
Oncolytic Viruses in Ovarian Cancer: Where Do We Stand? A Narrative Review

 ,
,  ,
,  , , ,
, , ,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
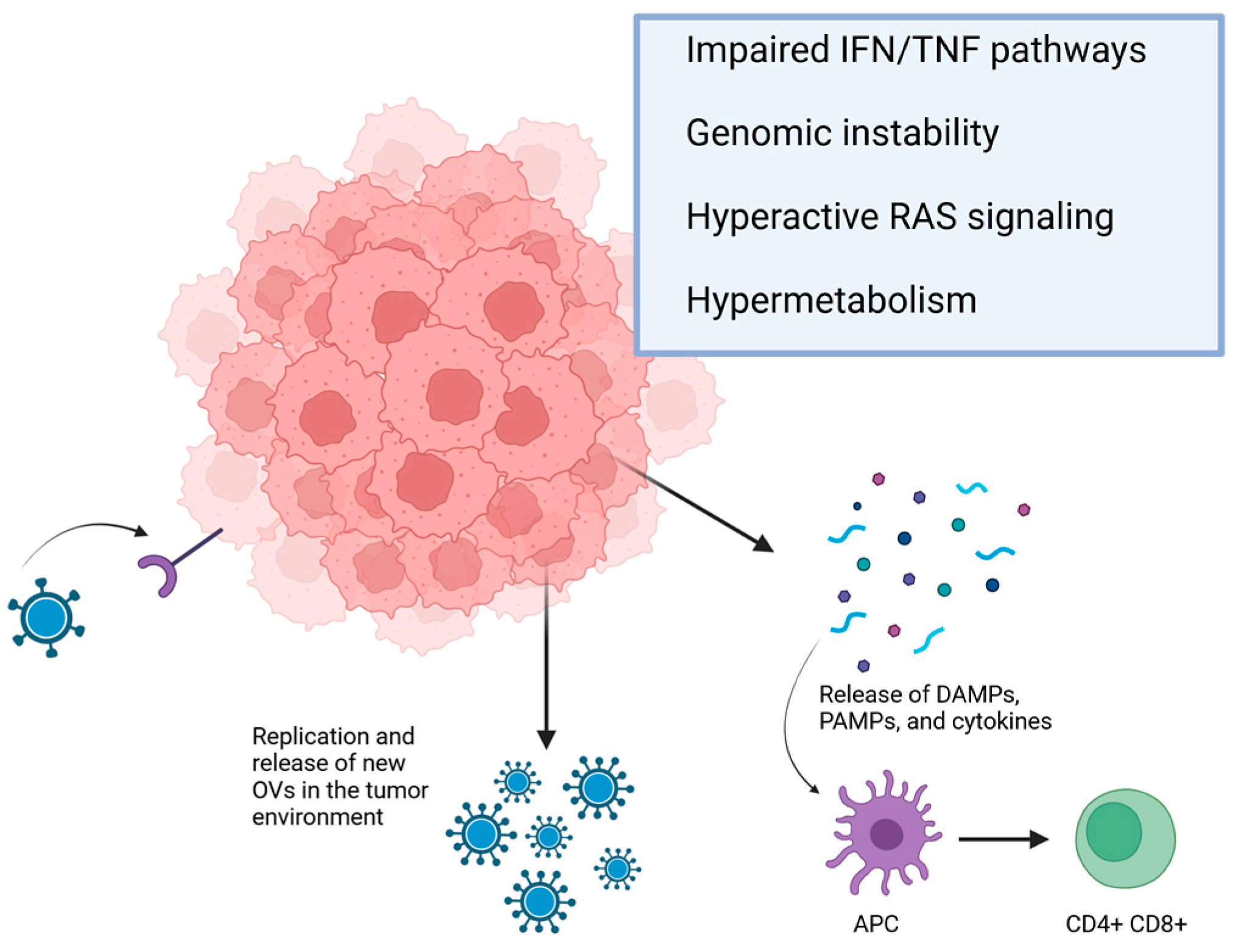
2. Overview of Mechanism of Action
3. Preclinical Studies on Oncolytic Viruses in Ovarian Cancer
3.1. Adenovirus
3.1.1. Immune Response Modulation
3.1.2. Coxsackievirus and Adenovirus Receptor (CAR)
3.1.3. Retinoblastoma (Rb) Pathway
3.1.4. Smac/DIABLO
3.1.5. Homologous Recombination Deficiency (HRD) Status
3.1.6. Using Neural Stem Cells to Deliver Adenovirus
3.1.7. Angiogenesis
3.2. Vesicular Stomatitis Virus
3.2.1. Immune Response Modulation
3.2.2. VSV Matrix Protein (MP)
3.3. Herpes Simplex Virus
3.3.1. Immune Response Modulation
3.3.2. Angiogenesis
3.3.3. HER2 Receptor
3.4. Measles Virus
3.4.1. Immune Response Modulation
3.4.2. Oncolytic Activity
3.5. Vaccinia Virus
3.5.1. Immune Response Modulation
3.5.2. Oncolytic Activity
3.6. Myxoma Virus
3.6.1. Immune Response Modulation
3.6.2. Oncolytic Activity
3.7. Reovirus
3.7.1. Immune Response Modulation
3.7.2. Oncolytic Activity
4. Clinical Trials Evaluating Oncolytic Virus for Treating Ovarian Cancer

{kind=link}
| Ref. and Year of Publication | Treatment | Study Design | Clinical Setting | Number of Patients | Clinical Endpoints | Results |
|---|---|---|---|---|---|---|
| Vasey et al., 2002 [177] | Intraperitoneal E1B-55-kd-Gene–Deleted Adenovirus ONYX-015 (dl1520) | Phase I | Recurrent/refractory EOC | 16 | Feasibility, safety | No evidence of clinical response |
| Kimball et al., 2010 [179] | CRAd, Ad5-Δ24-RGD | Phase I | Persistent/recurrent EOC | 21 | Feasibility, safety | 15 SD |
| Cerullo et al., 2010 [70] | Ad5-D24-GMCSF | Phase I | Refractory solid tumors, compassionate use | 4 | Feasibility, safety | 1 CR, 1 SD, 1 PR |
| Koski et al., 2010 [178] | Ad5/3-D24-GMCSF | Phase I | Refractory solid tumors, compassionate use | 4 | Feasibility, safety | 1 SD |
| Galanis et al., 2010 [154] | MV-CEA | Phase I | Progressive/recurrent/refractory EOC | 21 | Objective response | 12 SD, median survival 12.5 months |
| Breitbach et al., 2011 [180] | JX-594 Pox-virus | Phase I | Refractory solid tumors | 2 | Feasibility, safety | 2 SD |
| Kim et al.2013 [95] | Intraperitoneal Ad5/3-Δ24 | Phase I | Recurrent EOC | 10 | Feasibility, safety | 6 SD, 2 PD |
| Cohn et al., 2017 [186] | Reolysin + paclitaxel vs. paclitaxel alone | Phase IIB | Recurrent or Persistent Ovarian Epithelial, Fallopian Tube, or Primary Peritoneal Cancer | 108 | PFS | Median PFS: 4.3 months for paclitaxel; 4.4 months for paclitaxel + reovirus |
| Lauer et al., 2018 [181] | Vaccinia virus GL-ONC1 | Phase I | Advanced peritoneal carcinomatosis | 3 | Feasibility, safety | 2 PD, 1 NA |
| Holloway et al., 2018 [182] | VACV GL-ONC1 | Phase I | Heavy pretreated platinum-refractory/resistant EOC | 11 | Safety, survival | DCR (PR + SD): 55% |
| Moreno et al., 2021 [187] | Enadenotucirev + paclitaxel | Phase I | Recurrent pretreated platinum-resistant EOC | 38 | PFS | 4-month PFS rate: 64% (median 6.2 months); ORR: 10% |
| Pakola et al., 2024 [183] | TILT-123 adenovirus | Phase I | Refractory solid tumors | 3 | Safety, tumor response | NA |
| Michael et al., 2024 [185] | MVA-5T4 vs. placebo | Phase II | Relapsed EOC | 94 | PFS | 3 months with MVA-5T4; 3 months with placebo |
| Trial | Study Start | Treatment | Study Design | Clinical Setting | Enrollment | Clinical Endpoints |
|---|---|---|---|---|---|---|
| NCT02068794 | 2014 April 25 | MV-NIS infected mesenchymal stem cells | Phase I/II | Recurrent EOC | 34 | Safety/mean 12-months OS |
| NCT02364713 | 13 March 2015 | MV-NIS vs. standard chemotherapy | Phase II | Platinum-resistant EOC | 66 | PFS |
| NCT02759588 | May 2016 | VACV GL-ONC1 | Phase I/II | Recurrent or Refractory EOC | 46 | Safety, PFS |
| NCT02963831 | 7 September 2017 | ONCOS-102; Ad5/3-D24-GMCSF + durvalumab | Phase I/II | Advanced peritoneal malignancies | 67 | ORR |
| NCT03225989 | 1 March 2018 | LOAd703 | Phase I/II | Advanced cancers (including EOC) | 47 | Safety, PFS, OS |
| NCT05180851 | 30 November 2021 | L-IFN adenovirus | Phase I | Relapsed/refractory solid cancers (including EOC) | 28 | Safety |
| NCT05271318 | 17 May 2022 | Adenovirus (TILT-123) with pembrolizumab or pembrolizumab and pegylated liposomal doxorubicin | Phase I | Platinum-resistant or refractory EOC | 29 | Safety |
| NCT05281471 | 31 August 2022 | VACV + chemotherapy + bevacizumab | Phase III | Platinum-resistant/Refractory EOC | 186 | PFS |
| NCT05801783 | 2 December 2022 | HSV-1 R130 | Phase I | Relapsed/refractory EOC | 10 | Safety |
| NCT05684731 | 1 February 2023 | VACV K1 | Phase I | Recurrent or refractory EOC | 30 | Safety, efficacy |
| NCT06508307 | 26 April 2023 | VACV GC001 | Phase I | Advanced solid tumors (including EOC) | 21 | Safety |
5. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. The Dualistic Model of Ovarian Carcinogenesis. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Meinhold-Heerlein, I.; Fotopoulou, C.; Harter, P.; Kurzeder, C.; Mustea, A.; Wimberger, P.; Hauptmann, S.; Sehouli, J. The New WHO Classification of Ovarian, Fallopian Tube, and Primary Peritoneal Cancer and Its Clinical Implications. Arch. Gynecol. Obstet. 2016, 293, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Duska, L.R.; Kohn, E.C. The New Classifications of Ovarian, Fallopian Tube, and Primary Peritoneal Cancer and Their Clinical Implications. Ann. Oncol. 2017, 28, viii8–viii12. [Google Scholar] [CrossRef]
- Lliberos, C.; Richardson, G.; Papa, A. Oncogenic Pathways and Targeted Therapies in Ovarian Cancer. Biomolecules 2024, 14, 585. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial Ovarian Cancer: Evolution of Management in the Era of Precision Medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Kroeger, P.T.; Drapkin, R. Pathogenesis and Heterogeneity of Ovarian Cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef]
- Pesenti, C.; Beltrame, L.; Velle, A.; Fruscio, R.; Jaconi, M.; Borella, F.; Cribiù, F.M.; Calura, E.; Venturini, L.V.; Lenoci, D.; et al. Copy Number Alterations in Stage I Epithelial Ovarian Cancer Highlight Three Genomic Patterns Associated with Prognosis. Eur. J. Cancer 2022, 171, 85–95. [Google Scholar] [CrossRef]
- D’Orazi, G. Recent Advances in P53. Biomolecules 2021, 11, 211. [Google Scholar] [CrossRef]
- McConechy, M.K.; Ding, J.; Senz, J.; Yang, W.; Melnyk, N.; Tone, A.A.; Prentice, L.M.; Wiegand, K.C.; McAlpine, J.N.; Shah, S.P.; et al. Ovarian and Endometrial Endometrioid Carcinomas Have Distinct CTNNB1 and PTEN Mutation Profiles. Mod. Pathol. 2014, 27, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Itamochi, H.; Oishi, T.; Oumi, N.; Takeuchi, S.; Yoshihara, K.; Mikami, M.; Yaegashi, N.; Terao, Y.; Takehara, K.; Ushijima, K.; et al. Whole-Genome Sequencing Revealed Novel Prognostic Biomarkers and Promising Targets for Therapy of Ovarian Clear Cell Carcinoma. Br. J. Cancer 2017, 117, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Driva, T.S.; Schatz, C.; Haybaeck, J. Endometriosis-Associated Ovarian Carcinomas: How PI3K/AKT/mTOR Pathway Affects Their Pathogenesis. Biomolecules 2023, 13, 1253. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Luvero, D.; Shafer, A.; O’Connor, D.; Mangili, G.; Friedlander, M.; Pfisterer, J.; Mirza, M.R.; Kim, J.-W.; Alexandre, J.; et al. Gynecologic Cancer InterGroup (GCIG) Consensus Review for Mucinous Ovarian Carcinoma. Int. J. Gynecol. Cancer 2014, 24, S14–S19. [Google Scholar] [CrossRef]
- Borella, F.; Mitidieri, M.; Cosma, S.; Benedetto, C.; Bertero, L.; Fucina, S.; Ray-Coquard, I.; Carapezzi, A.; Ferraioli, D. Update on Prognostic and Predictive Markers in Mucinous Ovarian Cancer. Cancers 2023, 15, 1172. [Google Scholar] [CrossRef]
- Slomovitz, B.; Gourley, C.; Carey, M.S.; Malpica, A.; Shih, I.-M.; Huntsman, D.; Fader, A.N.; Grisham, R.N.; Schlumbrecht, M.; Sun, C.C.; et al. Low-Grade Serous Ovarian Cancer: State of the Science. Gynecol. Oncol. 2020, 156, 715–725. [Google Scholar] [CrossRef]
- Perrone, C.; Angioli, R.; Luvero, D.; Giannini, A.; Di Donato, V.; Cuccu, I.; Muzii, L.; Raspagliesi, F.; Bogani, G. Targeting BRAF Pathway in Low-Grade Serous Ovarian Cancer. J. Gynecol. Oncol. 2024, 35, e104. [Google Scholar] [CrossRef]
- Yoneoka, Y.; Ishikawa, M.; Uehara, T.; Shimizu, H.; Uno, M.; Murakami, T.; Kato, T. Treatment Strategies for Patients with Advanced Ovarian Cancer Undergoing Neoadjuvant Chemotherapy: Interval Debulking Surgery or Additional Chemotherapy? J. Gynecol. Oncol. 2019, 30, e81. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Raja, F.A.; Fotopoulou, C.; Gonzalez-Martin, A.; Colombo, N.; Sessa, C. Newly Diagnosed and Relapsed Epithelial Ovarian Carcinoma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2013, 24, vi24–vi32. [Google Scholar] [CrossRef]
- Mao, C.-L.; Seow, K.-M.; Chen, K.-H. The Utilization of Bevacizumab in Patients with Advanced Ovarian Cancer: A Systematic Review of the Mechanisms and Effects. Int. J. Mol. Sci. 2022, 23, 6911. [Google Scholar] [CrossRef]
- Liu, S.; Kasherman, L.; Fazelzad, R.; Wang, L.; Bouchard-Fortier, G.; Lheureux, S.; Krzyzanowska, M.K. The Use of Bevacizumab in the Modern Era of Targeted Therapy for Ovarian Cancer: A Systematic Review and Meta-Analysis. Gynecol. Oncol. 2021, 161, 601–612. [Google Scholar] [CrossRef] [PubMed]
- DiSilvestro, P.; Colombo, N.; Harter, P.; González-Martín, A.; Ray-Coquard, I.; Coleman, R.L. Maintenance Treatment of Newly Diagnosed Advanced Ovarian Cancer: Time for a Paradigm Shift? Cancers 2021, 13, 5756. [Google Scholar] [CrossRef] [PubMed]
- Goh, J.C.H.; Gourley, C.; Tan, D.S.P.; Nogueira-Rodrigues, A.; Elghazaly, H.; Edy Pierre, M.; Giornelli, G.; Kim, B.-G.; Morales–Vasquez, F.; Tyulyandina, A. Optimizing Treatment Selection and Sequencing Decisions for First-Line Maintenance Therapy of Newly Diagnosed Advanced Ovarian Cancer—International Considerations amongst Upper Middle- and High-Income Countries (UMIC and HIC). Gynecol. Oncol. Rep. 2022, 42, 101028. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Ou, S.; Wei, H.; Qin, X.; Jiang, Q. Comparative Efficacy and Safety of Poly (ADP-Ribose) Polymerase Inhibitors in Patients With Ovarian Cancer: A Systematic Review and Network Meta-Analysis. Front. Oncol. 2022, 12, 815265. [Google Scholar] [CrossRef]
- Tattersall, A.; Ryan, N.; Wiggans, A.J.; Rogozińska, E.; Morrison, J. Poly(ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer. Cochrane Database Syst. Rev. 2022, 2022, CD007929. [Google Scholar] [CrossRef]
- Nag, S.; Aggarwal, S.; Rauthan, A.; Warrier, N. Maintenance Therapy for Newly Diagnosed Epithelial Ovarian Cancer– a Review. J. Ovarian Res. 2022, 15, 88. [Google Scholar] [CrossRef]
- Borella, F.; Ghisoni, E.; Giannone, G.; Cosma, S.; Benedetto, C.; Valabrega, G.; Katsaros, D. Immune Checkpoint Inhibitors in Epithelial Ovarian Cancer: An Overview on Efficacy and Future Perspectives. Diagnostics 2020, 10, 146. [Google Scholar] [CrossRef]
- Dumitru, A.; Dobrica, E.-C.; Croitoru, A.; Cretoiu, S.M.; Gaspar, B.S. Focus on PD-1/PD-L1 as a Therapeutic Target in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 12067. [Google Scholar] [CrossRef]
- Peng, Z.; Li, M.; Li, H.; Gao, Q. PD-1/PD-L1 Immune Checkpoint Blockade in Ovarian Cancer: Dilemmas and Opportunities. Drug Discov. Today 2023, 28, 103666. [Google Scholar] [CrossRef]
- Yoon, W.-H.; DeFazio, A.; Kasherman, L. Immune Checkpoint Inhibitors in Ovarian Cancer: Where Do We Go from Here? Cancer Drug Resist. 2023, 6, 358–377. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Li, Z.; Chiocca, E.A.; Caligiuri, M.A.; Yu, J. The Emerging Field of Oncolytic Virus-Based Cancer Immunotherapy. Trends Cancer 2023, 9, 122–139. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Shen, Y.; Liang, T. Oncolytic Virotherapy: Basic Principles, Recent Advances and Future Directions. Signal Transduct. Target. Ther. 2023, 8, 156. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors in Gene Therapy: Where Do We Stand in 2023? Viruses 2023, 15, 698. [Google Scholar] [CrossRef]
- Volovat, S.R.; Scripcariu, D.V.; Vasilache, I.A.; Stolniceanu, C.R.; Volovat, C.; Augustin, I.G.; Volovat, C.C.; Ostafe, M.-R.; Andreea-Voichița, S.-G.; Bejusca-Vieriu, T.; et al. Oncolytic Virotherapy: A New Paradigm in Cancer Immunotherapy. Int. J. Mol. Sci. 2024, 25, 1180. [Google Scholar] [CrossRef]
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The Advent of Oncolytic Virotherapy in Oncology: The Rigvir® Story. Eur. J. Clin. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and Its Update in China. Curr. Cancer Drug Targets 2018, 18, 171–176. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Li, S.; Tong, J.; Rahman, M.M.; Shepherd, T.G.; McFadden, G. Oncolytic Virotherapy for Ovarian Cancer. Oncolytic Virother. 2012, 2012, 1–21. [Google Scholar] [CrossRef]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical Landscape of Oncolytic Virus Research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic Virus Therapy: A New Era of Cancer Treatment at Dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.-C.; Cho, C.-F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting Tumor-Specific Defects in the Interferon Pathway with a Previously Unknown Oncolytic Virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Li, Q.; Tan, F.; Wang, Y.; Liu, X.; Kong, X.; Meng, J.; Yang, L.; Cen, S. The Gamble between Oncolytic Virus Therapy and IFN. Front. Immunol. 2022, 13, 971674. [Google Scholar] [CrossRef]
- Lemos De Matos, A.; Franco, L.S.; McFadden, G. Oncolytic Viruses and the Immune System: The Dynamic Duo. Mol. Ther. Methods Clin. Dev. 2020, 17, 349–358. [Google Scholar] [CrossRef]
- Wojton, J.; Kaur, B. Impact of Tumor Microenvironment on Oncolytic Viral Therapy. Cytokine Growth Factor Rev. 2010, 21, 127–134. [Google Scholar] [CrossRef]
- Rahman, M.M.; McFadden, G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers 2021, 13, 5452. [Google Scholar] [CrossRef]
- Garant, K.A.; Shmulevitz, M.; Pan, L.; Daigle, R.M.; Ahn, D.-G.; Gujar, S.A.; Lee, P.W.K. Oncolytic Reovirus Induces Intracellular Redistribution of Ras to Promote Apoptosis and Progeny Virus Release. Oncogene 2016, 35, 771–782. [Google Scholar] [CrossRef]
- Thirukkumaran, C.; Morris, D.G. Oncolytic Viral Therapy Using Reovirus. In Gene Therapy of Solid Cancers; Walther, W., Stein, U., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; Volume 1317, pp. 187–223. ISBN 978-1-4939-2726-5. [Google Scholar]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental Therapy of Human Glioma by Means of a Genetically Engineered Virus Mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef]
- Todo, T.; Rabkin, S.D.; Chahlavi, A.; Martuza, R.L. Corticosteroid Administration Does Not Affect Viral Oncolytic Activity, but Inhibits Antitumor Immunity in Replication-Competent Herpes Simplex Virus Tumor Therapy. Hum. Gene Ther. 1999, 10, 2869–2878. [Google Scholar] [CrossRef] [PubMed]
- Todo, T.; Rabkin, S.D.; Sundaresan, P.; Wu, A.; Meehan, K.R.; Herscowitz, H.B.; Martuza, R.L. Systemic Antitumor Immunity in Experimental Brain Tumor Therapy Using a Multimutated, Replication-Competent Herpes Simplex Virus. Hum. Gene Ther. 1999, 10, 2741–2755. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; Fishbein, M.; Echavarria, M. Adenovirus. Semin. Respir. Crit. Care Med. 2011, 32, 494–511. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An Adenovirus Mutant That Replicates Selectively in P53- Deficient Human Tumor Cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef]
- Peng, Y.; Bai, J.; Li, W.; Su, Z.; Cheng, X. Advancements in P53-Based Anti-Tumor Gene Therapy Research. Molecules 2024, 29, 5315. [Google Scholar] [CrossRef]
- Cui, Y.; Chen, X.; Li, W.; Li, S.; Jin, N.; Li, X.; Li, Y.; Yue, Y. Ad-VT Causes Ovarian Cancer A2780 Cell Death via Mitochondrial Apoptosis and Autophagy Pathways. Transl. Oncol. 2024, 48, 102067. [Google Scholar] [CrossRef]
- Dai, Y.; Zhao, X.-J.; Li, F.; Yuan, Y.; Yan, D.-M.; Cao, H.; Huang, X.-Y.; Hu, Z.; Ma, D.; Gao, Q.-L. Truncated Bid Regulates Cisplatin Response via Activation of Mitochondrial Apoptosis Pathway in Ovarian Cancer. Hum. Gene Ther. 2020, 31, 325–338. [Google Scholar] [CrossRef]
- Zhao, T.; Ye, W.; Zhang, R.; Zhu, X.; Shi, Q.; Xu, X.; Chen, W.; Xu, L.; Meng, Y. Dual-regulated Oncolytic Adenovirus Carrying ERCC1 -siRNA Gene Possesses Potent Antitumor Effect on Ovarian Cancer Cells. Mol. Med. Rep. 2024, 30, 120. [Google Scholar] [CrossRef]
- Chen, Y.; Li, N.; Yang, J.; Li, K.; Tang, M.; Zhao, X.; Guo, W.; Tong, A.; Nie, C.; Peng, Y.; et al. PUMA Overexpression Dissociates Thioredoxin from ASK1 to Activate the JNK/BCL-2/BCL-XL Pathway Augmenting Apoptosis in Ovarian Cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166553. [Google Scholar] [CrossRef]
- Heise, C.; Ganly, I.; Kim, Y.T.; Sampson-Johannes, A.; Brown, R.; Kirn, D. Efficacy of a Replication-Selective Adenovirus against Ovarian Carcinomatosis Is Dependent on Tumor Burden, Viral Replication and P53 Status. Gene Ther. 2000, 7, 1925–1929. [Google Scholar] [CrossRef]
- Taipale, K.; Tähtinen, S.; Havunen, R.; Koski, A.; Liikanen, I.; Pakarinen, P.; Koivisto-Korander, R.; Kankainen, M.; Joensuu, T.; Kanerva, A.; et al. Interleukin 8 Activity Influences the Efficacy of Adenoviral Oncolytic Immunotherapy in Cancer Patients. Oncotarget 2018, 9, 6320–6335. [Google Scholar] [CrossRef] [PubMed]
- Salako, M.A.; Kulbe, H.; Ingemarsdotter, C.K.; Pirlo, K.J.; Williams, S.L.; Lockley, M.; Balkwill, F.R.; McNeish, I.A. Inhibition of the Inflammatory Cytokine TNF-α Increases Adenovirus Activity in Ovarian Cancer via Modulation of cIAP1/2 Expression. Mol. Ther. 2011, 19, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Browne, A.; Tookman, L.A.; Ingemarsdotter, C.K.; Bouwman, R.D.; Pirlo, K.; Wang, Y.; McNeish, I.A.; Lockley, M. Pharmacological Inhibition of Β3 Integrin Reduces the Inflammatory Toxicities Caused by Oncolytic Adenovirus without Compromising Anticancer Activity. Cancer Res. 2015, 75, 2811–2821. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef]
- Havunen, R.; Siurala, M.; Sorsa, S.; Grönberg-Vähä-Koskela, S.; Behr, M.; Tähtinen, S.; Santos, J.M.; Karell, P.; Rusanen, J.; Nettelbeck, D.M.; et al. Oncolytic Adenoviruses Armed with Tumor Necrosis Factor Alpha and Interleukin-2 Enable Successful Adoptive Cell Therapy. Mol. Ther. Oncolytics 2017, 4, 77–86. [Google Scholar] [CrossRef]
- Santos, J.M.; Heiniö, C.; Cervera-Carrascon, V.; Quixabeira, D.C.A.; Siurala, M.; Havunen, R.; Butzow, R.; Zafar, S.; De Gruijl, T.; Lassus, H.; et al. Oncolytic Adenovirus Shapes the Ovarian Tumor Microenvironment for Potent Tumor-Infiltrating Lymphocyte Tumor Reactivity. J. Immunother. Cancer 2020, 8, e000188. [Google Scholar] [CrossRef]
- Gonzalez-Pastor, R.; Goedegebuure, P.S.; Curiel, D.T. Understanding and Addressing Barriers to Successful Adenovirus-Based Virotherapy for Ovarian Cancer. Cancer Gene Ther. 2021, 28, 375–389. [Google Scholar] [CrossRef]
- Heiniö, C.; Havunen, R.; Santos, J.; De Lint, K.; Cervera-Carrascon, V.; Kanerva, A.; Hemminki, A. TNFa and IL2 Encoding Oncolytic Adenovirus Activates Pathogen and Danger-Associated Immunological Signaling. Cells 2020, 9, 798. [Google Scholar] [CrossRef]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Särkioja, M.; et al. Oncolytic Adenovirus Coding for Granulocyte Macrophage Colony-Stimulating Factor Induces Antitumoral Immunity in Cancer Patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef]
- Vassilev, L.; Ranki, T.; Joensuu, T.; Jäger, E.; Karbach, J.; Wahle, C.; Partanen, K.; Kairemo, K.; Alanko, T.; Turkki, R.; et al. Repeated Intratumoral Administration of ONCOS-102 Leads to Systemic Antitumor CD8+ T-Cell Response and Robust Cellular and Transcriptional Immune Activation at Tumor Site in a Patient with Ovarian Cancer. Oncoimmunology 2015, 4, e1017702. [Google Scholar] [CrossRef]
- Shaw, A.R.; Suzuki, M. Immunology of Adenoviral Vectors in Cancer Therapy. Mol. Ther. Methods Clin. Dev. 2019, 15, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lv, S.; Liu, P.; Ye, Z.; Yang, H.; Li, L.; Zhu, H.; Wang, Y.; Cui, L.; Jiang, D.; et al. A SIRPα-Fc Fusion Protein Enhances the Antitumor Effect of Oncolytic Adenovirus against Ovarian Cancer. Mol. Oncol. 2020, 14, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.Y.L.; Ennis, D.P.; Kennedy, P.R.; Hansell, C.; Dowson, S.; Farquharson, M.; Spiliopoulou, P.; Nautiyal, J.; McNamara, S.; Carlin, L.M.; et al. NK Cells Augment Oncolytic Adenovirus Cytotoxicity in Ovarian Cancer. Mol. Ther. Oncolytics 2020, 16, 289–301. [Google Scholar] [CrossRef]
- Basnet, S.; Van Der Heijden, M.; Quixabeira, D.C.A.; Jirovec, E.; Grönberg-Vähä-Koskela, S.A.M.; Clubb, J.H.A.; Kanerva, A.; Pakola, S.; Haybout, L.; Arias, V.; et al. Overcoming Effector T Cell Exhaustion in Ovarian Cancer Ascites with a Novel Adenovirus Encoding for a MUC1 Bispecific Antibody Engager and IL-2 Cytokine. Mol. Ther. 2024, 32, 3114–3127. [Google Scholar] [CrossRef]
- Basnet, S.; Santos, J.M.; Quixabeira, D.C.A.; Clubb, J.H.A.; Grönberg-Vähä-Koskela, S.A.M.; Arias, V.; Pakola, S.; Kudling, T.V.; Heiniö, C.; Havunen, R.; et al. Oncolytic Adenovirus Coding for Bispecific T Cell Engager against Human MUC-1 Potentiates T Cell Response against Solid Tumors. Mol. Ther. Oncolytics 2023, 28, 59–73. [Google Scholar] [CrossRef]
- Quixabeira, D.C.A.; Jirovec, E.; Pakola, S.; Havunen, R.; Basnet, S.; Santos, J.M.; Kudling, T.V.; Clubb, J.H.A.; Haybout, L.; Arias, V.; et al. Improving the Cytotoxic Response of Tumor-Infiltrating Lymphocytes towards Advanced Stage Ovarian Cancer with an Oncolytic Adenovirus Expressing a Human vIL-2 Cytokine. Cancer Gene Ther. 2023, 30, 1543–1553. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, X.; Wu, H.; Zhao, C.; Chen, J.; Li, R.; Yan, S.; Li, Y.; Zhang, Q.; Song, K.; et al. Oncolytic Adenovirus with MUC16-BiTE Shows Enhanced Antitumor Immune Response by Reversing the Tumor Microenvironment in PDX Model of Ovarian Cancer. OncoImmunology 2022, 11, 2096362. [Google Scholar] [CrossRef]
- Heiniö, C.; Clubb, J.; Kudling, T.; Quixabeira, D.; Cervera-Carrascon, V.; Havunen, R.; Grönberg-Vähä-Koskela, S.; Santos, J.M.; Tapper, J.; Kanerva, A.; et al. Effective Combination Immunotherapy with Oncolytic Adenovirus and Anti-PD-1 for Treatment of Human and Murine Ovarian Cancers. Diseases 2022, 10, 52. [Google Scholar] [CrossRef]
- Shi, G.; Shi, P.; Yu, Y.; Xu, J.; Ma, J.; Zhang, Y.; Dong, Z.; Shen, L.; Dai, L.; Cheng, L.; et al. Oncolytic Adenovirus Inhibits Malignant Ascites of Advanced Ovarian Cancer by Reprogramming the Ascitic Immune Microenvironment. Mol. Ther. Oncolytics 2021, 23, 488–500. [Google Scholar] [CrossRef]
- Kim, J.-S.; Lee, S.-H.; Cho, Y.-S.; Choi, J.-J.; Kim, Y.H.; Lee, J.-H. Enhancement of the Adenoviral Sensitivity of Human Ovarian Cancer Cells by Transient Expression of Coxsackievirus and Adenovirus Receptor (CAR). Gynecol. Oncol. 2002, 85, 260–265. [Google Scholar] [CrossRef]
- Zeimet, A.; Müller-Holzner, E.; Schuler, A.; Hartung, G.; Berger, J.; Hermann, M.; Widschwendter, M.; Bergelson, J.; Marth, C. Determination of Molecules Regulating Gene Delivery Using Adenoviral Vectors in Ovarian Carcinomas. Gene Ther. 2002, 9, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Vanderkwaak, T.J.; Wang, M.; Gómez-Navarro, J.; Rancourt, C.; Dmitriev, I.; Krasnykh, V.; Barnes, M.; Siegal, G.P.; Alvarez, R.; Curiel, D.T. An Advanced Generation of Adenoviral Vectors Selectively Enhances Gene Transfer for Ovarian Cancer Gene Therapy Approaches. Gynecol. Oncol. 1999, 74, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An Adenovirus Vector with Genetically Modified Fibers Demonstrates Expanded Tropism via Utilization of a Coxsackievirus and Adenovirus Receptor-Independent Cell Entry Mechanism. J. Virol. 1998, 72, 9706–9713. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Han, T.; Lam, J.T.; Leath, C.A.; Dmitriev, I.; Kashentseva, E.; Barnes, M.N.; Alvarez, R.D.; Curiel, D.T. Preclinical Evaluation of a Class of Infectivity-Enhanced Adenoviral Vectors in Ovarian Cancer Gene Therapy. Gene Ther. 2004, 11, 874–878. [Google Scholar] [CrossRef]
- Gamble, L.J.; Ugai, H.; Wang, M.; Borovjagin, A.V.; Matthews, Q.L. Therapeutic Efficacy of an Oncolytic Adenovirus Containing RGD Ligand in Minor Capsid Protein IX and Fiber, Δ24DoubleRGD, in an Ovarian Cancer Model. J. Mol. Biochem. 2012, 1, 26–39. [Google Scholar]
- Bauerschmitz, G.J.; Lam, J.T.; Kanerva, A.; Suzuki, K.; Nettelbeck, D.M.; Dmitriev, I.; Krasnykh, V.; Mikheeva, G.V.; Barnes, M.N.; Alvarez, R.D.; et al. Treatment of Ovarian Cancer with a Tropism Modified Oncolytic Adenovirus. Cancer Res. 2002, 62, 1266–1270. [Google Scholar]
- Strauss, R.; Sova, P.; Liu, Y.; Li, Z.Y.; Tuve, S.; Pritchard, D.; Brinkkoetter, P.; Möller, T.; Wildner, O.; Pesonen, S.; et al. Epithelial Phenotype Confers Resistance of Ovarian Cancer Cells to Oncolytic Adenoviruses. Cancer Res. 2009, 69, 5115–5125. [Google Scholar] [CrossRef]
- Wang, H.; Li, Z.-Y.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 Is a Receptor for Adenovirus Serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef]
- Kuryk, L.; Møller, A.-S.W. Chimeric Oncolytic Ad5/3 Virus Replicates and Lyses Ovarian Cancer Cells through Desmoglein-2 Cell Entry Receptor. J. Med. Virol. 2020, 92, 1309–1315. [Google Scholar] [CrossRef]
- Kanerva, A.; Mikheeva, G.V.; Krasnykh, V.; Coolidge, C.J.; Lam, J.T.; Mahasreshti, P.J.; Barker, S.D.; Straughn, M.; Barnes, M.N.; Alvarez, R.D.; et al. Targeting Adenovirus to the Serotype 3 Receptor Increases Gene Transfer Efficiency to Ovarian Cancer Cells. Clin. Cancer Res. 2002, 8, 275–280. [Google Scholar]
- Kanerva, A.; Wang, M.; Bauerschmitz, G.J.; Lam, J.T.; Desmond, R.A.; Bhoola, S.M.; Barnes, M.N.; Alvarez, R.D.; Siegal, G.P.; Curiel, D.T.; et al. Gene Transfer to Ovarian Cancer Versus Normal Tissues with Fiber-Modified Adenoviruses. Mol. Ther. 2002, 5, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, A.; Zinn, K.R.; Chaudhuri, T.R.; Lam, J.T.; Suzuki, K.; Uil, T.G.; Hakkarainen, T.; Bauerschmitz, G.J.; Wang, M.; Liu, B.; et al. Enhanced Therapeutic Efficacy for Ovarian Cancer with a Serotype 3 Receptor-Targeted Oncolytic Adenovirus. Mol. Ther. 2003, 8, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Raki, M.; Kanerva, A.; Ristimaki, A.; Desmond, R.A.; Chen, D.-T.; Ranki, T.; Sarkioja, M.; Kangasniemi, L.; Hemminki, A. Combination of Gemcitabine and Ad5/3-Δ24, a Tropism Modified Conditionally Replicating Adenovirus, for the Treatment of Ovarian Cancer. Gene Ther. 2005, 12, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Dmitriev, I.P.; Saddekni, S.; Kashentseva, E.A.; Harris, R.D.; Aurigemma, R.; Bae, S.; Singh, K.P.; Siegal, G.P.; Curiel, D.T.; et al. A Phase I Clinical Trial of Ad5/3-Δ24, a Novel Serotype-Chimeric, Infectivity-Enhanced, Conditionally-Replicative Adenovirus (CRAd), in Patients with Recurrent Ovarian Cancer. Gynecol. Oncol. 2013, 130, 518–524. [Google Scholar] [CrossRef]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.-X.; Levin, V.A.; Yung, W.K.A.; Kyritsis, A.P. A Mutant Oncolytic Adenovirus Targeting the Rb Pathway Produces Anti-Glioma Effect in Vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A.; Hawkins, L.; Kirn, D. An Adenovirus E1A Mutant That Demonstrates Potent and Selective Systemic Anti-Tumoral Efficacy. Nat. Med. 2000, 6, 1134–1139. [Google Scholar] [CrossRef]
- Felsani, A.; Mileo, A.M.; Paggi, M.G. Retinoblastoma Family Proteins as Key Targets of the Small DNA Virus Oncoproteins. Oncogene 2006, 25, 5277–5285. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and P53 Pathways in Cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef]
- Lockley, M.; Fernandez, M.; Wang, Y.; Li, N.F.; Conroy, S.; Lemoine, N.; McNeish, I. Activity of the Adenoviral E1A Deletion Mutant Dl 922–947 in Ovarian Cancer: Comparison with E1A Wild-Type Viruses, Bioluminescence Monitoring, and Intraperitoneal Delivery in Icodextrin. Cancer Res. 2006, 66, 989–998. [Google Scholar] [CrossRef]
- Baird, S.K.; Aerts, J.L.; Eddaoudi, A.; Lockley, M.; Lemoine, N.R.; McNeish, I.A. Oncolytic Adenoviral Mutants Induce a Novel Mode of Programmed Cell Death in Ovarian Cancer. Oncogene 2008, 27, 3081–3090. [Google Scholar] [CrossRef]
- Milanesio, M.C.; Giordano, S.; Valabrega, G. Clinical Implications of DNA Repair Defects in High-Grade Serous Ovarian Carcinomas. Cancers 2020, 12, 1315. [Google Scholar] [CrossRef] [PubMed]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat. Rev. Cancer 2023, 23, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Tookman, L.A.; Browne, A.K.; Connell, C.M.; Bridge, G.; Ingemarsdotter, C.K.; Dowson, S.; Shibata, A.; Lockley, M.; Martin, S.A.; McNeish, I.A. RAD51 and BRCA2 Enhance Oncolytic Adenovirus Type 5 Activity in Ovarian Cancer. Mol. Cancer Res. 2016, 14, 44–55. [Google Scholar] [CrossRef]
- Loboda, A.P.; Adonin, L.S.; Zvereva, S.D.; Guschin, D.Y.; Korneenko, T.V.; Telegina, A.V.; Kondratieva, O.K.; Frolova, S.E.; Pestov, N.B.; Barlev, N.A. BRCA Mutations—The Achilles Heel of Breast, Ovarian and Other Epithelial Cancers. Int. J. Mol. Sci. 2023, 24, 4982. [Google Scholar] [CrossRef]
- Jin, C.; Yuan, M.; Bu, H.; Jin, C. Antiangiogenic Strategies in Epithelial Ovarian Cancer: Mechanism, Resistance, and Combination Therapy. J. Oncol. 2022, 2022, 1–15. [Google Scholar] [CrossRef]
- Tuppurainen, L.; Sallinen, H.; Karvonen, A.; Valkonen, E.; Laakso, H.; Liimatainen, T.; Hytönen, E.; Hämäläinen, K.; Kosma, V.-M.; Anttila, M.; et al. Combined Gene Therapy Using AdsVEGFR2 and AdsTie2 With Chemotherapy Reduces the Growth of Human Ovarian Cancer and Formation of Ascites in Mice. Int. J. Gynecol. Cancer 2017, 27, 879–886. [Google Scholar] [CrossRef]
- Kujala, A.; Valkonen, E.; Sallinen, H.; Tuppurainen, L.; Laakso, H.; Ylä-Herttuala, E.; Liimatainen, T.; Kujala, J.; Jokelainen, O.; Sironen, R.; et al. AAV8-Mediated sVEGFR2 and sVEGFR3 Gene Therapy Combined with Chemotherapy Reduces the Growth and Microvasculature of Human Ovarian Cancer and Prolongs the Survival in Mice. Front. Med. 2022, 9, 1018208. [Google Scholar] [CrossRef]
- Holbrook, M.C.; Goad, D.W.; Grdzelishvili, V.Z. Expanding the Spectrum of Pancreatic Cancers Responsive to Vesicular Stomatitis Virus-Based Oncolytic Virotherapy: Challenges and Solutions. Cancers 2021, 13, 1171. [Google Scholar] [CrossRef]
- Zhang, Y.; Nagalo, B.M. Immunovirotherapy Based on Recombinant Vesicular Stomatitis Virus: Where Are We? Front. Immunol. 2022, 13, 898631. [Google Scholar] [CrossRef]
- Ahmed, M.M.; Okesanya, O.J.; Ukoaka, B.M.; Ibrahim, A.M.; Lucero-Prisno, D.E. Vesicular Stomatitis Virus: Insights into Pathogenesis, Immune Evasion, and Technological Innovations in Oncolytic and Vaccine Development. Viruses 2024, 16, 1933. [Google Scholar] [CrossRef]
- Tober, R.; Banki, Z.; Egerer, L.; Muik, A.; Behmüller, S.; Kreppel, F.; Greczmiel, U.; Oxenius, A.; Von Laer, D.; Kimpel, J. VSV-GP: A Potent Viral Vaccine Vector That Boosts the Immune Response upon Repeated Applications. J. Virol. 2014, 88, 4897–4907. [Google Scholar] [CrossRef] [PubMed]
- Muik, A.; Stubbert, L.J.; Jahedi, R.Z.; Geiβ, Y.; Kimpel, J.; Dold, C.; Tober, R.; Volk, A.; Klein, S.; Dietrich, U.; et al. Re-Engineering Vesicular Stomatitis Virus to Abrogate Neurotoxicity, Circumvent Humoral Immunity, and Enhance Oncolytic Potency. Cancer Res. 2014, 74, 3567–3578. [Google Scholar] [CrossRef] [PubMed]
- Capo-chichi, C.D.; Yeasky, T.M.; Heiber, J.F.; Wang, Y.; Barber, G.N.; Xu, X.-X. Explicit Targeting of Transformed Cells by VSV in Ovarian Epithelial Tumor-Bearing Wv Mouse Models. Gynecol. Oncol. 2010, 116, 269–275. [Google Scholar] [CrossRef]
- Manova, K.; Nocka, K.; Besmer, P.; Bachvarova, R.F. Gonadal Expression of C-Kit Encoded at the W Locus of the Mouse. Development 1990, 110, 1057–1069. [Google Scholar] [CrossRef]
- Reith, A.D.; Rottapel, R.; Giddens, E.; Brady, C.; Forrester, L.; Bernstein, A. W Mutant Mice with Mild or Severe Developmental Defects Contain Distinct Point Mutations in the Kinase Domain of the C-Kit Receptor. Genes Dev. 1990, 4, 390–400. [Google Scholar] [CrossRef]
- Dold, C.; Rodriguez Urbiola, C.; Wollmann, G.; Egerer, L.; Muik, A.; Bellmann, L.; Fiegl, H.; Marth, C.; Kimpel, J.; Von Laer, D. Application of Interferon Modulators to Overcome Partial Resistance of Human Ovarian Cancers to VSV-GP Oncolytic Viral Therapy. Mol. Ther. Oncolytics 2016, 3, 16021. [Google Scholar] [CrossRef]
- Geoffroy, K.; Mullins-Dansereau, V.; Leclerc-Desaulniers, K.; Viens, M.; Bourgeois-Daigneault, M.-C. Oncolytic Vesicular Stomatitis Virus Alone or in Combination with JAK Inhibitors Is Effective against Ovarian Cancer. Mol. Ther. Oncol. 2024, 32, 200826. [Google Scholar] [CrossRef]
- Nguyên, T.L.-A.; Abdelbary, H.; Arguello, M.; Breitbach, C.; Leveille, S.; Diallo, J.-S.; Yasmeen, A.; Bismar, T.A.; Kirn, D.; Falls, T.; et al. Chemical Targeting of the Innate Antiviral Response by Histone Deacetylase Inhibitors Renders Refractory Cancers Sensitive to Viral Oncolysis. Proc. Natl. Acad. Sci. USA 2008, 105, 14981–14986. [Google Scholar] [CrossRef]
- Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in Cancer Therapy. Viruses 2018, 10, 90. [Google Scholar] [CrossRef]
- Lin, X.; Chen, X.; Wei, Y.; Zhao, J.; Fan, L.; Wen, Y.; Wu, H.; Zhao, X. Efficient Inhibition of Intraperitoneal Human Ovarian Cancer Growth and Prolonged Survival by Gene Transfer of Vesicular Stomatitis Virus Matrix Protein in Nude Mice. Gynecol. Oncol. 2007, 104, 540–546. [Google Scholar] [CrossRef]
- Zhong, Q.; Wen, Y.-J.; Yang, H.-S.; Luo, H.; Fu, A.-F.; Yang, F.; Chen, L.-J.; Chen, X.; Qi, X.-R.; Lin, H.-G.; et al. Efficient Inhibition of Cisplatin-Resistant Human Ovarian Cancer Growth and Prolonged Survival by Gene Transferred Vesicular Stomatitis Virus Matrix Protein in Nude Mice. Ann. Oncol. 2008, 19, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Du, L.; Chen, X.; Chen, L.; Yi, T.; Chen, X.; Wen, Y.; Wei, Y.; Zhao, X. VEGF-D-Enhanced Lymph Node Metastasis of Ovarian Cancer Is Reversed by Vesicular Stomatitis Virus Matrix Protein. Int. J. Oncol. 2016, 49, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, L.A.; Upadhyay, R.; Greeley, Z.W.; Margulies, B.J. Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2. Viruses 2021, 13, 1228. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and Virulence of Herpes Simplex Virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef]
- Aldrak, N.; Alsaab, S.; Algethami, A.; Bhere, D.; Wakimoto, H.; Shah, K.; Alomary, M.N.; Zaidan, N. Oncolytic Herpes Simplex Virus-Based Therapies for Cancer. Cells 2021, 10, 1541. [Google Scholar] [CrossRef]
- Menotti, L.; Avitabile, E. Herpes Simplex Virus Oncolytic Immunovirotherapy: The Blossoming Branch of Multimodal Therapy. Int. J. Mol. Sci. 2020, 21, 8310. [Google Scholar] [CrossRef]
- Sanchala, D.S.; Bhatt, L.K.; Prabhavalkar, K.S. Oncolytic Herpes Simplex Viral Therapy: A Stride toward Selective Targeting of Cancer Cells. Front. Pharmacol. 2017, 8, 270. [Google Scholar] [CrossRef]
- Nakamori, M.; Fu, X.; Meng, F.; Jin, A.; Tao, L.; Bast, R.C.; Zhang, X. Effective Therapy of Metastatic Ovarian Cancer with an Oncolytic Herpes Simplex Virus Incorporating Two Membrane Fusion Mechanisms. Clin. Cancer Res. 2003, 9, 2727–2733. [Google Scholar]
- Nawa, A.; Nozawa, N.; Goshima, F.; Nagasaka, T.; Kikkawa, F.; Niwa, Y.; Nakanishi, T.; Kuzuya, K.; Nishiyama, Y. Oncolytic Viral Therapy for Human Ovarian Cancer Using a Novel Replication-Competent Herpes Simplex Virus Type I Mutant in a Mouse Model. Gynecol. Oncol. 2003, 91, 81–88. [Google Scholar] [CrossRef]
- Fu, X.; Tao, L.; Zhang, X. An Oncolytic Virus Derived from Type 2 Herpes Simplex Virus Has Potent Therapeutic Effect against Metastatic Ovarian Cancer. Cancer Gene Ther. 2007, 14, 480–487. [Google Scholar] [CrossRef]
- Coukos, G.; Makrigiannakis, A.; Kang, E.H.; Rubin, S.C.; Albelda, S.M.; Molnar-Kimber, K.L. Oncolytic Herpes Simplex Virus-1 Lacking ICP34.5 Induces P53-Independent Death and Is Efficacious against Chemotherapy-Resistant Ovarian Cancer. Clin. Cancer Res. 2000, 6, 3342–3353. [Google Scholar] [PubMed]
- Benencia, F.; Courrèges, M.C.; Conejo-García, J.R.; Mohamed-Hadley, A.; Zhang, L.; Buckanovich, R.J.; Carroll, R.; Fraser, N.; Coukos, G. HSV Oncolytic Therapy Upregulates Interferon-Inducible Chemokines and Recruits Immune Effector Cells in Ovarian Cancer. Mol. Ther. 2005, 12, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.D.; Meza-Perez, S.; Bevis, K.S.; Randall, T.D.; Gillespie, G.Y.; Langford, C.; Alvarez, R.D. IL-12 Expressing Oncolytic Herpes Simplex Virus Promotes Anti-Tumor Activity and Immunologic Control of Metastatic Ovarian Cancer in Mice. J. Ovarian Res. 2016, 9, 70. [Google Scholar] [CrossRef]
- Goshima, F.; Esaki, S.; Luo, C.; Kamakura, M.; Kimura, H.; Nishiyama, Y. Oncolytic Viral Therapy with a Combination of HF10, a Herpes Simplex Virus Type 1 Variant and Granulocyte-Macrophage Colony-Stimulating Factor for Murine Ovarian Cancer. Int. J. Cancer 2014, 134, 2865–2877. [Google Scholar] [CrossRef]
- Schreiber, L.M.; Urbiola, C.; Das, K.; Spiesschaert, B.; Kimpel, J.; Heinemann, F.; Stierstorfer, B.; Müller, P.; Petersson, M.; Erlmann, P.; et al. The lytic activity of VSV-GP treatment dominates the therapeutic effects in a syngeneic model of lung cancer. Br. J. Cancer 2019, 121, 647–658. [Google Scholar] [CrossRef]
- Benencia, F.; Courreges, M.C.; Conejo-García, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV Exerts Direct Antiangiogenic Activity in Ovarian Carcinoma. Hum. Gene Ther. 2005, 16, 765–778. [Google Scholar] [CrossRef]
- Benencia, F.; Courrèges, M.C.; Fraser, N.W.; Coukos, G. Herpes Virus Oncolytic Therapy Reverses Tumor Immune Dysfunction and Facilitates Tumor Antigen Presentation. Cancer Biol. Ther. 2008, 7, 1194–1205. [Google Scholar] [CrossRef]
- Omar, N.; Yan, B.; Salto-Tellez, M. HER2: An Emerging Biomarker in Non-Breast and Non-Gastric Cancers. Pathogenesis 2015, 2, 1–9. [Google Scholar] [CrossRef]
- Menotti, L.; Nicoletti, G.; Gatta, V.; Croci, S.; Landuzzi, L.; De Giovanni, C.; Nanni, P.; Lollini, P.-L.; Campadelli-Fiume, G. Inhibition of Human Tumor Growth in Mice by an Oncolytic Herpes Simplex Virus Designed to Target Solely HER-2-Positive Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9039–9044. [Google Scholar] [CrossRef]
- Nanni, P.; Gatta, V.; Menotti, L.; De Giovanni, C.; Ianzano, M.; Palladini, A.; Grosso, V.; Dall’ora, M.; Croci, S.; Nicoletti, G.; et al. Preclinical Therapy of Disseminated HER-2+ Ovarian and Breast Carcinomas with a HER-2-Retargeted Oncolytic Herpesvirus. PLoS Pathog. 2013, 9, e1003155. [Google Scholar] [CrossRef]
- Griffin, D.E. Measles Virus Persistence and Its Consequences. Curr. Opin. Virol. 2020, 41, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Leber, M.F.; Neault, S.; Jirovec, E.; Barkley, R.; Said, A.; Bell, J.C.; Ungerechts, G. Engineering and Combining Oncolytic Measles Virus for Cancer Therapy. Cytokine Growth Factor Rev. 2020, 56, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Ungerechts, G. Measles Virus as an Oncolytic Immunotherapy. Cancers 2021, 13, 544. [Google Scholar] [CrossRef] [PubMed]
- Grossardt, C.; Engeland, C.E.; Bossow, S.; Halama, N.; Zaoui, K.; Leber, M.F.; Springfeld, C.; Jaeger, D.; Von Kalle, C.; Ungerechts, G. Granulocyte-Macrophage Colony-Stimulating Factor-Armed Oncolytic Measles Virus Is an Effective Therapeutic Cancer Vaccine. Hum. Gene Ther. 2013, 24, 644–654. [Google Scholar] [CrossRef]
- Aref, S.; Bailey, K.; Fielding, A. Measles to the Rescue: A Review of Oncolytic Measles Virus. Viruses 2016, 8, 294. [Google Scholar] [CrossRef]
- Li, H.; Peng, K.-W.; Dingli, D.; Kratzke, R.A.; Russell, S.J. Oncolytic Measles Viruses Encoding Interferon β and the Thyroidal Sodium Iodide Symporter Gene for Mesothelioma Virotherapy. Cancer Gene Ther. 2010, 17, 550–558. [Google Scholar] [CrossRef]
- Iankov, I.D.; Allen, C.; Federspiel, M.J.; Myers, R.M.; Peng, K.W.; Ingle, J.N.; Russell, S.J.; Galanis, E. Expression of Immunomodulatory Neutrophil-Activating Protein of Helicobacter Pylori Enhances the Antitumor Activity of Oncolytic Measles Virus. Mol. Ther. 2012, 20, 1139–1147. [Google Scholar] [CrossRef]
- Allen, C.; Paraskevakou, G.; Iankov, I.; Giannini, C.; Schroeder, M.; Sarkaria, J.; Puri, R.K.; Russell, S.J.; Galanis, E. Interleukin-13 Displaying Retargeted Oncolytic Measles Virus Strains Have Significant Activity Against Gliomas With Improved Specificity. Mol. Ther. 2008, 16, 1556–1564. [Google Scholar] [CrossRef]
- Liu, C.; Erlichman, C.; McDonald, C.J.; Ingle, J.N.; Zollman, P.; Iankov, I.; Russell, S.J.; Galanis, E. Heat Shock Protein Inhibitors Increase the Efficacy of Measles Virotherapy. Gene Ther. 2008, 15, 1024–1034. [Google Scholar] [CrossRef]
- Peng, K.-W.; TenEyck, C.J.; Galanis, E.; Kalli, K.R.; Hartmann, L.C.; Russell, S.J. Intraperitoneal Therapy of Ovarian Cancer Using an Engineered Measles Virus. Cancer Res. 2002, 62, 4656–4662. [Google Scholar]
- Peng, K.-W.; Hadac, E.M.; Anderson, B.D.; Myers, R.; Harvey, M.; Greiner, S.M.; Soeffker, D.; Federspiel, M.J.; Russell, S.J. Pharmacokinetics of Oncolytic Measles Virotherapy: Eventual Equilibrium between Virus and Tumor in an Ovarian Cancer Xenograft Model. Cancer Gene Ther. 2006, 13, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Atherton, P.J.; Maurer, M.J.; Knutson, K.L.; Dowdy, S.C.; Cliby, W.A.; Haluska, P.; Long, H.J.; Oberg, A.; Aderca, I.; et al. Oncolytic Measles Virus Expressing the Sodium Iodide Symporter to Treat Drug-Resistant Ovarian Cancer. Cancer Res. 2015, 75, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.D.; Bossow, S.; Lampe, J.; Zimmermann, M.; Taran, F.-A.; Wallwiener, D.; Fehm, T.; Bitzer, M.; Lauer, U.M. Enhanced Killing of Ovarian Carcinoma Using Oncolytic Measles Vaccine Virus Armed with a Yeast Cytosine Deaminase and Uracil Phosphoribosyltransferase. Gynecol. Oncol. 2013, 130, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Mader, E.K.; Maeyama, Y.; Lin, Y.; Butler, G.W.; Russell, H.M.; Galanis, E.; Russell, S.J.; Dietz, A.B.; Peng, K.-W. Mesenchymal Stem Cell Carriers Protect Oncolytic Measles Viruses from Antibody Neutralization in an Orthotopic Ovarian Cancer Therapy Model. Clin. Cancer Res. 2009, 15, 7246–7255. [Google Scholar] [CrossRef]
- Xu, L.; Sun, H.; Lemoine, N.R.; Xuan, Y.; Wang, P. Oncolytic Vaccinia Virus and Cancer Immunotherapy. Front. Immunol. 2024, 14, 1324744. [Google Scholar] [CrossRef]
- Zhao, Y.; Adams, Y.F.; Croft, M. Preferential Replication of Vaccinia Virus in the Ovaries Is Independent of Immune Regulation Through IL-10 and TGF-β. Viral Immunol. 2011, 24, 387–396. [Google Scholar] [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational Combination of Oncolytic Vaccinia Virus and PD-L1 Blockade Works Synergistically to Enhance Therapeutic Efficacy. Nat. Commun. 2017, 8, 14754. [Google Scholar] [CrossRef]
- Hung, C.-F.; Tsai, Y.-C.; He, L.; Coukos, G.; Fodor, I.; Qin, L.; Levitsky, H.; Wu, T.-C. Vaccinia Virus Preferentially Infects and Controls Human and Murine Ovarian Tumors in Mice. Gene Ther. 2007, 14, 20–29. [Google Scholar] [CrossRef]
- Horita, K.; Kurosaki, H.; Nakatake, M.; Kuwano, N.; Oishi, T.; Itamochi, H.; Sato, S.; Kono, H.; Ito, M.; Hasegawa, K.; et al. lncRNA UCA1-Mediated Cdc42 Signaling Promotes Oncolytic Vaccinia Virus Cell-to-Cell Spread in Ovarian Cancer. Mol. Ther. Oncolytics 2019, 13, 35–48. [Google Scholar] [CrossRef]
- Whilding, L.M.; Archibald, K.M.; Kulbe, H.; Balkwill, F.R.; Öberg, D.; McNeish, I.A. Vaccinia Virus Induces Programmed Necrosis in Ovarian Cancer Cells. Mol. Ther. 2013, 21, 2074–2086. [Google Scholar] [CrossRef]
- Chalikonda, S.; Kivlen, M.H.; O’Malley, M.E.; Dong, X.D.; McCart, J.A.; Gorry, M.C.; Yin, X.-Y.; Brown, C.K.; Zeh, H.J.; Guo, Z.S.; et al. Oncolytic Virotherapy for Ovarian Carcinomatosis Using a Replication-Selective Vaccinia Virus Armed with a Yeast Cytosine Deaminase Gene. Cancer Gene Ther. 2008, 15, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yang, W.; Kim, D.K.; Kim, H.; Shin, M.; Choi, K.U.; Suh, D.S.; Kim, Y.H.; Hwang, T.-H.; Kim, J.H. Inhibition of MEK-ERK Pathway Enhances Oncolytic Vaccinia Virus Replication in Doxorubicin-Resistant Ovarian Cancer. Mol. Ther. Oncolytics 2022, 25, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-L.; Ma, B.; Pang, X.; Wu, T.-C.; Hung, C.-F. Treatment With Cyclooxygenase-2 Inhibitors Enables Repeated Administration of Vaccinia Virus for Control of Ovarian Cancer. Mol. Ther. 2009, 17, 1365–1372. [Google Scholar] [CrossRef]
- Mistarz, A.; Graczyk, M.; Winkler, M.; Singh, P.K.; Cortes, E.; Miliotto, A.; Liu, S.; Long, M.; Yan, L.; Stablewski, A.; et al. Induction of Cell Death in Ovarian Cancer Cells by Doxorubicin and Oncolytic Vaccinia Virus Is Associated with CREB3L1 Activation. Mol. Ther. Oncolytics 2021, 23, 38–50. [Google Scholar] [CrossRef]
- Chan, W.M.; Rahman, M.M.; McFadden, G. Oncolytic Myxoma Virus: The Path to Clinic. Vaccine 2013, 31, 4252–4258. [Google Scholar] [CrossRef]
- Nounamo, B.; Liem, J.; Cannon, M.; Liu, J. Myxoma Virus Optimizes Cisplatin for the Treatment of Ovarian Cancer In Vitro and in a Syngeneic Murine Dissemination Model. Mol. Ther. Oncolytics 2017, 6, 90–99. [Google Scholar] [CrossRef]
- Correa, R.J.M.; Komar, M.; Tong, J.G.K.; Sivapragasam, M.; Rahman, M.M.; McFadden, G.; DiMattia, G.E.; Shepherd, T.G. Myxoma Virus-Mediated Oncolysis of Ascites-Derived Human Ovarian Cancer Cells and Spheroids Is Impacted by Differential AKT Activity. Gynecol. Oncol. 2012, 125, 441–450. [Google Scholar] [CrossRef]
- Abad, A.T.; Danthi, P. Recognition of Reovirus RNAs by the Innate Immune System. Viruses 2020, 12, 667. [Google Scholar] [CrossRef]
- Bouziat, R.; Hinterleitner, R.; Brown, J.J.; Stencel-Baerenwald, J.E.; Ikizler, M.; Mayassi, T.; Meisel, M.; Kim, S.M.; Discepolo, V.; Pruijssers, A.J.; et al. Reovirus Infection Triggers Inflammatory Responses to Dietary Antigens and Development of Celiac Disease. Science 2017, 356, 44–50. [Google Scholar] [CrossRef]
- Jennings, V.A.; Ilett, E.J.; Scott, K.J.; West, E.J.; Vile, R.; Pandha, H.; Harrington, K.; Young, A.; Hall, G.D.; Coffey, M.; et al. Lymphokine-activated killer and dendritic cell carriage enhances oncolytic reovirus therapy for ovarian cancer by overcoming antibody neutralization in ascites. Int. J. Cancer 2014, 134, 1091–1101. [Google Scholar] [CrossRef]
- Hirasawa, K.; Nishikawa, S.G.; Norman, K.L.; Alain, T.; Kossakowska, A.; Lee, P.W.K. Oncolytic Reovirus against Ovarian and Colon Cancer. Cancer Res. 2002, 62, 1696–1701. [Google Scholar] [PubMed]
- An, Y.; Wang, X.; Wu, X.; Chen, L.; Yang, Y.; Lin, X.; Wang, N.; Duan, J.; Long, S.; Zhao, X. Oncolytic Reovirus Induces Ovarian Cancer Cell Apoptosis in a TLR3-Dependent Manner. Virus Res. 2021, 301, 198440. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.; Tyler, K.L. Down-Regulation of cFLIP Following Reovirus Infection Sensitizes Human Ovarian Cancer Cells to TRAIL-Induced Apoptosis. Apoptosis 2007, 12, 211–223. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, L.; Guo, T.; Huang, L.; Yang, Y.; Ye, R.; Zhang, Y.; Lin, X.; Fan, Y.; Gong, C.; et al. Cationic Liposomes Overcome Neutralizing Antibodies and Enhance Reovirus Efficacy in Ovarian Cancer. Virology 2024, 598, 110196. [Google Scholar] [CrossRef] [PubMed]
- Vasey, P.A.; Shulman, L.N.; Campos, S.; Davis, J.; Gore, M.; Johnston, S.; Kirn, D.H.; O’Neill, V.; Siddiqui, N.; Seiden, M.V.; et al. Phase I Trial of Intraperitoneal Injection of the E1B -55-Kd-Gene–Deleted Adenovirus ONYX-015 (Dl1520) Given on Days 1 Through 5 Every 3 Weeks in Patients With Recurrent/Refractory Epithelial Ovarian Cancer. J. Clin. Oncol. 2002, 20, 1562–1569. [Google Scholar] [CrossRef]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of Cancer Patients With a Serotype 5/3 Chimeric Oncolytic Adenovirus Expressing GMCSF. Mol. Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef]
- Kimball, K.J.; Preuss, M.A.; Barnes, M.N.; Wang, M.; Siegal, G.P.; Wan, W.; Kuo, H.; Saddekni, S.; Stockard, C.R.; Grizzle, W.E.; et al. A Phase I Study of a Tropism-Modified Conditionally Replicative Adenovirus for Recurrent Malignant Gynecologic Diseases. Clin. Cancer Res. 2010, 16, 5277–5287. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Burke, J.; Jonker, D.; Stephenson, J.; Haas, A.R.; Chow, L.Q.M.; Nieva, J.; Hwang, T.-H.; Moon, A.; Patt, R.; et al. Intravenous Delivery of a Multi-Mechanistic Cancer-Targeted Oncolytic Poxvirus in Humans. Nature 2011, 477, 99–102. [Google Scholar] [CrossRef]
- Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhöfer, U.; Glatzle, J.; Königsrainer, A.; Möhle, R.; Nann, D.; Fend, F.; et al. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clin. Cancer Res. 2018, 24, 4388–4398. [Google Scholar] [CrossRef]
- Holloway, R.W.; Kendrick, J.E.; Stephens, A.; Kennard, J.; Burt, J.; LeBlanc, J.; Sellers, K.; Smith, J.; Coakley, S. Phase 1b Study of Oncolytic Vaccinia Virus GL-ONC1 in Recurrent Ovarian Cancer (ROC). J. Clin. Oncol. 2018, 36, 5577. [Google Scholar] [CrossRef]
- Pakola, S.A.; Peltola, K.J.; Clubb, J.H.A.; Jirovec, E.; Haybout, L.; Kudling, T.V.; Alanko, T.; Korpisaari, R.; Juteau, S.; Jaakkola, M.; et al. Safety, Efficacy, and Biological Data of T-Cell–Enabling Oncolytic Adenovirus TILT-123 in Advanced Solid Cancers from the TUNIMO Monotherapy Phase I Trial. Clin. Cancer Res. 2024, 30, 3715–3725. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Hartmann, L.C.; Cliby, W.A.; Long, H.J.; Peethambaram, P.P.; Barrette, B.A.; Kaur, J.S.; Haluska, P.J.; Aderca, I.; Zollman, P.J.; et al. Phase I Trial of Intraperitoneal Administration of an Oncolytic Measles Virus Strain Engineered to Express Carcinoembryonic Antigen for Recurrent Ovarian Cancer. Cancer Res. 2010, 70, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Michael, A.; Wilson, W.; Sunshine, S.; Annels, N.; Harrop, R.; Blount, D.; Pandha, H.; Lord, R.; Ngai, Y.; Nicum, S.; et al. A Randomized Phase II Trial to Examine Modified Vaccinia Ankara-5T4 Vaccine in Patients with Relapsed Asymptomatic Ovarian Cancer (TRIOC). Int. J. Gynecol. Cancer 2024, 34, 1225–1231. [Google Scholar] [CrossRef]
- Cohn, D.E.; Sill, M.W.; Walker, J.L.; O’Malley, D.; Nagel, C.I.; Rutledge, T.L.; Bradley, W.; Richardson, D.L.; Moxley, K.M.; Aghajanian, C. Randomized Phase IIB Evaluation of Weekly Paclitaxel versus Weekly Paclitaxel with Oncolytic Reovirus (Reolysin®) in Recurrent Ovarian, Tubal, or Peritoneal Cancer: An NRG Oncology/Gynecologic Oncology Group Study. Gynecol. Oncol. 2017, 146, 477–483. [Google Scholar] [CrossRef]
- Moreno, V.; Barretina-Ginesta, M.P.; García-Donas, J.; Jayson, G.C.; Roxburgh, P.; Vázquez, R.M.; Michael, A.; Antón-Torres, A.; Brown, R.; Krige, D.; et al. Safety and efficacy of the tumor-selective adenovirus enadenotucirev with or without paclitaxel in platinum-resistant ovarian cancer: A phase 1 clinical trial. J. Immun. Cancer 2021, 9, e003645. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial Ovarian Cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef]
- Tong, J.G.; Valdes, Y.R.; Sivapragasam, M.; Barrett, J.W.; Bell, J.C.; Stojdl, D.; DiMattia, G.E.; Shepherd, T.G. Spatial and Temporal Epithelial Ovarian Cancer Cell Heterogeneity Impacts Maraba Virus Oncolytic Potential. BMC Cancer 2017, 17, 594. [Google Scholar] [CrossRef]
- Shen, Z.; Liu, X.; Fan, G.; Na, J.; Liu, Q.; Lin, F.; Zhang, Z.; Zhong, L. Improving the Therapeutic Efficacy of Oncolytic Viruses for Cancer: Targeting Macrophages. J. Transl. Med. 2023, 21, 842. [Google Scholar] [CrossRef]
- Chen, L.; Zuo, M.; Zhou, Q.; Wang, Y. Oncolytic Virotherapy in Cancer Treatment: Challenges and Optimization Prospects. Front. Immunol. 2023, 14, 1308890. [Google Scholar] [CrossRef]
- Veneziani, A.C.; Gonzalez-Ochoa, E.; Alqaisi, H.; Madariaga, A.; Bhat, G.; Rouzbahman, M.; Sneha, S.; Oza, A.M. Heterogeneity and Treatment Landscape of Ovarian Carcinoma. Nat. Rev. Clin. Oncol. 2023, 20, 820–842. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, Y.; Deng, M.; Huang, F.; Wang, J.; Blake, P.; Gabra, H.; Wang, Q. Abstract 6657: Oncolytic Vaccinia Virus Carrying OPCML Tumor Suppressor Is Active in Epithelial Ovarian Cancer. Cancer Res. 2024, 84, 6657. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borella, F.; Carosso, M.; Chiparo, M.P.; Ferraioli, D.; Bertero, L.; Gallio, N.; Preti, M.; Cusato, J.; Valabrega, G.; Revelli, A.; et al. Oncolytic Viruses in Ovarian Cancer: Where Do We Stand? A Narrative Review. Pathogens 2025, 14, 140. https://doi.org/10.3390/pathogens14020140
Borella F, Carosso M, Chiparo MP, Ferraioli D, Bertero L, Gallio N, Preti M, Cusato J, Valabrega G, Revelli A, et al. Oncolytic Viruses in Ovarian Cancer: Where Do We Stand? A Narrative Review. Pathogens. 2025; 14(2):140. https://doi.org/10.3390/pathogens14020140
Chicago/Turabian StyleBorella, Fulvio, Marco Carosso, Maria Pia Chiparo, Domenico Ferraioli, Luca Bertero, Niccolò Gallio, Mario Preti, Jessica Cusato, Giorgio Valabrega, Alberto Revelli, and et al. 2025. "Oncolytic Viruses in Ovarian Cancer: Where Do We Stand? A Narrative Review" Pathogens 14, no. 2: 140. https://doi.org/10.3390/pathogens14020140
APA StyleBorella, F., Carosso, M., Chiparo, M. P., Ferraioli, D., Bertero, L., Gallio, N., Preti, M., Cusato, J., Valabrega, G., Revelli, A., Marozio, L., & Cosma, S. (2025). Oncolytic Viruses in Ovarian Cancer: Where Do We Stand? A Narrative Review. Pathogens, 14(2), 140. https://doi.org/10.3390/pathogens14020140