hansson, B.G.; Widell, A.; Nordenfelt, E. Clinical Aspects of Delta Infection. BMJ 1983, 286, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Chen, T.-Z.; Huang, Y.-S.; Yen, F.-S.; Ting, L.-T.; Sheng, W.-Y.; Tsay, S.-H.; Lee, S.-D. Natural History of Hepatitis D Viral Superinfection: Significance of Viremia Detected by Polymerase Chain Reaction. Gastroenterology 1995, 108, 796–802. [Google Scholar] [CrossRef]
- Raimondo, G.; Brunetto, M.R.; Pontisso, P.; Smedile, A.; Maina, A.M.; Saitta, C.; Squadrito, G.; Tono, N.; The Associazione Italiana Studio Fegato (AISF) Cooperative Group. Longitudinal Evaluation Reveals a Complex Spectrum of Virological Profiles in Hepatitis B Virus/Hepatitis C Virus-Coinfected Patients. Hepatology 2006, 43, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Schaper, M.; Rodriguez-Frias, F.; Jardi, R.; Tabernero, D.; Homs, M.; Ruiz, G.; Quer, J.; Esteban, R.; Buti, M. Quantitative Longitudinal Evaluations of Hepatitis Delta Virus RNA and Hepatitis B Virus DNA Shows a Dynamic, Complex Replicative Profile in Chronic Hepatitis B and D. J. Hepatol. 2010, 52, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Lucifora, J.; Alfaiate, D.; Pons, C.; Michelet, M.; Ramirez, R.; Fusil, F.; Amirache, F.; Rossi, A.; Legrand, A.-F.; Charles, E.; et al. Hepatitis D Virus Interferes with Hepatitis B Virus RNA Production via Interferon-Dependent and -Independent Mechanisms. J. Hepatol. 2023, 78, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Kamal, H.; Westman, G.; Falconer, K.; Duberg, A.; Weiland, O.; Haverinen, S.; Wejstål, R.; Carlsson, T.; Kampmann, C.; Larsson, S.B.; et al. Long-Term Study of Hepatitis Delta Virus Infection at Secondary Care Centers: The Impact of Viremia on Liver-Related Outcomes. Hepatology 2020, 72, 1177–1190. [Google Scholar] [CrossRef]
- Roulot, D.; Brichler, S.; Layese, R.; BenAbdesselam, Z.; Zoulim, F.; Thibault, V.; Scholtes, C.; Roche, B.; Castelnau, C.; Poynard, T.; et al. Origin, HDV Genotype and Persistent Viremia Determine Outcome and Treatment Response in Patients with Chronic Hepatitis Delta. J. Hepatol. 2020, 73, 1046–1062. [Google Scholar] [CrossRef] [PubMed]
- Palom, A.; Rodríguez-Tajes, S.; Navascués, C.A.; García-Samaniego, J.; Riveiro-Barciela, M.; Lens, S.; Rodríguez, M.; Esteban, R.; Buti, M. Long-Term Clinical Outcomes in Patients with Chronic Hepatitis Delta: The Role of Persistent Viraemia. Aliment. Pharmacol. Ther. 2020, 51, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Foglieni, B.; Casazza, G.; Spreafico, M.; Colombo, M.; Prati, D. High Serum Levels of HDV RNA Are Predictors of Cirrhosis and Liver Cancer in Patients with Chronic Hepatitis Delta. PLoS ONE 2014, 9, e92062. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Brown, T.L.; Colan, E.J.; Wignall, F.S.; Gerin, J.L. A Genotype of Hepatitis D Virus That Occurs in Northern South America. Proc. Natl. Acad. Sci. USA 1993, 90, 9016–9020. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Chen, T.-Z.; Huo, T.-I.; Lee, S.; Choo, K.-B.; Chen, C.-M. Genotyping of Hepatitis D Virus by Restriction-Fragment Length Polymorphism and Relation to Outcome of Hepatitis D. Lancet 1995, 346, 939–941. [Google Scholar] [CrossRef]
- Niro, G.A.; Smedile, A.; Andriulli, A.; Rizzetto, M.; Gerin, J.L.; Casey, J.L. The Predominance of Hepatitis Delta Virus Genotype I among Chronically Infected Italian Patients. Hepatology 1997, 25, 728–734. [Google Scholar] [CrossRef]
- Su, C.; Huang, Y.; Huo, T.; Shih, H.H.; Sheen, I.; Chen, S.; Lee, P.; Lee, S.; Wu, J. Genotypes and Viremia of Hepatitis B and D Viruses Are Associated With Outcomes of Chronic Hepatitis D Patients. Gastroenterology 2006, 130, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Melo Da Silva, E.; Kay, A.; Lobato, C.; Muwonge, R.; Zoulim, F.; Brites, C.; Parana, R.; Trepo, C. Non-F HBV/HDV-3 Coinfection Is Associated with Severe Liver Disease in Western Brazilian Amazon. J. Med. Virol. 2019, 91, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Spaan, M.; Carey, I.; Bruce, M.; Shang, D.; Horner, M.; Dusheiko, G.; Agarwal, K. Hepatitis Delta Genotype 5 Is Associated with Favourable Disease Outcome and Better Response to Treatment Compared to Genotype 1. J. Hepatol. 2020, 72, 1097–1104. [Google Scholar] [CrossRef]
- Saracco, G.; Rosina, F.; Brunetto, M.R.; Amoroso, P.; Caredda, F.; Farci, P.; Piantino, P.; Bonino, F.; Rizzetto, M. Rapidly Progressive HBsAg-Positive Hepatitis in Italy. The Role of Hepatitis Delta Virus Infection. J. Hepatol. 1987, 5, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Smedile, A.; Rosina, F.; Saracco, G.; Chiaberge, E.; Lattore, V.; Fabino, A.; Brunetto, M.R.; Verme, G.; Rizzetto, M.; Bonino, F. Hepatitis B Virus Replication Modulates Pathogenesis of Hepatitis D Virus in Chronic Hepatitis D. Hepatology 1991, 13, 413–416. [Google Scholar] [CrossRef]
- Lozano, J.L.; Crespo, J.; De La Cruz, F.; Casafont, F.; Lopez-Arias, M.J.; Martín-Ramos, L.; Pons-Romero, F. Correlation between Hepatitis B Viremia and the Clinical and Histological Activity of Chronic Delta Hepatitis. Med. Microbiol. Immunol. 1994, 183, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Del Ninno, E.; Rumi, M.; Russo, A.; Sangiovanni, A.; De Franchis, R.; Ronchi, G.; Colombo, M. A 28-Year Study of the Course of Hepatitis Δ Infection: A Risk Factor for Cirrhosis and Hepatocellular Carcinoma. Gastroenterology 2009, 136, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, C.; Moradpour, D.; Sahli, R.; Suter-Riniker, F.; Lüthi, A.; Cavassini, M.; Günthard, H.F.; Battegay, M.; Bernasconi, E.; Schmid, P.; et al. Hepatitis Delta-Associated Mortality in HIV/HBV-Coinfected Patients. J. Hepatol. 2017, 66, 297–303. [Google Scholar] [CrossRef] [PubMed]
- McNair, A.N.B.; Cheng, D.; Monjardino, J.; Thomas, H.C.; Kerr, I.M. Hepatitis Delta Virus Replication in Vitro Is Not Affected by Interferon- or—Despite Intact Cellular Responses to Interferon and dsRNA. J. Gen. Virol. 1994, 75, 1371–1378. [Google Scholar] [CrossRef]
- Williams, V.; Brichler, S.; Radjef, N.; Lebon, P.; Goffard, A.; Hober, D.; Fagard, R.; Kremsdorf, D.; Dény, P.; Gordien, E. Hepatitis Delta Virus Proteins Repress Hepatitis B Virus Enhancers and Activate the Alpha/Beta Interferon-Inducible MxA Gene. J. Gen. Virol. 2009, 90, 2759–2767. [Google Scholar] [CrossRef]
- Heinicke, L.A.; Bevilacqua, P.C. Activation of PKR by RNA Misfolding: HDV Ribozyme Dimers Activate PKR. RNA 2012, 18, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Alfaiate, D.; Lucifora, J.; Abeywickrama-Samarakoon, N.; Michelet, M.; Testoni, B.; Cortay, J.-C.; Sureau, C.; Zoulim, F.; Dény, P.; Durantel, D. HDV RNA Replication Is Associated with HBV Repression and Interferon-Stimulated Genes Induction in Super-Infected Hepatocytes. Antivir. Res. 2016, 136, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Filzmayer, C.; Ni, Y.; Sültmann, H.; Mutz, P.; Hiet, M.-S.; Vondran, F.W.R.; Bartenschlager, R.; Urban, S. Hepatitis D Virus Replication Is Sensed by MDA5 and Induces IFN-β/λ Responses in Hepatocytes. J. Hepatol. 2018, 69, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Chida, T.; Ishida, Y.; Morioka, S.; Sugahara, G.; Han, C.; Lam, B.; Yamasaki, C.; Sugahara, R.; Li, M.; Tanaka, Y.; et al. Persistent Hepatic IFN System Activation in HBV-HDV Infection Determines Viral Replication Dynamics and Therapeutic Response. JCI Insight 2023, 8, e162404. [Google Scholar] [CrossRef]
- Gillich, N.; Zhang, Z.; Binder, M.; Urban, S.; Bartenschlager, R. Effect of Variants in LGP2 on MDA5-Mediated Activation of Interferon Response and Suppression of Hepatitis D Virus Replication. J. Hepatol. 2023, 78, 78–89. [Google Scholar] [CrossRef]
- Lange, F.; Garn, J.; Anagho, H.A.; Vondran, F.W.R.; Von Hahn, T.; Pietschmann, T.; Carpentier, A. Hepatitis D Virus Infection, Innate Immune Response and Antiviral Treatments in Stem Cell-derived Hepatocytes. Liver Int. 2023, 43, 2116–2129. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Allweiss, L.; Volz, T.; Helbig, M.; Bierwolf, J.; Lohse, A.W.; Pollok, J.M.; Petersen, J.; Dandri, M.; Lütgehetmann, M. Hepatitis Delta Co-Infection in Humanized Mice Leads to Pronounced Induction of Innate Immune Responses in Comparison to HBV Mono-Infection. J. Hepatol. 2015, 63, 346–353. [Google Scholar] [CrossRef]
- He, W.; Ren, B.; Mao, F.; Jing, Z.; Li, Y.; Liu, Y.; Peng, B.; Yan, H.; Qi, Y.; Sun, Y.; et al. Hepatitis D Virus Infection of Mice Expressing Human Sodium Taurocholate Co-Transporting Polypeptide. PLoS Pathog. 2015, 11, e1004840. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Amarán, L.; Usai, C.; Di Scala, M.; Godoy, C.; Ni, Y.; Hommel, M.; Palomo, L.; Segura, V.; Olagüe, C.; Vales, A.; et al. A New HDV Mouse Model Identifies Mitochondrial Antiviral Signaling Protein (MAVS) as a Key Player in IFN-β Induction. J. Hepatol. 2017, 67, 669–679. [Google Scholar] [CrossRef]
- Winer, B.Y.; Shirvani-Dastgerdi, E.; Bram, Y.; Sellau, J.; Low, B.E.; Johnson, H.; Huang, T.; Hrebikova, G.; Heller, B.; Sharon, Y.; et al. Preclinical Assessment of Antiviral Combination Therapy in a Genetically Humanized Mouse Model for Hepatitis Delta Virus Infection. Sci. Transl. Med. 2018, 10, eaap9328. [Google Scholar] [CrossRef]
- Racanelli, V.; Rehermann, B. The Liver as an Immunological Organ. Hepatology 2006, 43, S54–S62. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of Natural Killer Cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Lunemann, S.; Malone, D.F.G.; Grabowski, J.; Port, K.; Béziat, V.; Bremer, B.; Malmberg, K.-J.; Manns, M.P.; Sandberg, J.K.; Cornberg, M.; et al. Effects of HDV Infection and Pegylated Interferon α Treatment on the Natural Killer Cell Compartment in Chronically Infected Individuals. Gut 2015, 64, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Maric, J.; Garcés Lázaro, I.; Hofman, T.; Zhang, Z.; Ni, Y.; Keller, F.; Seufert, I.; Hofmann, M.; Neumann-Haefelin, C.; et al. Hepatitis D Infection Induces IFN-β-Mediated NK Cell Activation and TRAIL-Dependent Cytotoxicity. Front. Immunol. 2023, 14, 1287367. [Google Scholar] [CrossRef] [PubMed]
- Provine, N.M.; Klenerman, P. MAIT Cells in Health and Disease. Annu. Rev. Immunol. 2020, 38, 203–228. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.; Hengst, J.; Parrot, T.; Leeansyah, E.; Lunemann, S.; Malone, D.F.G.; Hardtke, S.; Strauss, O.; Zimmer, C.L.; Berglin, L.; et al. Chronic Hepatitis Delta Virus Infection Leads to Functional Impairment and Severe Loss of MAIT Cells. J. Hepatol. 2019, 71, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Pugnale, P.; Pazienza, V.; Guilloux, K.; Negro, F. Hepatitis Delta Virus Inhibits Alpha Interferon Signaling. Hepatology 2009, 49, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Gocke, D.J.; Verme, G.; Shih, J.W.-K.; Purcell, R.H.; Gerin, J.L. Incidence and significance of antibodies to delta antigen in hepatitis B virus infection. Lancet 1979, 314, 986–990. [Google Scholar] [CrossRef]
- Urban, S.; Neumann-Haefelin, C.; Lampertico, P. Hepatitis D Virus in 2021: Virology, Immunology and New Treatment Approaches for a Difficult-to-Treat Disease. Gut 2021, 70, 1782–1794. [Google Scholar] [CrossRef]
- Nisini, R.; Paroli, M.; Accapezzato, D.; Bonino, F.; Rosina, F.; Santantonio, T.; Sallusto, F.; Amoroso, A.; Houghton, M.; Barnaba, V. Human CD4+ T-Cell Response to Hepatitis Delta Virus: Identification of Multiple Epitopes and Characterization of T-Helper Cytokine Profiles. J. Virol. 1997, 71, 2241–2251. [Google Scholar] [CrossRef]
- Landahl, J.; Bockmann, J.H.; Scheurich, C.; Ackermann, C.; Matzat, V.; Heide, J.; Nuurei, T.; D’Antonio, G.; Von Felden, J.; Sette, A.; et al. Detection of a Broad Range of Low-Level Major Histocompatibility Complex Class II–Restricted, Hepatitis Delta Virus (HDV)–Specific T-Cell Responses Regardless of Clinical Status. J. Infect. Dis. 2019, 219, 568–577. [Google Scholar] [CrossRef]
- Karimzadeh, H.; Kiraithe, M.M.; Oberhardt, V.; Salimi Alizei, E.; Bockmann, J.; Schulze Zur Wiesch, J.; Budeus, B.; Hoffmann, D.; Wedemeyer, H.; Cornberg, M.; et al. Mutations in Hepatitis D Virus Allow It to Escape Detection by CD8+ T Cells and Evolve at the Population Level. Gastroenterology 2019, 156, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Kefalakes, H.; Koh, C.; Sidney, J.; Amanakis, G.; Sette, A.; Heller, T.; Rehermann, B. Hepatitis D Virus-Specific CD8+ T Cells Have a Memory-Like Phenotype Associated With Viral Immune Escape in Patients With Chronic Hepatitis D Virus Infection. Gastroenterology 2019, 156, 1805–1819.e9. [Google Scholar] [CrossRef]
- Schirdewahn, T.; Grabowski, J.; Owusu Sekyere, S.; Bremer, B.; Wranke, A.; Lunemann, S.; Schlaphoff, V.; Kirschner, J.; Hardtke, S.; Manns, M.P.; et al. The Third Signal Cytokine Interleukin 12 Rather Than Immune Checkpoint Inhibitors Contributes to the Functional Restoration of Hepatitis D Virus–Specific T Cells. J. Infect. Dis. 2017, 215, 139–149. [Google Scholar] [CrossRef]
- Karimzadeh, H.; Kiraithe, M.M.; Kosinska, A.D.; Glaser, M.; Fiedler, M.; Oberhardt, V.; Salimi Alizei, E.; Hofmann, M.; Mok, J.Y.; Nguyen, M.; et al. Amino Acid Substitutions within HLA-B*27-Restricted T Cell Epitopes Prevent Recognition by Hepatitis Delta Virus-Specific CD8+ T Cells. J. Virol. 2018, 92, e01891-17. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, C.; Xue, R.; Liu, M.; Bai, J.; Bao, J.; Wang, Y.; Jiang, N.; Li, Z.; Wang, W.; et al. Deep Whole-Genome Analysis of 494 Hepatocellular Carcinomas. Nature 2024, 627, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Guilhot, S.; Huang, S.N.; Xia, Y.P.; La Monica, N.; Lai, M.M.; Chisari, F.V. Expression of the Hepatitis Delta Virus Large and Small Antigens in Transgenic Mice. J. Virol. 1994, 68, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Maestro, S.; Gómez-Echarte, N.; Camps, G.; Usai, C.; Suárez, L.; Vales, Á.; Olagüe, C.; Aldabe, R.; González-Aseguinolaza, G. AAV-HDV: An Attractive Platform for the In Vivo Study of HDV Biology and the Mechanism of Disease Pathogenesis. Viruses 2021, 13, 788. [Google Scholar] [CrossRef] [PubMed]
- Usai, C.; Maestro, S.; Camps, G.; Olague, C.; Suárez-Amaran, L.; Vales, A.; Aragon, T.; Hommel, M.; Aldabe, R.; Gonzalez-Aseguinolaza, G. TNF-Alpha Inhibition Ameliorates HDV-Induced Liver Damage in a Mouse Model of Acute Severe Infection. JHEP Rep. 2020, 2, 100098. [Google Scholar] [CrossRef]
- Park, C.-Y.; Oh, S.-H.; Kang, S.M.; Lim, Y.-S.; Hwang, S.B. Hepatitis Delta Virus Large Antigen Sensitizes to TNF-α-Induced NF-κB Signaling. Mol. Cells 2009, 28, 49–56. [Google Scholar] [CrossRef]
- Yang, Y.M.; Seki, E. TNFα in Liver Fibrosis. Curr. Pathobiol. Rep. 2015, 3, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Townsend, E.C.; Zhang, G.Y.; Ali, R.; Firke, M.; Moon, M.S.; Han, M.A.T.; Fram, B.; Glenn, J.S.; Kleiner, D.E.; Koh, C.; et al. The Balance of Type 1 and Type 2 Immune Responses in the Contexts of Hepatitis B Infection and Hepatitis D Infection. J. Gastroenterol. Hepatol. 2019, 34, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Costante, F.; Stella, L.; Santopaolo, F.; Gasbarrini, A.; Pompili, M.; Asselah, T.; Ponziani, F.R. Molecular and Clinical Features of Hepatocellular Carcinoma in Patients with HBV-HDV Infection. J. Hepatocell. Carcinoma 2023, 10, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.B.; Park, K.J. Cell Cycle Arrest Mediated by Hepatitis Delta Antigen. FEBS Lett. 1999, 449, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pearlberg, J.; Liu, Y.-T.; Ganem, D. Deleterious Effects of Hepatitis Delta Virus Replication on Host Cell Proliferation. J. Virol. 2001, 75, 3600–3604. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Curley, S.A.; Wu, X.; Brown, P.; Hwang, J.P.; Shetty, K.; Yao, Z.-X.; He, A.R.; Li, S.; Katz, L.; et al. Hepatic Stem Cells and Transforming Growth Factor β in Hepatocellular Carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. TGF-β SIGNAL TRANSDUCTION. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Choi, S.; Jeong, S.; Hwang, S.B. Large Hepatitis Delta Antigen Modulates Transforming Growth Factor-β Signaling Cascades: Implication of Hepatitis Delta Virus–Induced Liver Fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef]
- Williams, V.; Brichler, S.; Khan, E.; Chami, M.; Dény, P.; Kremsdorf, D.; Gordien, E. Large Hepatitis Delta Antigen Activates STAT-3 and NF-κB via Oxidative Stress. J. Viral Hepat. 2012, 19, 744–753. [Google Scholar] [CrossRef]
- He, G.; Karin, M. NF-κB and STAT3—Key Players in Liver Inflammation and Cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Goto, T.; Kato, N.; Ono-Nita, S.K.; Yoshida, H.; Otsuka, M.; Shiratori, Y.; Omata, M. Large Isoform of Hepatitis Delta Antigen Activates Serum Response Factor-Associated Transcription. J. Biol. Chem. 2000, 275, 37311–37316. [Google Scholar] [CrossRef]
- Chen, M.; Du, D.; Zheng, W.; Liao, M.; Zhang, L.; Liang, G.; Gong, M. Small Hepatitis Delta Antigen Selectively Binds to Target mRNA in Hepatic Cells: A Potential Mechanism by Which Hepatitis D Virus Downregulates Glutathione S-Transferase P1 and Induces Liver Injury and Hepatocarcinogenesis. Biochem. Cell Biol. 2019, 97, 130–139. [Google Scholar] [CrossRef]
- Chan, A.O.O.; Rashid, A. CpG Island Methylation in Precursors of Gastrointestinal Malignancies. Curr. Mol. Med. 2006, 6, 401–408. [Google Scholar] [CrossRef]
- Villanueva, A.; Portela, A.; Sayols, S.; Battiston, C.; Hoshida, Y.; Méndez-González, J.; Imbeaud, S.; Letouzé, E.; Hernandez-Gea, V.; Cornella, H.; et al. DNA Methylation-based Prognosis and Epidrivers in Hepatocellular Carcinoma. Hepatology 2015, 61, 1945–1956. [Google Scholar] [CrossRef]
- Benegiamo, G.; Vinciguerra, M.; Guarnieri, V.; Niro, G.A.; Andriulli, A.; Pazienza, V. Hepatitis Delta Virus Induces Specific DNA Methylation Processes in Huh-7 Liver Cancer Cells. FEBS Lett. 2013, 587, 1424–1428. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Hong, S.W.; Lee, H.; Kim, W.H. Overexpression of Clusterin in Human Hepatocellular Carcinoma. Human Pathol. 2004, 35, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.H.; Sham, J.S.T.; Xie, D.; Tzang, C.-H.; Tang, D.; Ma, N.; Hu, L.; Wang, Y.; Wen, J.-M.; Xiao, G.; et al. Clusterin Plays an Important Role in Hepatocellular Carcinoma Metastasis. Oncogene 2006, 25, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.-T.; Lee, Y.-J.; Ko, J.-L.; Tsai, C.-C.; Tseng, C.-J.; Sheu, G.-T. Hepatitis Delta Virus Epigenetically Enhances Clusterin Expression via Histone Acetylation in Human Hepatocellular Carcinoma Cells. J. Gen. Virol. 2009, 90, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Matsuura, K.; Kleiner, D.E.; Zamboni, F.; Alter, H.J.; Farci, P. Analysis of Long Noncoding RNA Expression in Hepatocellular Carcinoma of Different Viral Etiology. J. Transl. Med. 2016, 14, 328. [Google Scholar] [CrossRef]
- Beeharry, Y.; Goodrum, G.; Imperiale, C.J.; Pelchat, M. The Hepatitis Delta Virus Accumulation Requires Paraspeckle Components and Affects NEAT1 Level and PSP1 Localization. Sci. Rep. 2018, 8, 6031. [Google Scholar] [CrossRef]
- Mendes, M.; Pérez-Hernandez, D.; Vázquez, J.; Coelho, A.V.; Cunha, C. Proteomic Changes in HEK-293 Cells Induced by Hepatitis Delta Virus Replication. J. Proteom. 2013, 89, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Diaz, G.; Engle, R.E.; Tice, A.; Melis, M.; Montenegro, S.; Rodriguez-Canales, J.; Hanson, J.; Emmert-Buck, M.R.; Bock, K.W.; Moore, I.N.; et al. Molecular Signature and Mechanisms of Hepatitis D Virus-Associated Hepatocellular Carcinoma. Mol. Cancer Res. 2018, 16, 1406–1419. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Ma, X.; Zhang, W.; Chang, X.; An, L.; Niu, M.; Chen, Y.; Sun, C.; Yang, Y. Microarray Data Mining and Preliminary Bioinformatics Analysis of Hepatitis D Virus-Associated Hepatocellular Carcinoma. Biomed. Res. Int. 2021, 2021, 1093702. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Homs, M.; Rodriguez-Frias, F.; Funalleras, G.; Jardí, R.; Sauleda, S.; Tabernero, D.; Schaper, M.; Esteban, R. Clinical Outcome of Acute and Chronic Hepatitis Delta over Time: A Long-Term Follow-up Study. J. Viral Hepat. 2011, 18, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Wranke, A.; Pinheiro Borzacov, L.M.; Parana, R.; Lobato, C.; Hamid, S.; Ceausu, E.; Dalekos, G.N.; Rizzetto, M.; Turcanu, A.; Niro, G.A.; et al. Clinical and Virological Heterogeneity of Hepatitis Delta in Different Regions World-wide: The Hepatitis Delta International Network (HDIN). Liver Int. 2018, 38, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Verme, G.; Recchia, S.; Bonino, F.; Farci, P.; Aricò, S.; Calzia, R.; Picciotto, A.; Colombo, M.; Popper, H. Chronic Hepatitis in Carriers of Hepatitis B Surface Antigen, with Intrahepatic Expression of the Delta Antigen. An Active and Progressive Disease Unresponsive to Immunosuppressive Treatment. Ann. Intern. Med. 1983, 98, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Rosina, F.; Conoscitore, P.; Cuppone, R.; Rocca, G.; Giuliani, A.; Cozzolongo, R.; Niro, G.; Smedile, A.; Saracco, G.; Andriulli, A.; et al. Changing Pattern of Chronic Hepatitis D in Southern Europe. Gastroenterology 1999, 117, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Manesis, E.K.; Vourli, G.; Dalekos, G.; Vasiliadis, T.; Manolaki, N.; Hounta, A.; Koutsounas, S.; Vafiadis, I.; Nikolopoulou, G.; Giannoulis, G.; et al. Prevalence and Clinical Course of Hepatitis Delta Infection in Greece: A 13-Year Prospective Study. J. Hepatol. 2013, 59, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Amougou, M.A.; Noah, D.N.; Moundipa, P.F.; Pineau, P.; Njouom, R. A Prominent Role of Hepatitis D Virus in Liver Cancers Documented in Central Africa. BMC Infect. Dis. 2016, 16, 647. [Google Scholar] [CrossRef]
- Brancaccio, G.; Fasano, M.; Grossi, A.; Santantonio, T.A.; Gaeta, G.B. Clinical Outcomes in Patients with Hepatitis D, Cirrhosis and Persistent Hepatitis B Virus Replication, and Receiving Long-Term Tenofovir or Entecavir. Aliment. Pharmacol. Ther. 2019, 49, 1071–1076. [Google Scholar] [CrossRef]
- Fattovich, G. Influence of Hepatitis Delta Virus Infection on Morbidity and Mortality in Compensated Cirrhosis Type B. Gut 2000, 46, 420–426. [Google Scholar] [CrossRef]
- Gheorghe, L.; Iacob, S.; Simionov, I.; Vãdan, R.; Gheorghe, C.; Iacob, R.; Pârvulescu, I.; Constantinescu, I. Natural History of Compensated Viral B and D Cirrhosis. Rom. J. Gastroenterol. 2005, 14, 329–335. [Google Scholar]
- Niro, G.A.; Smedile, A.; Ippolito, A.M.; Ciancio, A.; Fontana, R.; Olivero, A.; Valvano, M.R.; Abate, M.L.; Gioffreda, D.; Caviglia, G.P.; et al. Outcome of Chronic Delta Hepatitis in Italy: A Long-Term Cohort Study. J. Hepatol. 2010, 53, 834–840. [Google Scholar] [CrossRef]
- Jang, T.-Y.; Wei, Y.-J.; Liu, T.-W.; Yeh, M.-L.; Liu, S.-F.; Hsu, C.-T.; Hsu, P.-Y.; Lin, Y.-H.; Liang, P.-C.; Hsieh, M.-H.; et al. Role of Hepatitis D Virus Infection in Development of Hepatocellular Carcinoma among Chronic Hepatitis B Patients Treated with Nucleotide/Nucleoside Analogues. Sci. Rep. 2021, 11, 8184. [Google Scholar] [CrossRef] [PubMed]
- Kushner, T.; Serper, M.; Kaplan, D.E. Delta Hepatitis within the Veterans Affairs Medical System in the United States: Prevalence, Risk Factors, and Outcomes. J. Hepatol. 2015, 63, 586–592. [Google Scholar] [CrossRef]
- Ji, J.; Sundquist, K.; Sundquist, J. A Population-Based Study of Hepatitis D Virus as Potential Risk Factor for Hepatocellular Carcinoma. JNCI J. Natl. Cancer Inst. 2012, 104, 790–792. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-E.; Su, C.-W.; Huang, Y.-S.; Huang, Y.-H.; Hou, M.-C.; Wu, J.-C. Hepatitis D Virus Dual Infection Increased the Risk of Hepatocellular Carcinoma Compared with Hepatitis B Virus Mono Infection: A Meta-Analysis. J. Chin. Med. Assoc. 2022, 85, 30–41. [Google Scholar] [CrossRef]
- Kamal, H.; Fornes, R.; Simin, J.; Stål, P.; Duberg, A.-S.; Brusselaers, N.; Aleman, S. Risk of Hepatocellular Carcinoma in Hepatitis B and D Virus Co-Infected Patients: A Systematic Review and Meta-Analysis of Longitudinal Studies. J. Viral Hepat. 2021, 28, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z.; Qureshi, M.; Hamid, S.; Jafri, W. Hepatocellular Carcinoma in Hepatitis D: Does It Differ from Hepatitis B Monoinfection? Saudi J. Gastroenterol. 2012, 18, 18–22. [Google Scholar] [CrossRef]
- Wranke, A.; Heidrich, B.; Deterding, K.; Hupa-Breier, K.L.; Kirschner, J.; Bremer, B.; Cornberg, M.; Wedemeyer, H. Clinical Long-Term Outcome of Hepatitis D Compared to Hepatitis B Monoinfection. Hepatol. Int. 2023, 17, 1359–1367. [Google Scholar] [CrossRef]
- Kamal, H.; Aleman, S. D-SOLVE Consortium Natural History of Untreated HDV Patients: Always a Progressive Disease? Liver Int. 2023, 43 (Suppl. S1), 5–21. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardo, D.; Franzè, M.S.; Caminiti, G.; Pollicino, T. Hepatitis Delta Virus and Hepatocellular Carcinoma. Pathogens 2024, 13, 362. https://doi.org/10.3390/pathogens13050362
Lombardo D, Franzè MS, Caminiti G, Pollicino T. Hepatitis Delta Virus and Hepatocellular Carcinoma. Pathogens. 2024; 13(5):362. https://doi.org/10.3390/pathogens13050362
Chicago/Turabian StyleLombardo, Daniele, Maria Stella Franzè, Giuseppe Caminiti, and Teresa Pollicino. 2024. "Hepatitis Delta Virus and Hepatocellular Carcinoma" Pathogens 13, no. 5: 362. https://doi.org/10.3390/pathogens13050362
APA StyleLombardo, D., Franzè, M. S., Caminiti, G., & Pollicino, T. (2024). Hepatitis Delta Virus and Hepatocellular Carcinoma. Pathogens, 13(5), 362. https://doi.org/10.3390/pathogens13050362