
Insights into the Naso-Oropharyngeal Bacterial Composition in Suspected SARS-CoV-2 Cases

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sample Collection
2.3. SARS-CoV-2 Detection
2.4. DNA Extraction and Amplification
2.5. Library Preparation
2.6. Data Analyses
3. Results
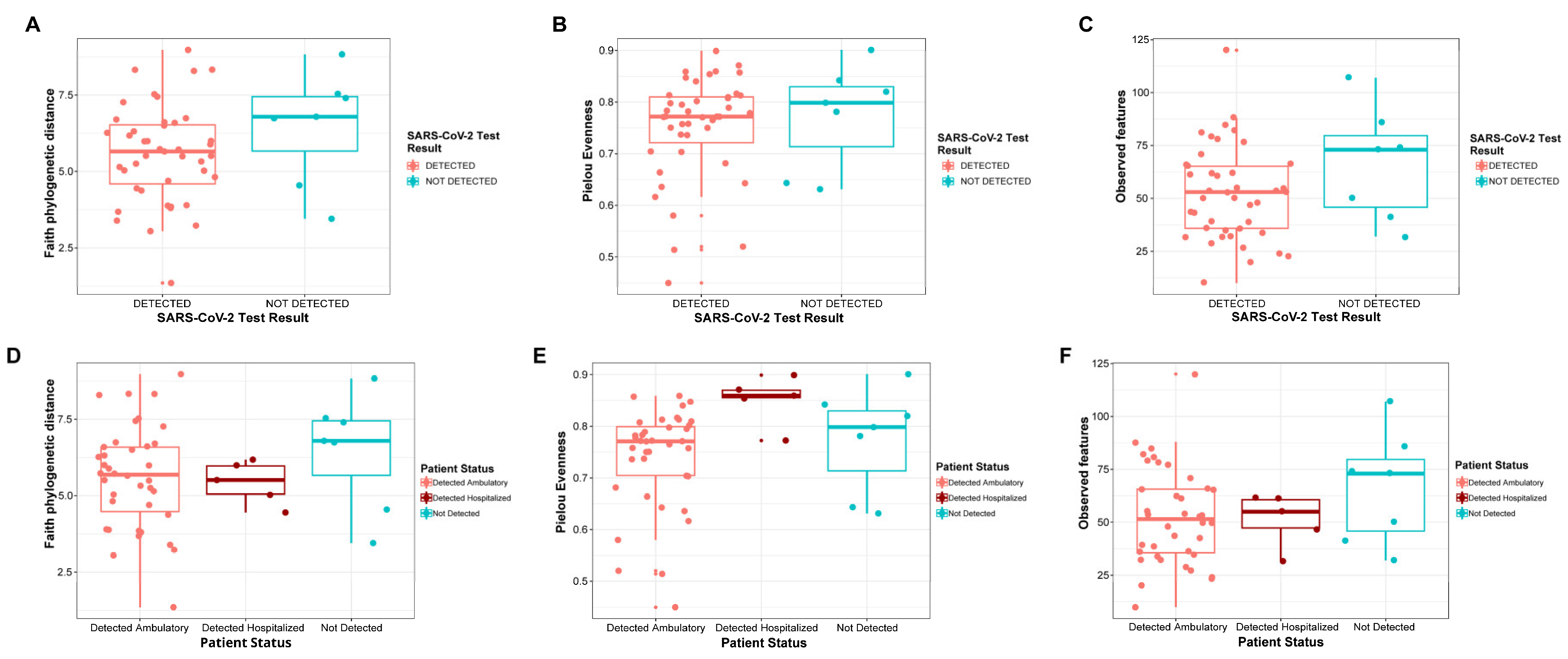
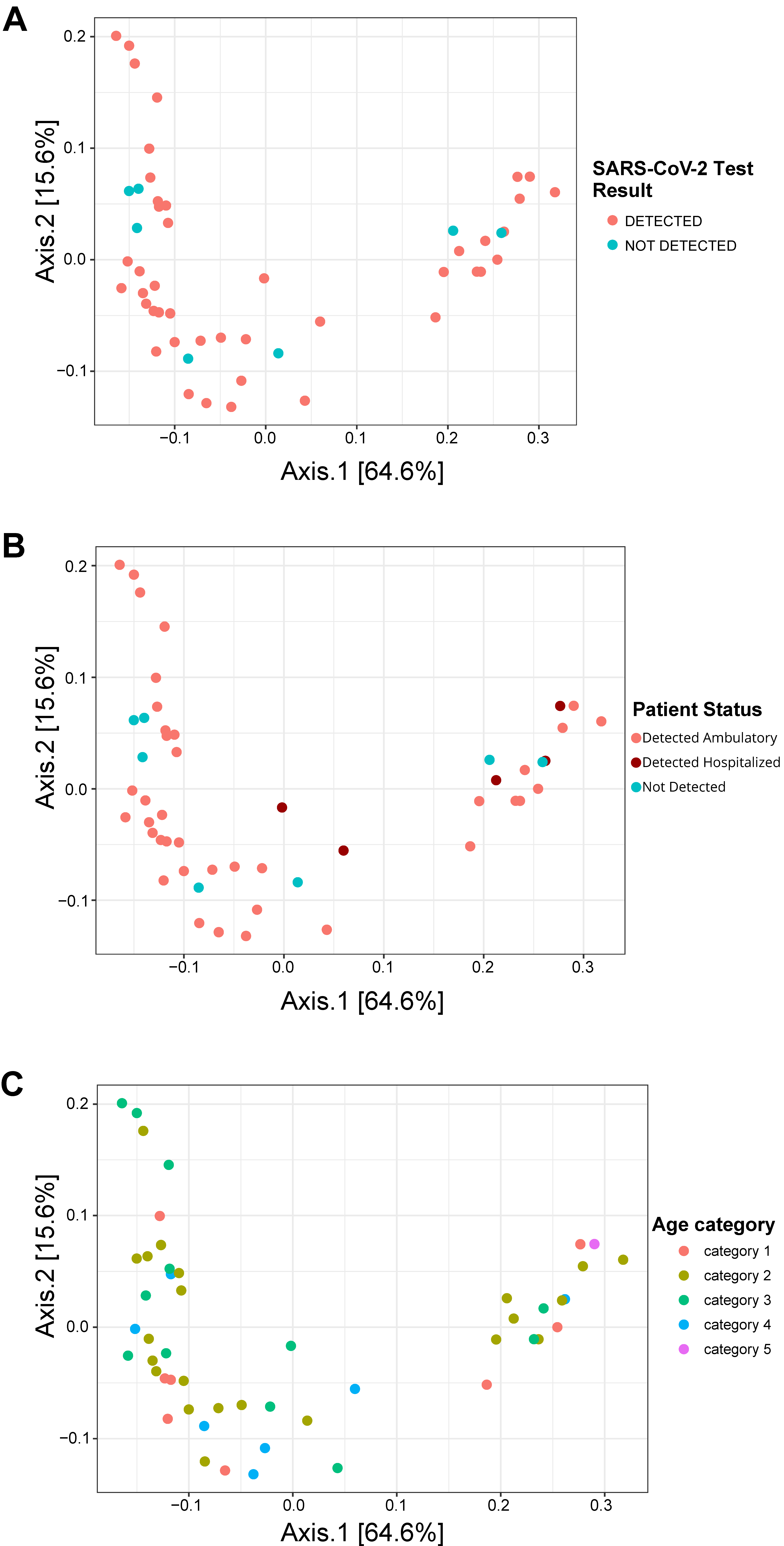
3.1. Alpha and Beta Diversity
3.2. Relative Abundance
3.3. Linear Discriminant Analysis Effect Size (LEfSe)
3.4. Bacterial Functional Analysis in the SARS-CoV-2 Test Result Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, H.; Liu, L.; Zhang, D.; Xu, J.; Dai, H.; Tang, N.; Su, X.; Cao, B. SARS-CoV-2 and Viral Sepsis: Observations and Hypotheses. Lancet 2020, 395, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.K.C.; Cheung, M.K.; Lui, G.C.Y.; Ling, L.; Chan, J.Y.K.; Ng, R.W.Y.; Chan, H.C.; Yeung, A.C.M.; Ho, W.C.S.; Boon, S.S.; et al. Limited Impact of SARS-CoV-2 on the Human Naso-Oropharyngeal Microbiota in Hospitalized Patients. Microbiol. Spectr. 2022, 10, e0219622. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, C.; Scaglione, G.L.; Testa, D.; Setaro, M.; Russo, F.; Di Domenico, C.; Atripaldi, L.; Zollo, M.; Corrado, F.; Salvatore, P.; et al. Nasal Microbiome in COVID-19: A Potential Role of Corynebacterium in Anosmia. Curr. Microbiol. 2023, 80, 53. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Ogunrinola, G.A.; Oyewale, J.O.; Oshamika, O.O.; Olasehinde, G.I. The Human Microbiome and Its Impacts on Health. Int. J. Microbiol. 2020, 2020, 8045646. [Google Scholar] [CrossRef] [PubMed]
- Man, W.H.; de Steenhuijsen Piters, W.A.A.; Bogaert, D. The Microbiota of the Respiratory Tract: Gatekeeper to Respiratory Health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Karyakarte, R.; Joshi, S.; Das, R.; Jani, K.; Shouche, Y.; Sharma, A. Nasopharyngeal Microbiome Reveals the Prevalence of Opportunistic Pathogens in SARS-CoV-2 Infected Individuals and Their Association with Host Types. Microbes Infect. 2022, 24, 104880. [Google Scholar] [CrossRef] [PubMed]
- De Steenhuijsen Piters, W.A.A.; Binkowska, J.; Bogaert, D. Early Life Microbiota and Respiratory Tract Infections. Cell Host Microbe 2020, 28, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Engen, P.A.; Naqib, A.; Jennings, C.; Green, S.J.; Landay, A.; Keshavarzian, A.; Voigt, R.M. Nasopharyngeal Microbiota in SARS-CoV-2 Positive and Negative Patients. Biol. Proced. Online 2021, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Shilts, M.H.; Rosas-Salazar, C.; Strickland, B.A.; Kimura, K.S.; Asad, M.; Sehanobish, E.; Freeman, M.H.; Wessinger, B.C.; Gupta, V.; Brown, H.M.; et al. Severe COVID-19 Is Associated with an Altered Upper Respiratory Tract Microbiome. Front. Cell. Infect. Microbiol. 2022, 11, 781968. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, X.; Liu, W.; Yang, C.; Jia, R.; Ke, Y.; Guo, J.; Jia, L.; Wang, C.; Chen, Y. Comparison of the Respiratory Tract Microbiome in Hospitalized COVID-19 Patients with Different Disease Severity. J. Med. Virol. 2022, 94, 5284–5293. [Google Scholar] [CrossRef] [PubMed]
- Merenstein, C.; Liang, G.; Whiteside, S.A.; Cobián-Güemes, A.G.; Merlino, M.S.; Taylor, L.J.; Glascock, A.; Bittinger, K.; Tanes, C.; Graham-Wooten, J.; et al. Signatures of COVID-19 Severity and Immune Response in the Respiratory Tract Microbiome. mBio 2021, 12, e01777-21. [Google Scholar] [CrossRef] [PubMed]
- Franco, D.; Gonzalez, C.; Abrego, L.E.; Carrera, J.-P.; Diaz, Y.; Caicedo, Y.; Moreno, A.; Chavarria, O.; Gondola, J.; Castillo, M.; et al. Early Transmission Dynamics, Spread, and Genomic Characterization of SARS-CoV-2 in Panama. Emerg. Infect. Dis. 2021, 27, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Vigil-De Gracia, P.; Guerrero, E.; Gaitán, M.; Fu, C.; Chen-Germán, M.; Villalobos, R.; Coronado, L.; Martínez, A.A.; Araúz, D.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 Detected in Placentas of 2 Coronavirus Disease 2019–Positive Asymptomatic Pregnant Women—Case Report. AJOG Glob. Rep. 2021, 1, 100001. [Google Scholar] [CrossRef] [PubMed]
- Díaz, Y.; Ortiz, A.; Weeden, A.; Castillo, D.; González, C.; Moreno, B.; Martínez-Montero, M.; Castillo, M.; Vasquez, G.; Sáenz, L.; et al. SARS-CoV-2 Reinfection with a Virus Harboring Mutation in the Spike and the Nucleocapsid Proteins in Panama. Int. J. Infect. Dis. 2021, 108, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Minich, J.J.; Ali, F.; Marotz, C.; Belda-Ferre, P.; Chiang, L.; Shaffer, J.P.; Carpenter, C.S.; McDonald, D.; Gilbert, J.; Allard, S.M.; et al. Feasibility of Using Alternative Swabs and Storage Solutions for Paired SARS-CoV-2 Detection and Microbiome Analysis in the Hospital Environment. Microbiome 2021, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-High-Throughput Microbial Community Analysis on the Illumina HiSeq and MiSeq Platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Quintero, I.J.; Castillo, A.M.; Mejía, L.C. Diversity and Taxonomy of Soil Bacterial Communities in Urban and Rural Mangrove Forests of the Panama Bay. Microorganisms 2022, 10, 2191. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, G.; Ewald, J.; Pang, Z.; Shiri, T.; Xia, J. MicrobiomeAnalyst 2.0: Comprehensive Statistical, Functional and Integrative Analysis of Microbiome Data. Nucleic Acids Res. 2013, 1, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for Prediction of Metagenome Functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Mai, J.; Cao, X.; Burberry, A.; Cominelli, F.; Zhang, L. Ggpicrust2: An R Package for PICRUSt2 Predicted Functional Profile Analysis and Visualization. Bioinformatics 2023, 39, btad470. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Halevi, S.; Hadar, R.; Efroni, G.; Glick Saar, E.; Keller, N.; Amir, A.; Amit, S.; Haberman, Y. SARS-CoV-2 Does Not Have a Strong Effect on the Nasopharyngeal Microbial Composition. Sci. Rep. 2021, 11, 8922. [Google Scholar] [CrossRef] [PubMed]
- De Maio, F.; Posteraro, B.; Ponziani, F.R.; Cattani, P.; Gasbarrini, A.; Sanguinetti, M. Nasopharyngeal Microbiota Profiling of SARS-CoV-2 Infected Patients. Biol. Proced. Online 2020, 22, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Wang, Y.; Shi, Z.; Zhang, L.; Ren, H.; He, W.; Zhang, Z.; Zhu, A.; Zhao, J.; Xiao, F.; et al. Characterization of Respiratory Microbial Dysbiosis in Hospitalized COVID-19 Patients. Cell Discov. 2021, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, H.H.; Fissel, J.A.; Fanelli, B.; Bergman, Y.; Gniazdowski, V.; Dadlani, M.; Carroll, K.C.; Colwell, R.R.; Simner, P.J. Metagenomic Next-Generation Sequencing of Nasopharyngeal Specimens Collected from Confirmed and Suspect COVID-19 Patients. mBio 2020, 11, e01969-20. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E. Commensal Bacteria in the Upper Respiratory Tract Regulate Susceptibility to Infection. Curr. Opin. Immunol. 2020, 66, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Ventero, M.P.; Moreno-Perez, O.; Molina-Pardines, C.; Paytuví-Gallart, A.; Boix, V.; Escribano, I.; Galan, I.; González-delaAleja, P.; López-Pérez, M.; Sánchez-Martínez, R.; et al. Nasopharyngeal Microbiota as an Early Severity Biomarker in COVID-19 Hospitalised Patients. J. Infect. 2022, 84, 329. [Google Scholar] [CrossRef] [PubMed]
- Crovetto, F.; Selma-Royo, M.; Crispi, F.; Carbonetto, B.; Pascal, R.; Larroya, M.; Casas, I.; Tortajada, M.; Escudero, N.; Muñoz-Almagro, C.; et al. Nasopharyngeal Microbiota Profiling of Pregnant Women with SARS-CoV-2 Infection. Sci. Rep. 2022, 12, 13404. [Google Scholar] [CrossRef] [PubMed]
- Schenck, L.P.; Surette, M.G.; Bowdish, D.M.E. Composition and Immunological Significance of the Upper Respiratory Tract Microbiota. FEBS Lett. 2016, 590, 3705–3720. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Song, T.; Zhou, B.; Geber, A.; Ma, Y.; Zhang, L.; Volk, M.; Kapadia, S.N.; Jenkins, S.G.; Salvatore, M.; et al. Microbial Composition of the Human Nasopharynx Varies According to Influenza Virus Type and Vaccination Status. mBio 2019, 10, e01296-19. [Google Scholar] [CrossRef]
- Ventero, M.P.; Cuadrat, R.R.C.; Vidal, I.; Andrade, B.G.N.; Molina-Pardines, C.; Haro-Moreno, J.M.; Coutinho, F.H.; Merino, E.; Regitano, L.C.A.; Silveira, C.B.; et al. Nasopharyngeal Microbial Communities of Patients Infected With SARS-CoV-2 That Developed COVID-19. Front. Microbiol. 2021, 12, 560. [Google Scholar] [CrossRef] [PubMed]
- Rueca, M.; Fontana, A.; Bartolini, B.; Piselli, P.; Mazzarelli, A.; Copetti, M.; Binda, E.; Perri, F.; Gruber, C.E.M.; Nicastri, E.; et al. Investigation of Nasal/Oropharyngeal Microbial Community of COVID-19 Patients by 16S rDNA Sequencing. Int. J. Environ. Res. Public Health 2021, 18, 2174. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, L.; Favero, C.; Solazzo, G.; Mariani, J.; Luganini, A.; Ferraroni, M.; Montomoli, E.; Milani, G.P.; Bollati, V. Nasopharyngeal Bacterial Microbiota Composition and SARS-CoV-2 IgG Antibody Maintenance in Asymptomatic/Paucisymptomatic Subjects. Front. Cell. Infect. Microbiol. 2022, 12, 882302. [Google Scholar] [CrossRef] [PubMed]
- Edouard, S.; Million, M.; Bachar, D.; Dubourg, G.; Michelle, C.; Ninove, L.; Charrel, R.; Raoult, D. The Nasopharyngeal Microbiota in Patients with Viral Respiratory Tract Infections Is Enriched in Bacterial Pathogens. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, S.; Zhang, Z.; Lee, X.; Wu, W.; Huang, Z.; Lei, Z.; Xu, W.; Chen, D.; Wu, X.; et al. Association between the Nasopharyngeal Microbiome and Metabolome in Patients with COVID-19. Synth. Syst. Biotechnol. 2021, 6, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-W.; Xu, H.-S.; Liu, S.-J. COVID-19 and Neurodegenerative Diseases. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 4535–4544. [Google Scholar] [CrossRef] [PubMed]
- Kumpitsch, C.; Koskinen, K.; Schöpf, V.; Moissl-Eichinger, C. The Microbiome of the Upper Respiratory Tract in Health and Disease. BMC Biol. 2019, 17, 87. [Google Scholar] [CrossRef] [PubMed]
- Brealey, J.C.; Sly, P.D.; Young, P.R.; Chappell, K.J. Viral Bacterial Co-Infection of the Respiratory Tract during Early Childhood. FEMS Microbiol. Lett. 2015, 362, fnv062. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.K.-K.; Zhou, J.-W.; Guan, W.; Li, S.-K.; Yang, Z.-F.; Tsui, S.K.-W. Modulation of Potential Respiratory Pathogens by pH1N1 Viral Infection. Clin. Microbiol. Infect. 2013, 19, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Liu, P.; Zhang, T.; Wu, Q.; Zeng, M.; Ma, Y.; Jin, X.; Xu, J.; Zhang, Z.; Zhang, C. Progressive Deterioration of the Upper Respiratory Tract and the Gut Microbiomes in Children during the Early Infection Stages of COVID-19. J. Genet. Genom. 2021, 48, 803–814. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atencio, L.A.; Quintero, I.J.; Almanza, A.; Eskildsen, G.; Sánchez-Gallego, J.; Herrera, M.; Fernández-Marín, H.; Loaiza, J.R.; Mejía, L.C. Insights into the Naso-Oropharyngeal Bacterial Composition in Suspected SARS-CoV-2 Cases. Pathogens 2024, 13, 615. https://doi.org/10.3390/pathogens13080615
Atencio LA, Quintero IJ, Almanza A, Eskildsen G, Sánchez-Gallego J, Herrera M, Fernández-Marín H, Loaiza JR, Mejía LC. Insights into the Naso-Oropharyngeal Bacterial Composition in Suspected SARS-CoV-2 Cases. Pathogens. 2024; 13(8):615. https://doi.org/10.3390/pathogens13080615
Chicago/Turabian StyleAtencio, Librada A., Indira J. Quintero, Alejandro Almanza, Gilberto Eskildsen, Joel Sánchez-Gallego, Mellissa Herrera, Hermógenes Fernández-Marín, José R. Loaiza, and Luis C. Mejía. 2024. "Insights into the Naso-Oropharyngeal Bacterial Composition in Suspected SARS-CoV-2 Cases" Pathogens 13, no. 8: 615. https://doi.org/10.3390/pathogens13080615
APA StyleAtencio, L. A., Quintero, I. J., Almanza, A., Eskildsen, G., Sánchez-Gallego, J., Herrera, M., Fernández-Marín, H., Loaiza, J. R., & Mejía, L. C. (2024). Insights into the Naso-Oropharyngeal Bacterial Composition in Suspected SARS-CoV-2 Cases. Pathogens, 13(8), 615. https://doi.org/10.3390/pathogens13080615