
Hybrid Capture-Based Sequencing Enables Highly Sensitive Zoonotic Virus Detection Within the One Health Framework


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Viruses and Probe Design
2.2. Viral Reference Samples for Testing
2.3. Animal-Origin Samples of Unknown Viruses
2.4. SARS-CoV-2 Throat Swab Samples
2.5. Metagenomic Next-Generation Sequencing and Hybrid Capture-Based Target Enrichment
2.6. Data Analysis and Bioinformatics Pipeline
3. Results
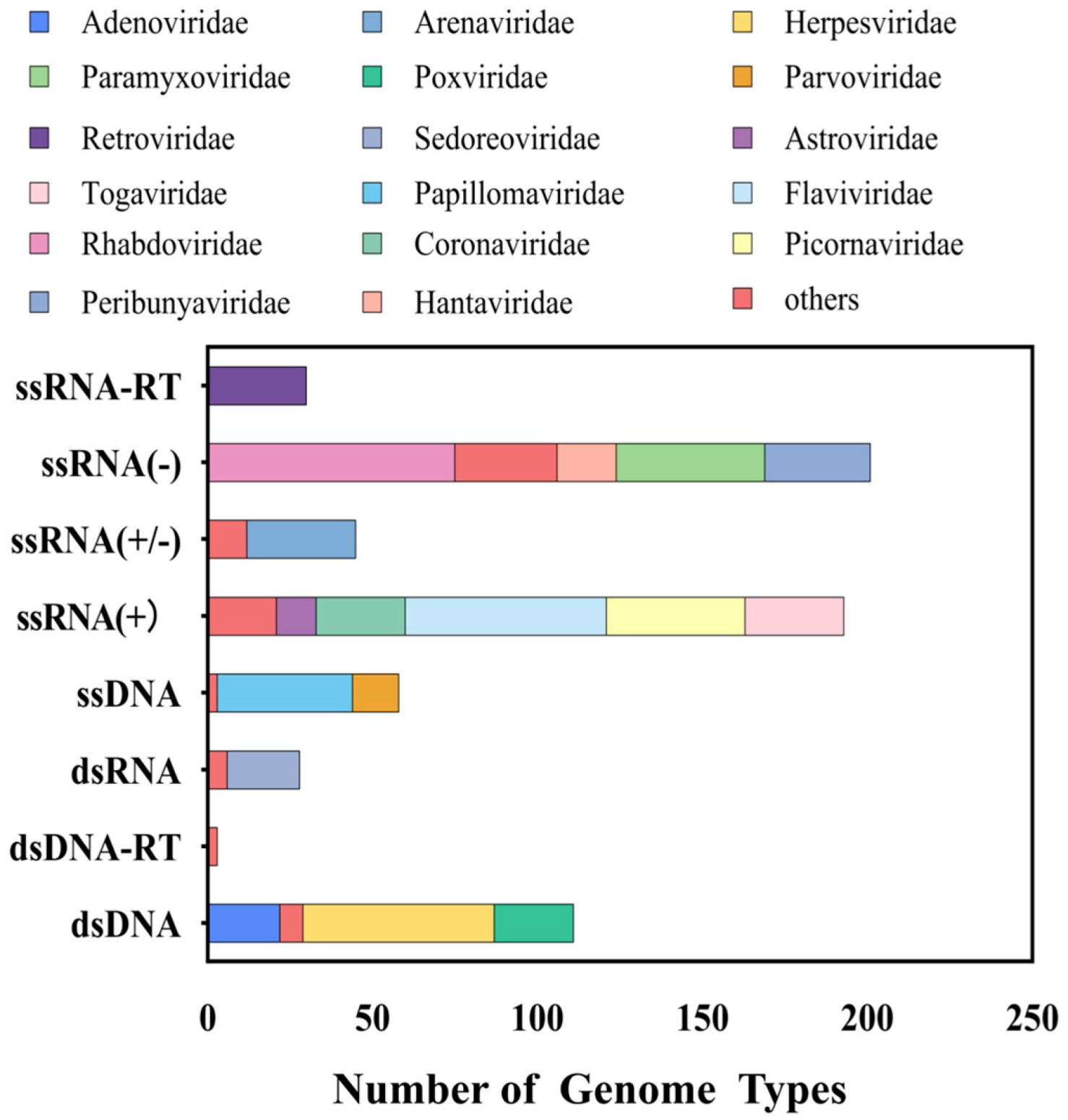
3.1. Viral Probe Library
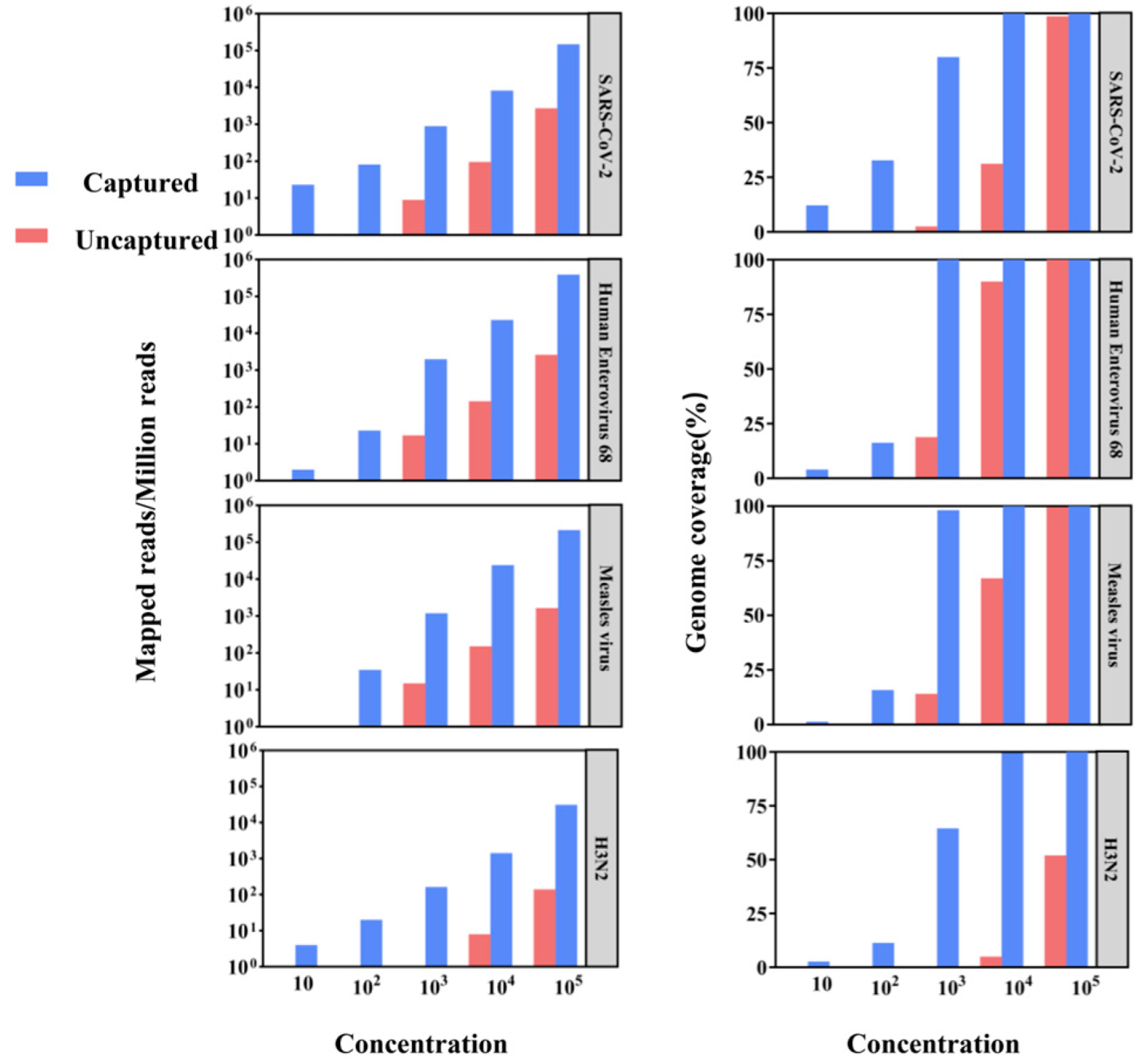
3.2. Test of Hybrid Capture-Based Sequencing in Viral Reference Samples
3.3. Application of Capture Sequencing for Identifying Unknown Viruses in Animal Samples
3.4. Assessment of Capture Sequencing in SARS-CoV-2 Throat Swab Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200.e5187. [Google Scholar] [CrossRef]
- Metsky, H.C.; Siddle, K.J.; Gladden-Young, A.; Qu, J.; Yang, D.K.; Brehio, P.; Goldfarb, A.; Piantadosi, A.; Wohl, S.; Carter, A.; et al. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat. Biotechnol. 2019, 37, 160–168. [Google Scholar] [CrossRef]
- Wylezich, C.; Calvelage, S.; Schlottau, K.; Ziegler, U.; Pohlmann, A.; Höper, D.; Beer, M. Next-generation diagnostics: Virus capture facilitates a sensitive viral diagnosis for epizootic and zoonotic pathogens including SARS-CoV-2. Microbiome 2021, 9, 51. [Google Scholar] [CrossRef]
- Fitzpatrick, A.H.; Rupnik, A.; O’Shea, H.; Crispie, F.; Keaveney, S.; Cotter, P. High Throughput Sequencing for the Detection and Characterization of RNA Viruses. Front. Microbiol. 2021, 12, 621719. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef]
- Kuchinski, K.S.; Duan, J.; Himsworth, C.; Hsiao, W.; Prystajecky, N.A. ProbeTools: Designing hybridization probes for targeted genomic sequencing of diverse and hypervariable viral taxa. BMC Genom. 2022, 23, 579. [Google Scholar] [CrossRef]
- Kuchinski, K.S.; Loos, K.D.; Suchan, D.M.; Russell, J.N.; Sies, A.N.; Kumakamba, C.; Muyembe, F.; Mbala Kingebeni, P.; Ngay Lukusa, I.; N’Kawa, F.; et al. Targeted genomic sequencing with probe capture for discovery and surveillance of coronaviruses in bats. Elife 2022, 11, e79777. [Google Scholar] [CrossRef]
- Gaudin, M.; Desnues, C. Hybrid Capture-Based Next Generation Sequencing and Its Application to Human Infectious Diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Mollentze, N.; DStreicker, D.G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 9423–9430. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, N.P.G.; Chorlton, S.D.; Krajden, M.; Manges, A.R. Agnostic Sequencing for Detection of Viral Pathogens. Clin. Microbiol. Rev. 2023, 36, e0011922. [Google Scholar] [CrossRef]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bártolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. mBio 2015, 6, e01491-15. [Google Scholar] [CrossRef] [PubMed]
- Mourik, K.; Sidorov, I.; Carbo, E.C.; van der Meer, D.; Boot, A.; Kroes, A.C.M.; Claas, E.C.J.; Boers, S.A.; de Vries, J.J.C. Comparison of the performance of two targeted metagenomic virus capture probe-based methods using reference control materials and clinical samples. J. Clin. Microbiol. 2024, 62, e0034524. [Google Scholar] [CrossRef]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef]
- Takano, T.; Yanai, Y.; Hiramatsu, K.; Doki, T.; Hohdatsu, T. Novel single-stranded, circular DNA virus identified in cats in Japan. Arch. Virol. 2018, 163, 3389–3393. [Google Scholar] [CrossRef]
- Bukasov, R.; Dossym, D.; Filchakova, O. Detection of RNA viruses from influenza and HIV to Ebola and SARS-CoV-2: A review. Anal. Methods 2021, 13, 34–55. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Grouping | Ct Value | Uncaptured Samples | Capture Enrichment Samples | ||
|---|---|---|---|---|---|
| Mapped Reads | Genome Coverage (%) | Mapped Reads | Genome Coverage (%) | ||
| Low Ct Value | 21 | 10,247 | 99.11 | 37,958 | 99.84 |
| 22 | 21,082 | 99.78 | 57,144 | 99.84 | |
| 24 | 6718 | 96.07 | 27,777 | 98.18 | |
| 25 | 4114 | 98.43 | 41,718 | 99.19 | |
| 25 | 5359 | 94.92 | 36,152 | 98.87 | |
| 25 | 3647 | 84.13 | 23,778 | 86.43 | |
| Medium Ct Value | 27 | 2611 | 75.43 | 58,249 | 88.54 |
| 28 | 1980 | 52.02 | 50,781 | 91.11 | |
| 30 | 898 | 12.57 | 49,459 | 54.21 | |
| High Ct Value | 32 | 0 | 0 | 401 | 24.47 |
| 33 | 0 | 0 | 105 | 8.11 | |
| 34 | 0 | 0 | 251 | 12.43 | |
| 35 | 0 | 0 | 285 | 7.60 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, W.; Wang, J.; Li, T.; Wu, J.; Wang, J.; Wen, S.; Huang, J.; Shi, Y.; Zheng, K.; Zhai, Y.; et al. Hybrid Capture-Based Sequencing Enables Highly Sensitive Zoonotic Virus Detection Within the One Health Framework. Pathogens 2025, 14, 264. https://doi.org/10.3390/pathogens14030264
Mao W, Wang J, Li T, Wu J, Wang J, Wen S, Huang J, Shi Y, Zheng K, Zhai Y, et al. Hybrid Capture-Based Sequencing Enables Highly Sensitive Zoonotic Virus Detection Within the One Health Framework. Pathogens. 2025; 14(3):264. https://doi.org/10.3390/pathogens14030264
Chicago/Turabian StyleMao, Weiya, Jin Wang, Ting Li, Jiani Wu, Jiangrong Wang, Shubo Wen, Jicheng Huang, Yongxia Shi, Kui Zheng, Yali Zhai, and et al. 2025. "Hybrid Capture-Based Sequencing Enables Highly Sensitive Zoonotic Virus Detection Within the One Health Framework" Pathogens 14, no. 3: 264. https://doi.org/10.3390/pathogens14030264
APA StyleMao, W., Wang, J., Li, T., Wu, J., Wang, J., Wen, S., Huang, J., Shi, Y., Zheng, K., Zhai, Y., Li, X., Long, Y., Lu, J., & Guo, C. (2025). Hybrid Capture-Based Sequencing Enables Highly Sensitive Zoonotic Virus Detection Within the One Health Framework. Pathogens, 14(3), 264. https://doi.org/10.3390/pathogens14030264