
Comparative Genomic Analysis of Prophages in Human Vaginal Isolates of Streptococcus agalactiae

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and DNA Isolation
2.2. Genome Sequencing, Assembly, and Prophage Isolation
2.3. GBS Prophage Database Creation and Genome Clustering
2.4. Genomic Analysis of Prophages
3. Results
3.1. Identification of Prophages in GBS Clinical Isolates
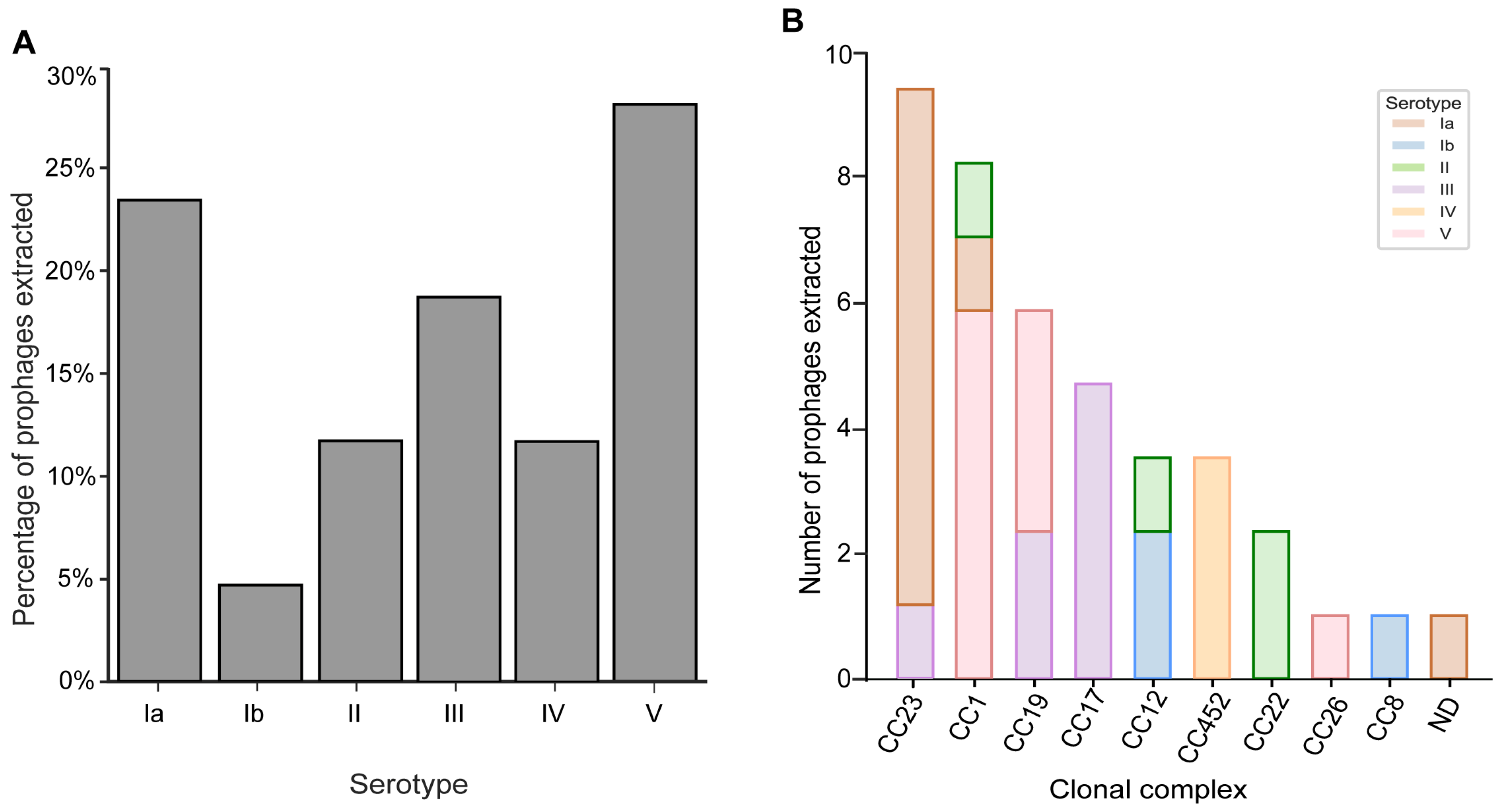
3.2. Prophages Are Prevalent across GBS Serotypes and Clonal Complexes
3.3. Prophages Integrate within Specific Regions of Their Streptococcal Host Genome
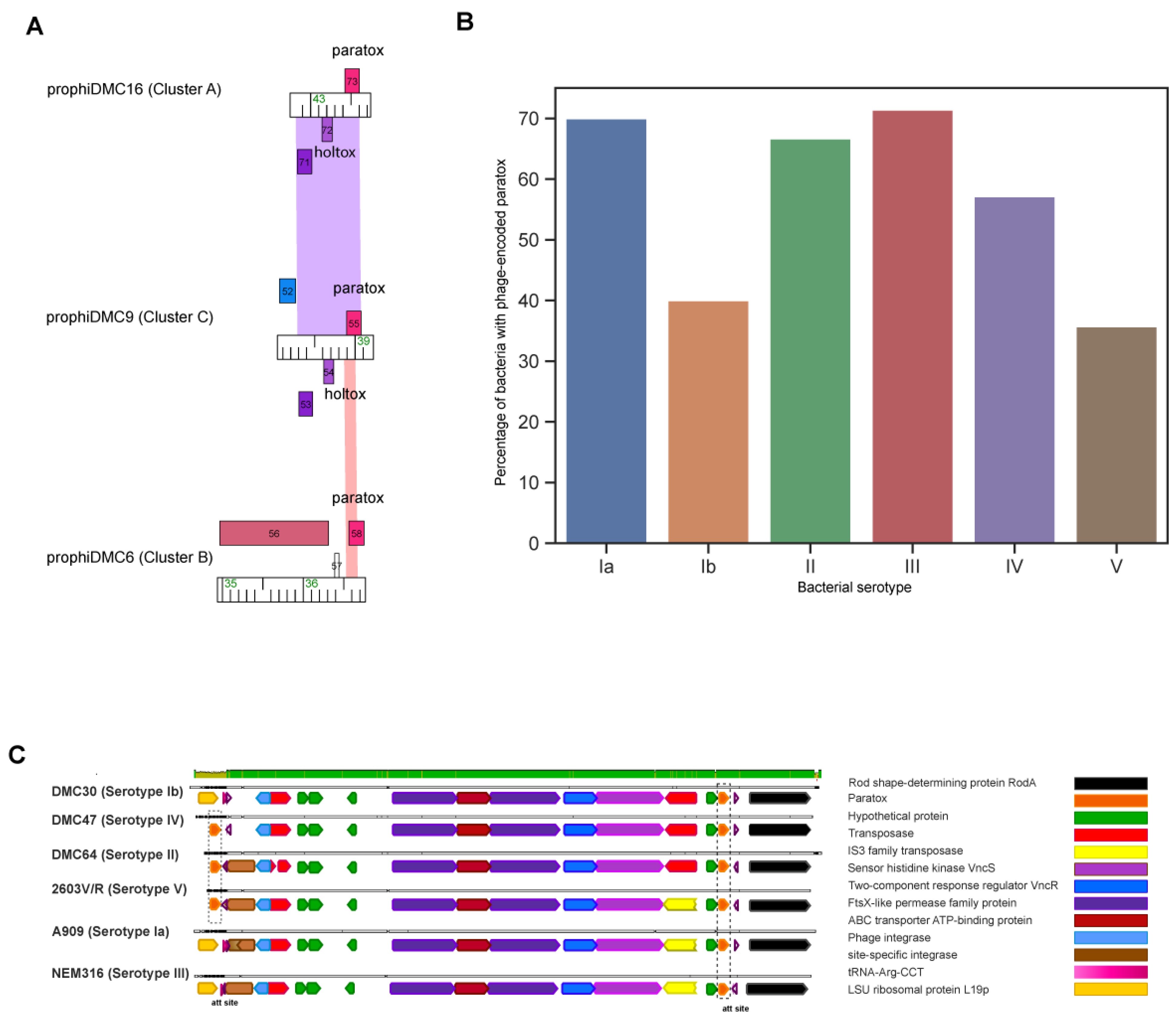
3.4. GBS Prophages Encode Multiple Toxin-Antitoxin Systems
3.5. Prx Is Encoded on the Bacterial Chromosome and Often in GBS Prophages
3.6. GBS Prophages Encode a Gene Upstream of Prx with Holin-like and Transmembrane Domains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Russell, N.J.; Seale, A.C.; O’Driscoll, M.; O’Sullivan, C.; Bianchi-Jassir, F.; Gonzalez-Guarin, J.; Lawn, J.E.; Baker, C.J.; Bartlett, L.; Cutland, C.; et al. Maternal Colonization with Group B Streptococcus and Serotype Distribution Worldwide: Systematic Review and Meta-Analyses. Clin. Infect. Dis. 2017, 65, S100–S111. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsen, C.; Mæland, J.A.; Lyng, R.V.; Radtke, A.; Afset, J.E. Molecular Characteristics of Streptococcus Agalactiae Strains Deficient in Alpha-like Protein Encoding Genes. J. Med. Microbiol. 2017, 66, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kwatra, G.; Adrian, P.V.; Shiri, T.; Buchmann, E.J.; Cutland, C.L.; Madhi, S.A. Natural Acquired Humoral Immunity against Serotype-Specific Group B Streptococcus Rectovaginal Colonization Acquisition in Pregnant Women. Clin. Microbiol. Infect. 2015, 21, 568.e13–568.e21. [Google Scholar] [CrossRef] [PubMed]
- Effectiveness of Intrapartum Antibiotic Prophylaxis for Early-Onset Group B Streptococcal Infection: An Integrative Review|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S1871519217301336?token=0255F68E13436531D872A4B2A17EE2BC79EE3C4A2D1567262FACF385B3568ED5F12BA824F0342FAA81B001639672742E&originRegion=us-east-1&originCreation=20221205181842 (accessed on 5 December 2022).
- Vornhagen, J.; Quach, P.; Boldenow, E.; Merillat, S.; Whidbey, C.; Ngo, L.Y.; Adams Waldorf, K.M.; Rajagopal, L. Bacterial Hyaluronidase Promotes Ascending GBS Infection and Preterm Birth. mBio 2016, 7, e00781-16. [Google Scholar] [CrossRef]
- Edmond, K.M.; Kortsalioudaki, C.; Scott, S.; Schrag, S.J.; Zaidi, A.K.M.; Cousens, S.; Heath, P.T. Group B Streptococcal Disease in Infants Aged Younger than 3 Months: Systematic Review and Meta-Analysis. Lancet 2012, 379, 547–556. [Google Scholar] [CrossRef]
- Penders, J.; Thijs, C.; van den Brandt, P.A.; Kummeling, I.; Snijders, B.; Stelma, F.; Adams, H.; van Ree, R.; Stobberingh, E.E. Gut Microbiota Composition and Development of Atopic Manifestations in Infancy: The KOALA Birth Cohort Study. Gut 2007, 56, 661–667. [Google Scholar] [CrossRef]
- Björkstén, B. Effects of Intestinal Microflora and the Environment on the Development of Asthma and Allergy. Springer Semin. Immunopathol. 2004, 25, 257–270. [Google Scholar] [CrossRef]
- Lundin, A.; Bok, C.M.; Aronsson, L.; Björkholm, B.; Gustafsson, J.-A.; Pott, S.; Arulampalam, V.; Hibberd, M.; Rafter, J.; Pettersson, S. Gut Flora, Toll-like Receptors and Nuclear Receptors: A Tripartite Communication That Tunes Innate Immunity in Large Intestine. Cell. Microbiol. 2008, 10, 1093–1103. [Google Scholar] [CrossRef]
- Hayes, K.; O’Halloran, F.; Cotter, L. A Review of Antibiotic Resistance in Group B Streptococcus: The Story so Far. Crit. Rev. Microbiol. 2020, 46, 253–269. [Google Scholar] [CrossRef]
- Tamura, G.S.; Kuypers, J.M.; Smith, S.; Raff, H.; Rubens, C.E. Adherence of Group B Streptococci to Cultured Epithelial Cells: Roles of Environmental Factors and Bacterial Surface Components. Infect. Immun. 1994, 62, 2450–2458. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Poyart, C.; Trieu-Cuot, P.; Lamberet, G.; Gruss, A.; Gaudu, P. Respiration Metabolism of Group B Streptococcus Is Activated by Environmental Haem and Quinone and Contributes to Virulence. Mol. Microbiol. 2005, 56, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Di Palo, B.; Rippa, V.; Santi, I.; Brettoni, C.; Muzzi, A.; Metruccio, M.M.E.; Grifantini, R.; Telford, J.L.; Paccani, S.R.; Soriani, M. Adaptive Response of Group B Streptococcus to High Glucose Conditions: New Insights on the CovRS Regulation Network. PLoS ONE 2013, 8, e61294. [Google Scholar] [CrossRef] [PubMed]
- Sitkiewicz, I.; Green, N.M.; Guo, N.; Bongiovanni, A.M.; Witkin, S.S.; Musser, J.M. Transcriptome Adaptation of Group B Streptococcus to Growth in Human Amniotic Fluid. PLoS ONE 2009, 4, e6114. [Google Scholar] [CrossRef]
- Landwehr-Kenzel, S.; Henneke, P. Interaction of Streptococcus Agalactiae and Cellular Innate Immunity in Colonization and Disease. Front. Immunol. 2014, 5, 519. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, R.A.; Borges, E.C.; Sutton, J.A.; Aronoff, D.M.; Gaddy, J.A.; Petroff, M.G.; Manning, S.D. Genetically Distinct Group B Streptococcus Strains Induce Varying Macrophage Cytokine Responses. PLoS ONE 2019, 14, e0222910. [Google Scholar] [CrossRef] [PubMed]
- McGee, L.; Chochua, S.; Li, Z.; Mathis, S.; Rivers, J.; Metcalf, B.; Ryan, A.; Alden, N.; Farley, M.M.; Harrison, L.H.; et al. Multistate, Population-Based Distributions of Candidate Vaccine Targets, Clonal Complexes, and Resistance Features of Invasive Group B Streptococci Within the United States, 2015–2017. Clin. Infect. Dis. 2021, 72, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Burcham, L.R.; Spencer, B.L.; Keeler, L.R.; Runft, D.L.; Patras, K.A.; Neely, M.N.; Doran, K.S. Determinants of Group B Streptococcal Virulence Potential amongst Vaginal Clinical Isolates from Pregnant Women. PLoS ONE 2019, 14, e0226699. [Google Scholar] [CrossRef]
- Gori, A.; Harrison, O.B.; Mlia, E.; Nishihara, Y.; Chan, J.M.; Msefula, J.; Mallewa, M.; Dube, Q.; Swarthout, T.D.; Nobbs, A.H.; et al. Pan-GWAS of Streptococcus Agalactiae Highlights Lineage-Specific Genes Associated with Virulence and Niche Adaptation. mBio 2020, 11, e00728-20. [Google Scholar] [CrossRef]
- Shabayek, S.; Spellerberg, B. Group B Streptococcal Colonization, Molecular Characteristics, and Epidemiology. Front. Microbiol. 2018, 9, 437. [Google Scholar] [CrossRef]
- Teatero, S.; Ferrieri, P.; Martin, I.; Demczuk, W.; McGeer, A.; Fittipaldi, N. Serotype Distribution, Population Structure, and Antimicrobial Resistance of Group B Streptococcus Strains Recovered from Colonized Pregnant Women. J. Clin. Microbiol. 2017, 55, 412–422. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome Analysis of Multiple Pathogenic Isolates of Streptococcus Agalactiae: Implications for the Microbial “Pan-Genome. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-Mediated Spread of Bacterial Virulence Genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brüssow, H. Prophage Genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef] [PubMed]
- van der Mee-Marquet, N.; Diene, S.M.; Barbera, L.; Courtier-Martinez, L.; Lafont, L.; Ouachée, A.; Valentin, A.-S.; Santos, S.D.; Quentin, R.; François, P. Analysis of the Prophages Carried by Human Infecting Isolates Provides New Insight into the Evolution of Group B Streptococcus Species. Clin. Microbiol. Infect. 2018, 24, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Domelier, A.-S.; van der Mee-Marquet, N.; Sizaret, P.-Y.; Héry-Arnaud, G.; Lartigue, M.-F.; Mereghetti, L.; Quentin, R. Molecular Characterization and Lytic Activities of Streptococcus Agalactiae Bacteriophages and Determination of Lysogenic-Strain Features. J. Bacteriol. 2009, 191, 4776–4785. [Google Scholar] [CrossRef] [PubMed]
- Lichvariková, A.; Soltys, K.; Szemes, T.; Slobodnikova, L.; Bukovska, G.; Turna, J.; Drahovska, H. Characterization of Clinical and Carrier Streptococcus Agalactiae and Prophage Contribution to the Strain Variability. Viruses 2020, 12, 1323. [Google Scholar] [CrossRef]
- Renard, A.; Barbera, L.; Courtier-Martinez, L.; Dos Santos, S.; Valentin, A.-S.; Mereghetti, L.; Quentin, R.; van der Mee-Marquet, N.L. phiD12-Like Livestock-Associated Prophages Are Associated with Novel Subpopulations of Streptococcus Agalactiae Infecting Neonates. Front. Cell. Infect. Microbiol. 2019, 9, 166. [Google Scholar] [CrossRef]
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R., Jr.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F.; et al. Whole Genome Comparison of a Large Collection of Mycobacteriophages Reveals a Continuum of Phage Genetic Diversity. eLife 2015, 4, e06416. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Aull, H.G.; Jacobs-Sera, D.; Garlena, R.A.; Russell, D.A.; Smith, B.E.; Mahalingam, V.; Abad, L.; Gauthier, C.H.; Hatfull, G.F. The Prophage and Plasmid Mobilome as a Likely Driver of Mycobacterium Abscessus Diversity. mBio 2021, 12, e03441-20. [Google Scholar] [CrossRef]
- Mashburn-Warren, L.; Goodman, S.D.; Federle, M.J.; Prehna, G. The Conserved Mosaic Prophage Protein Paratox Inhibits the Natural Competence Regulator ComR in Streptococcus. Sci. Rep. 2018, 8, 16535. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc/ (accessed on 4 September 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of Genome Assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Cresawn, S.G.; Bogel, M.; Day, N.; Jacobs-Sera, D.; Hendrix, R.W.; Hatfull, G.F. Phamerator: A Bioinformatic Tool for Comparative Bacteriophage Genomics. BMC Bioinform. 2011, 12, 395. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying Bacterial Genes and Endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J.; Borodovsky, M. GeneMark: Web Software for Gene Finding in Prokaryotes, Eukaryotes and Viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred Interactive Server for Protein Homology Detection and Structure Prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio Team: Boston, MA, USA, 2020. [Google Scholar]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the SciPy 2010 9th Python in Science Conference, Austin, TX, USA, 28–30 June 2010; pp. 56–61. [Google Scholar]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M.; Botvinnik, O.; O’Kane, D.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Cole, J.B.; Warmenhoven, J.; et al. mwaskom/Seaborn: V0.8.1 (September 2017). Zenodo 2017. [Google Scholar] [CrossRef]
- Rezaei Javan, R.; Ramos-Sevillano, E.; Akter, A.; Brown, J.; Brueggemann, A.B. Prophages and Satellite Prophages Are Widespread in Streptococcus and May Play a Role in Pneumococcal Pathogenesis. Nat. Commun. 2019, 10, 4852. [Google Scholar] [CrossRef] [PubMed]
- Farley, M.M.; Strasbaugh, L.J. Group B Streptococcal Disease in Nonpregnant Adults. Clin. Infect. Dis. 2001, 33, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Heath, P.T.; Jardine, L.A. Neonatal Infections: Group B Streptococcus. BMJ Clin. Evid. 2014, 2014, 0323. [Google Scholar] [PubMed]
- Luan, S.-L.; Granlund, M.; Sellin, M.; Lagergård, T.; Spratt, B.G.; Norgren, M. Multilocus Sequence Typing of Swedish Invasive Group B Streptococcus Isolates Indicates a Neonatally Associated Genetic Lineage and Capsule Switching. J. Clin. Microbiol. 2005, 43, 3727–3733. [Google Scholar] [CrossRef] [PubMed]
- Landy, A.; Ross, W. Viral Integration and Excision: Structure of the Lambda Att Sites. Science 1977, 197, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, L.; Sigal, N.; Borovok, I.; Nir-Paz, R.; Herskovits, A.A. Prophage Excision Activates Listeria Competence Genes That Promote Phagosomal Escape and Virulence. Cell 2012, 150, 792–802. [Google Scholar] [CrossRef] [PubMed]
- LeRoux, M.; Laub, M.T. Toxin-Antitoxin Systems as Phage Defense Elements. Annu. Rev. Microbiol. 2022, 76, 013730. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, S.; Lévesque, C.M. Characterization of a Streptococcus Mutans Intergenic Region Containing a Small Toxic Peptide and Its Cis-Encoded Antisense Small RNA Antitoxin. PLoS ONE 2013, 8, e54291. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Koonin, E.V. The HicAB Cassette, a Putative Novel, RNA-Targeting Toxin-Antitoxin System in Archaea and Bacteria. Bioinformatics 2006, 22, 2581–2584. [Google Scholar] [CrossRef]
- Lehnherr, H.; Maguin, E.; Jafri, S.; Yarmolinsky, M.B. Plasmid Addiction Genes of Bacteriophage P1: Doc, Which Causes Cell Death on Curing of Prophage, and Phd, Which Prevents Host Death When Prophage Is Retained. J. Mol. Biol. 1993, 233, 414–428. [Google Scholar] [CrossRef]
- Yang, Q.E.; Walsh, T.R. Toxin–Antitoxin Systems and Their Role in Disseminating and Maintaining Antimicrobial Resistance. FEMS Microbiol. Rev. 2017, 41, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Edwards, R.A.; Taylor, W.W.; Low, D.E.; McGeer, A.; Kotb, M. Mosaic Prophages with Horizontally Acquired Genes Account for the Emergence and Diversification of the Globally Disseminated M1T1 Clone of Streptococcus Pyogenes. J. Bacteriol. 2005, 187, 3311–3318. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, T.; Anthony, T.; Saranya, S.; Pushpam, P.L.; Gunasekaran, P. Functional Characterization of a New Holin-like Antibacterial Protein Coding Gene Tmp1 from Goat Skin Surface Metagenome. Appl. Microbiol. Biotechnol. 2011, 89, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Banks, D.J.; Beres, S.B.; Musser, J.M. The Fundamental Contribution of Phages to GAS Evolution, Genome Diversification and Strain Emergence. Trends Microbiol. 2002, 10, 515–521. [Google Scholar] [CrossRef]
- Cushman, J.; Freeman, E.; McCallister, S.; Schumann, A.; Hutchison, K.W.; Molloy, S.D. Increased whiB7 Expression and Antibiotic Resistance in Mycobacterium Chelonae Carrying Two Prophages. BMC Microbiol. 2021, 21, 176. [Google Scholar] [CrossRef] [PubMed]
- Argov, T.; Sapir, S.R.; Pasechnek, A.; Azulay, G.; Stadnyuk, O.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. Coordination of Cohabiting Phage Elements Supports Bacteria–Phage Cooperation. Nat. Commun. 2019, 10, 5288. [Google Scholar] [CrossRef]
- Lemire, S.; Figueroa-Bossi, N.; Bossi, L. Bacteriophage Crosstalk: Coordination of Prophage Induction by Trans-Acting Antirepressors. PLoS Genet. 2011, 7, e1002149. [Google Scholar] [CrossRef] [PubMed]
- Coleman, D.; Knights, J.; Russell, R.; Shanley, D.; Birkbeck, T.H.; Dougan, G.; Charles, I. Insertional Inactivation of the Staphylococcus Aureusβ-Toxin by Bacteriophage Φ13 Occurs by Site-and Orientation-Specific Integration of the φ 13 Genome. Mol. Microbiol. 1991, 5, 933–939. [Google Scholar] [CrossRef]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages Mediate Defense against Phage Infection through Diverse Mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef]
- Gross, M.; Marianovsky, I.; Glaser, G. MazG—A Regulator of Programmed Cell Death in Escherichia Coli. Mol. Microbiol. 2006, 59, 590–601. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prophage | Cluster | Genome Size (bp) | % GC | No. of Genes | Type of Integrase | Gene Upstream of Insertion Site (Att Site) | Inserts Into | Gene Downstream of Insertion Site (Att Site) | Attachment Site |
|---|---|---|---|---|---|---|---|---|---|
| Javan 5 (2603 V/R) | A | 40,574 | 35.3 | 78 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| Javan 7 (A909) | A | 37,225 | 37.1 | 62 | Tyrosine | hypothetical protein | N/A | HU family DNA-binding protein | TTATAGTTGGGGCGAATTTGGGGCATAA |
| prophigbs515 | A | 40,634 | 34.9 | 89 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC2-1 | A | 39,700 | 36.8 | 66 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC5 | A | 46,132 | 35.9 | 80 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC15 | A | 38,551 | 36.7 | 68 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC16 | A | 43,746 | 36.4 | 71 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC21-2 | A | 43,397 | 36.6 | 73 | Serine | N-acetylmannosamine kinase | acetyl xylan esterase | Sialic acid utilization regulator, RpiR family | GATTTTGATGACTTC |
| prophiDMC25 | A | 38,551 | 36.7 | 68 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC33-1 | A | 43,746 | 36.4 | 72 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC34 | A | 37,294 | 36.7 | 62 | Serine | ComGF | N/A | ComGB | TAAATTTTTC |
| prophiDMC43-1 | A | 43,738 | 38.6 | 58 | Tyrosine | bacterial ribosome SSU maturation protein RimP | tRNA-Ser-GGA | tRNA (guanine(46)-N(7))-methyltransferase | AATCCCCTCCTCTCCTTT |
| prophiDMC47 | A | 45,805 | 35.8 | 81 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC48 | A | 45,885 | 35.8 | 80 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC49 | A | 45,685 | 35.8 | 84 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC51 | A | 45,805 | 35.8 | 82 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC61 | A | 45,686 | 35.8 | 80 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC64 | A | 40,022 | 37 | 68 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC66-1 | A | 45,884 | 35.8 | 80 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC67 | A | 37,262 | 36.8 | 61 | Serine | ComGF | N/A | ComGB | TAAATTTTTC |
| prophiDMC68-1 | A | 45,777 | 35.8 | 80 | Tyrosine | integrase | tRNA-Arg | LSU ribosomal protein L19p | ATGTCCCCTGCC |
| prophiDMC6 | B | 36,849 | 39.7 | 57 | Serine | ComGD | ComGC | ComGB | TAAATTTTTC |
| prophiDMC17 | B | 36,582 | 39.7 | 54 | Serine | ComGD | ComGC | ComGB | TAAATTTTTC |
| prophiDMC30 | B | 36,585 | 39.7 | 56 | Serine | ComGD | ComGC | ComGB | TAAATTTTTC |
| prophiDMC36 | B | 36,581 | 39.6 | 54 | Serine | ComGD | ComGC | ComGB | TAAATTTTTC |
| prophiDMC62 | B | 36,534 | 39.7 | 54 | Serine | ComGD | ComGC | ComGB | TAAATTTTTC |
| prophiCNCTC 10/84 | C | 40,696 | 36.4 | 67 | Tyrosine | hypothetical protein | N/A | HU family DNA-binding protein | TTATAGTTGGGGCGAATTTGGGGCATAA |
| prophiDMC4 | C | 38,991 | 36.2 | 55 | Tyrosine | hypothetical protein | N/A | DNA binding protein HbSu | TTATGCCCCAAATTCGCCCCAACTATAA |
| prophiDMC9 | C | 38,963 | 36.2 | 55 | Tyrosine | hypothetical protein | N/A | DNA-binding protein HbSu | TTATGCCCCAAATTCGCCCCAACTATAA |
| prophiDMC69 | C | 38,991 | 36.2 | 65 | Tyrosine | hypothetical protein | N/A | DNA binding protein HbSu | TTATAGTTGGGGCGAATTTGGGGCATAA |
| Javan 8 (A909) | D | 45,841 | 42.2 | 43 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiCJBIII (CJBIII) | D | 48,336 | 41.8 | 46 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiDMC1 | D | 45,705 | 42.5 | 45 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiDMC2-2 | D | 46,693 | 42.5 | 49 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiDMC21-1 | D | 45,421 | 42.5 | 45 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiDMC24 | D | 43,168 | 43.9 | 46 | Serine | hypothetical protein | N/A | hydrolase (HAD superfamily) | TGGTATAAT |
| prophiDMC28 | D | 45,702 | 42.5 | 45 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiDMC33-2 | D | 46,690 | 42.5 | 45 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiDMC43-2 | D | 44,915 | 42.8 | 44 | Serine | transcriptional regulator AcrR family | N/A | hypothetical protein | ACTTTTGAAAAGGAGA |
| prophiDMC66-2 | D | 43,168 | 43.9 | 47 | Serine | hypothetical protein | N/A | hydrolase (HAD superfamily) | TGGTATAAT |
| Javan 6 (2603 V/R) | E | 34,100 | 40.2 | 40 | Tyrosine | alkyl hydroperoxide reductase protein F | tRNA-Cys | Na+/H+ antiporter | AATCCGTCTACCGCCT |
| prophiDMC20 | E | 36,343 | 40 | 53 | Tyrosine | alkyl hydroperoxide reductase protein F | tRNA-Cys | Na+/H+ antiporter | AATCCGTCTACCGCCT |
| prophiDMC27 | E | 36,093 | 40 | 53 | Tyrosine | alkyl hydroperoxide reductase protein F | tRNA-Cys | Na+/H+ antiporter | AATCCGTCTACCGCCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiafe-Kwakye, C.S.; Fournier, A.; Maurais, H.; Southworth, K.J.; Molloy, S.D.; Neely, M.N. Comparative Genomic Analysis of Prophages in Human Vaginal Isolates of Streptococcus agalactiae. Pathogens 2024, 13, 610. https://doi.org/10.3390/pathogens13080610
Wiafe-Kwakye CS, Fournier A, Maurais H, Southworth KJ, Molloy SD, Neely MN. Comparative Genomic Analysis of Prophages in Human Vaginal Isolates of Streptococcus agalactiae. Pathogens. 2024; 13(8):610. https://doi.org/10.3390/pathogens13080610
Chicago/Turabian StyleWiafe-Kwakye, Caitlin S., Andrew Fournier, Hannah Maurais, Katie J. Southworth, Sally D. Molloy, and Melody N. Neely. 2024. "Comparative Genomic Analysis of Prophages in Human Vaginal Isolates of Streptococcus agalactiae" Pathogens 13, no. 8: 610. https://doi.org/10.3390/pathogens13080610