
Emerging Strategies and Progress in the Medical Management of Marburg Virus Disease
 , , and
, , and 
Abstract
1. Introduction
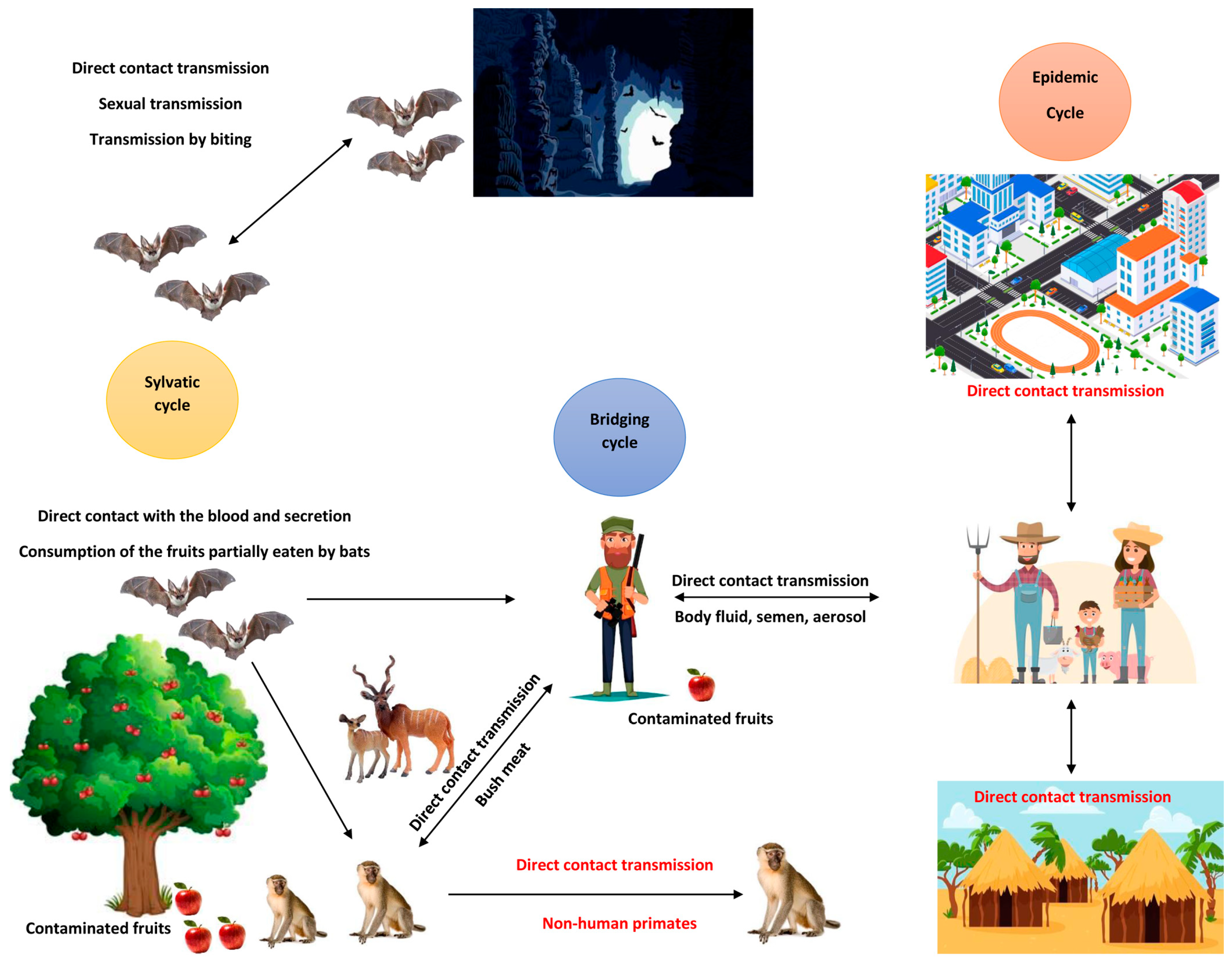
2. Route of Transmission and Clinical Presentation
3. Marburg Virus Genomic Characteristics Related to Virus Entry, Transmission, and Targets for Treatment and Vaccine Strategies
4. Supportive Treatment
5. Experimental Therapies
5.1. Antiviral Therapy
5.1.1. Favipiravir (T-705)
5.1.2. Galidesivir (BCX4430)
5.1.3. Remdesivir (GS-5734)
5.1.4. Obeldesivir (GS-5245)
5.2. Monoclonal and Polyclonal Antibodies
5.2.1. Polyclonal Antibodies
5.2.2. Monoclonal Antibodies
5.3. Marburg Virus Vaccine
5.3.1. Adenovirus-Vectored Vaccines
5.3.2. DNA Vaccines
5.3.3. Recombinant Vesicular Stomatitis Virus (rVSV) Vaccine
6. New Target for New Hope
6.1. Estradiol Benzoate (EB)
6.2. Small Interfering RNA (siRNA)
6.3. Interferon-β Therapy
6.4. Phosphorodiamidate Morpholino Oligomers
6.5. Eritoran Tetrasodium (E5564)
6.6. Glycoprotein (GP) Receptor Antagonists
7. Case Management of MVD During the Recent Outbreak in Rwanda
8. Future Directions and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brauburger, K.; Hume, A.J.; Mühlberger, E.; Olejnik, J. Forty-Five Years of Marburg Virus Research. Viruses 2012, 4, 1878–1927. [Google Scholar] [CrossRef] [PubMed]
- Muvunyi, C.M.; Ngabonziza, J.C.S.; Bigirimana, N.; Ndembi, N.; Siddig, E.E.; Kaseya, J.; Ahmed, A. Evidence-Based Guidance for One Health Preparedness, Prevention, and Response Strategies to Marburg Virus Disease Outbreaks. Diseases 2024, 12, 309. [Google Scholar] [CrossRef] [PubMed]
- Muvunyi, C.M.; Mohamed, N.S.; Siddig, E.E.; Ahmed, A. Genomic Evolution and Phylodynamics of the Species Orthomarburgvirus Marburgense (Marburg and Ravn Viruses) to Understand Viral Adaptation and Marburg Virus Disease’s Transmission Dynamics. Pathogens 2024, 13, 1107. [Google Scholar] [CrossRef] [PubMed]
- Biedenkopf, N.; Bukreyev, A.; Chandran, K.; Di Paola, N.; Formenty, P.B.H.; Griffiths, A.; Hume, A.J.; Mühlberger, E.; Netesov, S.V.; Palacios, G.; et al. ICTV Virus Taxonomy Profile: Filoviridae 2024. J. Gen. Virol. 2024, 105, 001955. [Google Scholar] [CrossRef]
- Okesanya, O.J.; Manirambona, E.; Olaleke, N.O.; Osumanu, H.A.; Faniyi, A.A.; Bouaddi, O.; Gbolahan, O.; Lasala, J.J.; Lucero-Prisno, D.E. Rise of Marburg Virus in Africa: A Call for Global Preparedness. Ann. Med. Surg. 2023, 85, 5285–5290. [Google Scholar] [CrossRef]
- Srivastava, S.; Kumar, S.; Ashique, S.; Sridhar, S.B.; Shareef, J.; Thomas, S. Novel Antiviral Approaches for Marburg: A Promising Therapeutics in the Pipeline. Front. Microbiol. 2024, 15, 1387628. [Google Scholar] [CrossRef]
- Mehedi, M.; Groseth, A.; Feldmann, H.; Ebihara, H. Clinical Aspects of Marburg Hemorrhagic Fever. Future Virol. 2011, 6, 1091–1106. [Google Scholar] [CrossRef]
- Hu, S.; Noda, T. Filovirus Helical Nucleocapsid Structures. Microscopy 2023, 72, 178–190. [Google Scholar] [CrossRef]
- Edwards, M.R.; Vogel, O.A.; Mori, H.; Davey, R.A.; Basler, C.F. Marburg Virus VP30 Is Required for Transcription Initiation at the Glycoprotein Gene. mBio 2022, 13, e0224322. [Google Scholar] [CrossRef]
- Feldmann, H.; Volchkov, V.E.; Volchkova, V.A.; Klenk, H.D. The Glycoproteins of Marburg and Ebola Virus and Their Potential Roles in Pathogenesis. Arch. Virol. Suppl. 1999, 15, 159–169. [Google Scholar] [CrossRef]
- Rigby, I.; Michelen, M.; Dagens, A.; Cheng, V.; Dahmash, D.; Harriss, E.; Webb, E.; Cai, E.; Lipworth, S.; Oti, A.; et al. Standard of Care for Viral Haemorrhagic Fevers (VHFs): A Systematic Review of Clinical Management Guidelines for High-Priority VHFs. Lancet Infect. Dis. 2023, 23, e240–e252. [Google Scholar] [CrossRef] [PubMed]
- The Africa Centres for Disease Control and Prevention (Africa CDC) Marburg Virus Disease (MVD). Available online: https://africacdc.org/disease/marburg-virus-disease-mvd/ (accessed on 27 October 2024).
- Cleveland Clinic. Marburg Virus Disease. Available online: https://my.clevelandclinic.org/health/diseases/25097-marburg-virus-disease (accessed on 27 October 2024).
- World Health Organization (WHO). Marburg Virus Disease. The Republic of Rwanda. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2024-DON537 (accessed on 27 October 2024).
- Albakri, K.; Al-Hajali, M.; Saleh, O.; Alkhalil, A.M.; Mohd, A.B.; Samain, C.A.; Abuasad, N.N.; Hasan, H.; Khaity, A.; Farahat, R.A. Marburg Virus Disease Treatments and Vaccines: Recent Gaps and Implications. Ann. Med. Surg. 2023, 85, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Zhang, Y.-J. Antisense Phosphorodiamidate Morpholino Oligomers as Novel Antiviral Compounds. Front. Microbiol. 2018, 9, 750. [Google Scholar] [CrossRef]
- Nakayama, E.; Tomabechi, D.; Matsuno, K.; Kishida, N.; Yoshida, R.; Feldmann, H.; Takada, A. Antibody-Dependent Enhancement of Marburg Virus Infection. J. Infect. Dis. 2011, 204 (Suppl. S3), S978–S985. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Holland, R.; Wood, M.; Pasetka, C.; Palmer, L.; Samaridou, E.; McClintock, K.; Borisevich, V.; Geisbert, T.W.; Cross, R.W.; et al. Combination Treatment of Mannose and GalNAc Conjugated Small Interfering RNA Protects against Lethal Marburg Virus Infection. Mol. Ther. 2023, 31, 269–281. [Google Scholar] [CrossRef]
- Smith, L.M.; Hensley, L.E.; Geisbert, T.W.; Johnson, J.; Stossel, A.; Honko, A.; Yen, J.Y.; Geisbert, J.; Paragas, J.; Fritz, E.; et al. Interferon-β Therapy Prolongs Survival in Rhesus Macaque Models of Ebola and Marburg Hemorrhagic Fever. J. Infect. Dis. 2013, 208, 310–318. [Google Scholar] [CrossRef]
- Jin, Z.; Smith, L.K.; Rajwanshi, V.K.; Kim, B.; Deval, J. The Ambiguous Base-Pairing and High Substrate Efficiency of T-705 (Favipiravir) Ribofuranosyl 5′-Triphosphate towards Influenza A Virus Polymerase. PLoS ONE 2013, 8, e68347. [Google Scholar] [CrossRef]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a Novel Viral RNA Polymerase Inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef]
- Oestereich, L.; Lüdtke, A.; Wurr, S.; Rieger, T.; Muñoz-Fontela, C.; Günther, S. Successful Treatment of Advanced Ebola Virus Infection with T-705 (Favipiravir) in a Small Animal Model. Antivir. Res. 2014, 105, 17–21. [Google Scholar] [CrossRef]
- Yen, H.-L. Current and Novel Antiviral Strategies for Influenza Infection. Curr. Opin. Virol. 2016, 18, 126–134. [Google Scholar] [CrossRef]
- Sissoko, D.; Laouenan, C.; Folkesson, E.; M’Lebing, A.-B.; Beavogui, A.-H.; Baize, S.; Camara, A.-M.; Maes, P.; Shepherd, S.; Danel, C.; et al. Experimental Treatment with Favipiravir for Ebola Virus Disease (the JIKI Trial): A Historically Controlled, Single-Arm Proof-of-Concept Trial in Guinea. PLoS Med. 2016, 13, e1001967. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Z.; He, S.; Wong, G.; Banadyga, L.; Qiu, X. Successful Treatment of Marburg Virus with Orally Administrated T-705 (Favipiravir) in a Mouse Model. Antivir. Res. 2018, 151, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bixler, S.L.; Bocan, T.M.; Wells, J.; Wetzel, K.S.; Van Tongeren, S.A.; Dong, L.; Garza, N.L.; Donnelly, G.; Cazares, L.H.; Nuss, J. Efficacy of Favipiravir (T-705) in Nonhuman Primates Infected with Ebola Virus or Marburg Virus. Antivir. Res. 2018, 151, 97–104. [Google Scholar] [PubMed]
- Warren, T.K.; Wells, J.; Panchal, R.G.; Stuthman, K.S.; Garza, N.L.; Van Tongeren, S.A.; Dong, L.; Retterer, C.J.; Eaton, B.P.; Pegoraro, G. Protection against Filovirus Diseases by a Novel Broad-Spectrum Nucleoside Analogue BCX4430. Nature 2014, 508, 402–405. [Google Scholar]
- Julander, J.G.; Demarest, J.F.; Taylor, R.; Gowen, B.B.; Walling, D.M.; Mathis, A.; Babu, Y.S. An Update on the Progress of Galidesivir (BCX4430), a Broad-Spectrum Antiviral. Antivir. Res. 2021, 195, 105180. [Google Scholar] [CrossRef]
- Cross, R.W.; Bornholdt, Z.A.; Prasad, A.N.; Woolsey, C.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Abelson, D.M.; Kim, D.H.; Shestowsky, W.S.; et al. Combination Therapy with Remdesivir and Monoclonal Antibodies Protects Nonhuman Primates against Advanced Sudan Virus Disease. JCI Insight 2022, 7, e159090. [Google Scholar] [CrossRef]
- Porter, D.P.; Weidner, J.M.; Gomba, L.; Bannister, R.; Blair, C.; Jordan, R.; Wells, J.; Wetzel, K.; Garza, N.; Van Tongeren, S.; et al. Remdesivir (GS-5734) Is Efficacious in Cynomolgus Macaques Infected With Marburg Virus. J. Infect. Dis. 2020, 222, 1894–1901. [Google Scholar] [CrossRef]
- Cross, R.W.; Bornholdt, Z.A.; Prasad, A.N.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Abelson, D.M.; Kim, D.H.; Shestowsky, W.S.; Campbell, L.A.; et al. Combination Therapy Protects Macaques against Advanced Marburg Virus Disease. Nat. Commun. 2021, 12, 1891. [Google Scholar] [CrossRef]
- Cao, L.; Li, Y.; Yang, S.; Li, G.; Zhou, Q.; Sun, J.; Xu, T.; Yang, Y.; Liao, R.; Shi, Y.; et al. The Adenosine Analog Prodrug ATV006 Is Orally Bioavailable and Has Preclinical Efficacy against Parental SARS-CoV-2 and Variants. Sci. Transl. Med. 2022, 14, eabm7621. [Google Scholar] [CrossRef]
- Gilead Sciences A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of GS-5245 for the Treatment of COVID-19 in Participants with High-Risk for Disease Progression. 2023. Available online: https://clinicaltrials.gov/ (accessed on 12 July 2024).
- Martinez, D.R.; Moreira, F.R.; Catanzaro, N.J.; Diefenbacher, M.V.; Zweigart, M.R.; Gully, K.L.; De la Cruz, G.; Brown, A.J.; Adams, L.E.; Yount, B.; et al. The Oral Nucleoside Prodrug GS-5245 Is Efficacious against SARS-CoV-2 and Other Endemic, Epidemic, and Enzootic Coronaviruses. Sci. Transl. Med. 2024, 16, eadj4504. [Google Scholar] [CrossRef]
- Sprecher, A.; Van Herp, M. An Oral Antiviral for Ebola Disease. Science 2024, 383, 1181–1182. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.W.; Woolsey, C.; Chu, V.C.; Babusis, D.; Bannister, R.; Vermillion, M.S.; Geleziunas, R.; Barrett, K.T.; Bunyan, E.; Nguyen, A.-Q.; et al. Oral Administration of Obeldesivir Protects Nonhuman Primates against Sudan Ebolavirus. Science 2024, 383, eadk6176. [Google Scholar] [CrossRef] [PubMed]
- Coffin, K.M.; Liu, J.; Warren, T.K.; Blancett, C.D.; Kuehl, K.A.; Nichols, D.K.; Bearss, J.J.; Schellhase, C.W.; Retterer, C.J.; Weidner, J.M.; et al. Persistent Marburg Virus Infection in the Testes of Nonhuman Primate Survivors. Cell Host Microbe 2018, 24, 405–416.e3. [Google Scholar] [CrossRef] [PubMed]
- Enria, D.A.; Briggiler, A.M.; Fernandez, N.J.; Levis, S.C.; Maiztegui, J.I. Importance of Dose of Neutralising Antibodies in Treatment of Argentine Haemorrhagic Fever with Immune Plasma. Lancet 1984, 2, 255–256. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Frame, J.D.; Rhoderick, J.B.; Monson, M.H. Endemic Lassa Fever in Liberia. IV. Selection of Optimally Effective Plasma for Treatment by Passive Immunization. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 380–384. [Google Scholar] [CrossRef]
- Jahrling, P.B.; Geisbert, J.B.; Swearengen, J.R.; Larsen, T.; Geisbert, T.W. Ebola Hemorrhagic Fever: Evaluation of Passive Immunotherapy in Nonhuman Primates. J. Infect. Dis. 2007, 196 (Suppl. S2), S400–S403. [Google Scholar] [CrossRef]
- Mupapa, K.; Massamba, M.; Kibadi, K.; Kuvula, K.; Bwaka, A.; Kipasa, M.; Colebunders, R.; Muyembe-Tamfum, J.J. Treatment of Ebola Hemorrhagic Fever with Blood Transfusions from Convalescent Patients. International Scientific and Technical Committee. J. Infect. Dis. 1999, 179 (Suppl. S1), S18–S23. [Google Scholar] [CrossRef]
- Cross, R.W.; Mire, C.E.; Feldmann, H.; Geisbert, T.W. Post-Exposure Treatments for Ebola and Marburg Virus Infections. Nat. Rev. Drug Discov. 2018, 17, 413–434. [Google Scholar] [CrossRef]
- Dye, J.M.; Herbert, A.S.; Kuehne, A.I.; Barth, J.F.; Muhammad, M.A.; Zak, S.E.; Ortiz, R.A.; Prugar, L.I.; Pratt, W.D. Postexposure Antibody Prophylaxis Protects Nonhuman Primates from Filovirus Disease. Proc. Natl. Acad. Sci. USA 2012, 109, 5034–5039. [Google Scholar] [CrossRef]
- Mire, C.E.; Geisbert, J.B.; Borisevich, V.; Fenton, K.A.; Agans, K.N.; Flyak, A.I.; Deer, D.J.; Steinkellner, H.; Bohorov, O.; Bohorova, N.; et al. Therapeutic Treatment of Marburg and Ravn Virus Infection in Nonhuman Primates with a Human Monoclonal Antibody. Sci. Transl. Med. 2017, 9, eaai8711. [Google Scholar] [CrossRef]
- The World Health Organization (WHO). MBP091 Update- WHO; WHO: Geneva, Switzerland, 2023. [Google Scholar]
- PLOS Global Public Health. Rwanda’s Marburg Outbreak: The Latest Test of Pandemic Preparedness and Global Health Resilience. In Speaking of Medicine and Health; PLOS: San Francisco, CA, USA, 2024; Available online: https://speakingofmedicine.plos.org/2024/10/09/rwandas-marburg-outbreak-the-latest-test-of-pandemic-preparedness-and-global-health-resilience/ (accessed on 7 December 2024).
- Mwesigwa, B.; Houser, K.V.; Hofstetter, A.R.; Ortega-Villa, A.M.; Naluyima, P.; Kiweewa, F.; Nakabuye, I.; Yamshchikov, G.V.; Andrews, C.; O’Callahan, M.; et al. Safety, Tolerability, and Immunogenicity of the Ebola Sudan Chimpanzee Adenovirus Vector Vaccine (cAd3-EBO S) in Healthy Ugandan Adults: A Phase 1, Open-Label, Dose-Escalation Clinical Trial. Lancet Infect. Dis. 2023, 23, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Feldmann, H. Recombinant Vesicular Stomatitis Virus-Based Vaccines against Ebola and Marburg Virus Infections. J. Infect. Dis. 2011, 204 (Suppl. S3), S1075–S1081. [Google Scholar] [CrossRef]
- Sarwar, U.N.; Costner, P.; Enama, M.E.; Berkowitz, N.; Hu, Z.; Hendel, C.S.; Sitar, S.; Plummer, S.; Mulangu, S.; Bailer, R.T.; et al. Safety and Immunogenicity of DNA Vaccines Encoding Ebolavirus and Marburgvirus Wild-Type Glycoproteins in a Phase I Clinical Trial. J. Infect. Dis. 2015, 211, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Suschak, J.J.; Schmaljohn, C.S. Vaccines against Ebola Virus and Marburg Virus: Recent Advances and Promising Candidates. Hum. Vaccines Immunother. 2019, 15, 2359–2377. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Liu, G.; Cao, W.; He, S.; Leung, A.; Ströher, U.; Fairchild, M.J.; Nichols, R.; Crowell, J.; Fusco, J.; et al. A Cloned Recombinant Vesicular Stomatitis Virus-Vectored Marburg Vaccine, PHV01, Protects Guinea Pigs from Lethal Marburg Virus Disease. Vaccines 2022, 10, 1004. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Bailey, M.; Geisbert, J.B.; Asiedu, C.; Roederer, M.; Grazia-Pau, M.; Custers, J.; Jahrling, P.; Goudsmit, J.; Koup, R.; et al. Vector Choice Determines Immunogenicity and Potency of Genetic Vaccines against Angola Marburg Virus in Nonhuman Primates. J. Virol. 2010, 84, 10386–10394. [Google Scholar] [CrossRef]
- Swenson, D.L.; Wang, D.; Luo, M.; Warfield, K.L.; Woraratanadharm, J.; Holman, D.H.; Dong, J.Y.; Pratt, W.D. Vaccine to Confer to Nonhuman Primates Complete Protection against Multistrain Ebola and Marburg Virus Infections. Clin. Vaccine Immunol. 2008, 15, 460–467. [Google Scholar] [CrossRef]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-Existing Immunity against Ad Vectors Humoral, Cellular, and Innate Response, What’s Important? Hum. Vaccines Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Bailey, M.; Hensley, L.; Asiedu, C.; Geisbert, J.; Stanley, D.; Honko, A.; Johnson, J.; Mulangu, S.; Pau, M.G.; et al. Recombinant Adenovirus Serotype 26 (Ad26) and Ad35 Vaccine Vectors Bypass Immunity to Ad5 and Protect Nonhuman Primates against Ebolavirus Challenge. J. Virol. 2011, 85, 4222–4233. [Google Scholar] [CrossRef]
- Stanley, D.A.; Honko, A.N.; Asiedu, C.; Trefry, J.C.; Lau-Kilby, A.W.; Johnson, J.C.; Hensley, L.; Ammendola, V.; Abbate, A.; Grazioli, F.; et al. Chimpanzee Adenovirus Vaccine Generates Acute and Durable Protective Immunity against Ebolavirus Challenge. Nat. Med. 2014, 20, 1126–1129. [Google Scholar] [CrossRef]
- DeZure, A.; Graham, B.S. Chapter 28—Vaccines for Emerging Viral Diseases. In The Vaccine Book, 2nd ed.; Bloom, B.R., Lambert, P.-H., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 543–560. ISBN 978-0-12-802174-3. [Google Scholar]
- Happe, M.; Hofstetter, A.R.; Wang, J.; Yamshchikov, G.V.; Holman, L.A.; Novik, L.; Strom, L.; Kiweewa, F.; Wakabi, S.; Millard, M.; et al. Heterologous cAd3-Ebola and MVA-EbolaZ Vaccines Are Safe and Immunogenic in US and Uganda Phase 1/1b Trials. NPJ Vaccines 2024, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Hamer, M.J.; Houser, K.V.; Hofstetter, A.R.; Ortega-Villa, A.M.; Lee, C.; Preston, A.; Augustine, B.; Andrews, C.; Yamshchikov, G.V.; Hickman, S.; et al. Safety, Tolerability, and Immunogenicity of the Chimpanzee Adenovirus Type 3-Vectored Marburg Virus (cAd3-Marburg) Vaccine in Healthy Adults in the USA: A First-in-Human, Phase 1, Open-Label, Dose-Escalation Trial. Lancet 2023, 401, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, S.; Grimes-Serrano, J.M. Current Progress of DNA Vaccine Studies in Humans. Expert. Rev. Vaccines 2008, 7, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, D.; Geisbert, T.W.; Feldmann, H. Progress in Filovirus Vaccine Development: Evaluating the Potential for Clinical Use. Expert. Rev. Vaccines 2011, 10, 63–77. [Google Scholar] [CrossRef]
- Riemenschneider, J.; Garrison, A.; Geisbert, J.; Jahrling, P.; Hevey, M.; Negley, D.; Schmaljohn, A.; Lee, J.; Hart, M.K.; Vanderzanden, L.; et al. Comparison of Individual and Combination DNA Vaccines for B. Anthracis, Ebola Virus, Marburg Virus and Venezuelan Equine Encephalitis Virus. Vaccine 2003, 21, 4071–4080. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-Dicaprio, K.M.; Geisbert, J.B.; Reed, D.S.; Feldmann, F.; Grolla, A.; Ströher, U.; Fritz, E.A.; Hensley, L.E.; Jones, S.M.; et al. Vesicular Stomatitis Virus-Based Vaccines Protect Nonhuman Primates against Aerosol Challenge with Ebola and Marburg Viruses. Vaccine 2008, 26, 6894–6900. [Google Scholar] [CrossRef]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Geisbert, J.B.; Ströher, U.; Hensley, L.E.; Grolla, A.; Fritz, E.A.; Feldmann, F.; Feldmann, H.; Jones, S.M. Cross-Protection against Marburg Virus Strains by Using a Live, Attenuated Recombinant Vaccine. J. Virol. 2006, 80, 9659–9666. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Geisbert, J.B.; Leung, A.; Daddario-DiCaprio, K.M.; Hensley, L.E.; Grolla, A.; Feldmann, H. Single-Injection Vaccine Protects Nonhuman Primates against Infection with Marburg Virus and Three Species of Ebola Virus. J. Virol. 2009, 83, 7296–7304. [Google Scholar] [CrossRef]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Ströher, U.; Geisbert, J.B.; Grolla, A.; Fritz, E.A.; Fernando, L.; Kagan, E.; Jahrling, P.B.; Hensley, L.E.; et al. Postexposure Protection against Marburg Haemorrhagic Fever with Recombinant Vesicular Stomatitis Virus Vectors in Non-Human Primates: An Efficacy Assessment. Lancet 2006, 367, 1399–1404. [Google Scholar] [CrossRef]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and Effectiveness of an rVSV-Vectored Vaccine in Preventing Ebola Virus Disease: Final Results from the Guinea Ring Vaccination, Open-Label, Cluster-Randomised Trial (Ebola Ça Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef]
- He, J.; Wu, J.; Chen, J.; Zhang, S.; Guo, Y.; Zhang, X.; Han, J.; Zhang, Y.; Guo, Y.; Lin, Y.; et al. Identification of Estradiol Benzoate as an Inhibitor of HBx Using Inducible Stably Transfected HepG2 Cells Expressing HiBiT Tagged HBx. Molecules 2022, 27, 5000. [Google Scholar] [CrossRef] [PubMed]
- Mahdian, S.; Zarrabi, M.; Panahi, Y.; Dabbagh, S. Repurposing FDA-Approved Drugs to Fight COVID-19 Using in Silico Methods: Targeting SARS-CoV-2 RdRp Enzyme and Host Cell Receptors (ACE2, CD147) through Virtual Screening and Molecular Dynamic Simulations. Inform. Med. Unlocked 2021, 23, 100541. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pan, X.; Huang, Y.; Cheng, C.; Xu, X.; Wu, Y.; Xu, Y.; Shang, W.; Niu, X.; Wan, Y.; et al. Drug Repurposing of Itraconazole and Estradiol Benzoate against COVID-19 by Blocking SARS-CoV-2 Spike Protein-Mediated Membrane Fusion. Adv. Ther. 2021, 4, 2000224. [Google Scholar] [CrossRef] [PubMed]
- Alsaady, I.M.; Bajrai, L.H.; Alandijany, T.A.; Gattan, H.S.; El-Daly, M.M.; Altwaim, S.A.; Alqawas, R.T.; Dwivedi, V.D.; Azhar, E.I. Cheminformatics Strategies Unlock Marburg Virus VP35 Inhibitors from Natural Compound Library. Viruses 2023, 15, 1739. [Google Scholar] [CrossRef]
- Caplen, N.J.; Zheng, Z.; Falgout, B.; Morgan, R.A. Inhibition of Viral Gene Expression and Replication in Mosquito Cells by dsRNA-Triggered RNA Interference. Mol. Ther. 2002, 6, 243–251. [Google Scholar] [CrossRef]
- Fowler, T.; Bamberg, S.; Möller, P.; Klenk, H.-D.; Meyer, T.F.; Becker, S.; Rudel, T. Inhibition of Marburg Virus Protein Expression and Viral Release by RNA Interference. J. Gen. Virol. 2005, 86, 1181–1188. [Google Scholar] [CrossRef]
- Müller, S.; Günther, S. Broad-Spectrum Antiviral Activity of Small Interfering RNA Targeting the Conserved RNA Termini of Lassa Virus. Antimicrob. Agents Chemother. 2007, 51, 2215–2218. [Google Scholar] [CrossRef]
- Gavrilov, K.; Saltzman, W.M. Therapeutic siRNA: Principles, Challenges, and Strategies. Yale J. Biol. Med. 2012, 85, 187–200. [Google Scholar]
- Ursic-Bedoya, R.; Mire, C.E.; Robbins, M.; Geisbert, J.B.; Judge, A.; MacLachlan, I.; Geisbert, T.W. Protection against Lethal Marburg Virus Infection Mediated by Lipid Encapsulated Small Interfering RNA. J. Infect. Dis. 2014, 209, 562–570. [Google Scholar] [CrossRef]
- Thi, E.P.; Mire, C.E.; Ursic-Bedoya, R.; Geisbert, J.B.; Lee, A.C.H.; Agans, K.N.; Robbins, M.; Deer, D.J.; Fenton, K.A.; MacLachlan, I.; et al. Marburg Virus Infection in Nonhuman Primates: Therapeutic Treatment by Lipid-Encapsulated siRNA. Sci. Transl. Med. 2014, 6, 250ra116. [Google Scholar] [CrossRef]
- Valmas, C.; Grosch, M.N.; Schümann, M.; Olejnik, J.; Martinez, O.; Best, S.M.; Krähling, V.; Basler, C.F.; Mühlberger, E. Marburg Virus Evades Interferon Responses by a Mechanism Distinct from Ebola Virus. PLoS Pathog. 2010, 6, e1000721. [Google Scholar] [CrossRef] [PubMed]
- Basler, C.F.; Amarasinghe, G.K. Evasion of Interferon Responses by Ebola and Marburg Viruses. J. Interferon Cytokine Res. 2009, 29, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Iversen, P.L.; Warren, T.K.; Wells, J.B.; Garza, N.L.; Mourich, D.V.; Welch, L.S.; Panchal, R.G.; Bavari, S. Discovery and Early Development of AVI-7537 and AVI-7288 for the Treatment of Ebola Virus and Marburg Virus Infections. Viruses 2012, 4, 2806–2830. [Google Scholar] [CrossRef]
- Heald, A.E.; Charleston, J.S.; Iversen, P.L.; Warren, T.K.; Saoud, J.B.; Al-Ibrahim, M.; Wells, J.; Warfield, K.L.; Swenson, D.L.; Welch, L.S.; et al. AVI-7288 for Marburg Virus in Nonhuman Primates and Humans. N. Engl. J. Med. 2015, 373, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Barochia, A.; Solomon, S.; Cui, X.; Natanson, C.; Eichacker, P.Q. Eritoran Tetrasodium (E5564) Treatment for Sepsis: Review of Preclinical and Clinical Studies. Expert Opin. Drug Metab. Toxicol. 2011, 7, 479–494. [Google Scholar] [CrossRef]
- Tidswell, M.; LaRosa, S.P. Toll-like Receptor-4 Antagonist Eritoran Tetrasodium for Severe Sepsis. Expert Rev. Anti-Infect. Ther. 2011, 9, 507–520. [Google Scholar] [CrossRef]
- Younan, P.; Ramanathan, P.; Graber, J.; Gusovsky, F.; Bukreyev, A. The Toll-Like Receptor 4 Antagonist Eritoran Protects Mice from Lethal Filovirus Challenge. mBio 2017, 8. [Google Scholar] [CrossRef]
- Bixler, S.L.; Goff, A.J. The Role of Cytokines and Chemokines in Filovirus Infection. Viruses 2015, 7, 5489–5507. [Google Scholar] [CrossRef]
- Guito, J.C.; Prescott, J.B.; Arnold, C.E.; Amman, B.R.; Schuh, A.J.; Spengler, J.R.; Sealy, T.K.; Harmon, J.R.; Coleman-McCray, J.D.; Kulcsar, K.A.; et al. Asymptomatic Infection of Marburg Virus Reservoir Bats Is Explained by a Strategy of Immunoprotective Disease Tolerance. Curr. Biol. 2021, 31, 257–270.e5. [Google Scholar] [CrossRef]
- Jarczak, D.; Nierhaus, A. Cytokine Storm—Definition, Causes, and Implications. Int. J. Mol. Sci. 2022, 23, 11740. [Google Scholar] [CrossRef]
- Dolmatova, E.V.; Wang, K.; Mandavilli, R.; Griendling, K.K. The Effects of Sepsis on Endothelium and Clinical Implications. Cardiovasc. Res. 2021, 117, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Schnittler, H.J.; Mahner, F.; Drenckhahn, D.; Klenk, H.D.; Feldmann, H. Replication of Marburg Virus in Human Endothelial Cells. A Possible Mechanism for the Development of Viral Hemorrhagic Disease. J. Clin. Investig. 1993, 91, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 Trafficking and Its Influence on LPS-Induced pro-Inflammatory Signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef] [PubMed]
- Mazgaeen, L.; Gurung, P. Recent Advances in Lipopolysaccharide Recognition Systems. Int. J. Mol. Sci. 2020, 21, 379. [Google Scholar] [CrossRef]
- Lucas, K.; Maes, M. Role of the Toll Like Receptor (TLR) Radical Cycle in Chronic Inflammation: Possible Treatments Targeting the TLR4 Pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef]
- Soares, J.-B.; Pimentel-Nunes, P.; Roncon-Albuquerque, R.; Leite-Moreira, A. The Role of Lipopolysaccharide/Toll-like Receptor 4 Signaling in Chronic Liver Diseases. Hepatol. Int. 2010, 4, 659–672. [Google Scholar] [CrossRef]
- Shirey, K.A.; Lai, W.; Scott, A.J.; Lipsky, M.; Mistry, P.; Pletneva, L.M.; Karp, C.L.; McAlees, J.; Gioannini, T.L.; Weiss, J.; et al. The TLR4 Antagonist Eritoran Protects Mice from Lethal Influenza Infection. Nature 2013, 497, 498–502. [Google Scholar] [CrossRef]
- Kim, H.M.; Park, B.S.; Kim, J.-I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal Structure of the TLR4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef]
- Korff, S.; Loughran, P.; Cai, C.; Lee, Y.S.; Scott, M.; Billiar, T.R. Eritoran Attenuates Tissue Damage and Inflammation in Hemorrhagic Shock/Trauma. J. Surg. Res. 2013, 184, e17–e25. [Google Scholar] [CrossRef]
- Kuzmich, N.N.; Sivak, K.V.; Chubarev, V.N.; Porozov, Y.B.; Savateeva-Lyubimova, T.N.; Peri, F. TLR4 Signaling Pathway Modulators as Potential Therapeutics in Inflammation and Sepsis. Vaccines 2017, 5, 34. [Google Scholar] [CrossRef]
- Cheng, H.; Lear-Rooney, C.M.; Johansen, L.; Varhegyi, E.; Chen, Z.W.; Olinger, G.G.; Rong, L. Inhibition of Ebola and Marburg Virus Entry by G Protein-Coupled Receptor Antagonists. J. Virol. 2015, 89, 9932–9938. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S. β-Arrestins and G Protein-Coupled Receptor Kinases in Viral Entry: A Graphical Review. Cell. Signal. 2023, 102, 110558. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, A.L.; Bond, A.C.S.; Bennett, L.J.; Craig, S.E.; Winski, D.P.; Kirkby, L.C.; Kraemer, A.R.; Kelly, K.G.; Hess, S.T.; Maginnis, M.S. GPCR Inhibitors Have Antiviral Properties against JC Polyomavirus Infection. Viruses 2024, 16, 1559. [Google Scholar] [CrossRef] [PubMed]
- Rwanda Biomedical Center (RBC). MARBURG Virus Information—Rwanda Biomedical Centre. Available online: https://rbc.gov.rw/marburg/ (accessed on 8 December 2024).
- Sibomana, J.P. Fight or Flight—Facing the Marburg Outbreak in Rwanda. N. Engl. J. Med. 2024, 391, 2070–2072. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Science. 16 October 2024. Available online: https://www.science.org/content/article/debate-erupts-about-how-deploy-experimental-marburg-drugs-and-vaccines-rwanda (accessed on 8 December 2024).
- Rwanda Ministry of Health. Rwanda National Guidelines for Management of Marburg Virus Disease; Rwanda Biomedical Center: Kigali, Rwanda, 2024. [Google Scholar]


{kind=link}
{kind=link}
| Antiviral Therapy | Type | Mechanism of Action | Mode of Administration | Efficacy Against MARV | Other Viral Targets | Regulatory Status |
|---|---|---|---|---|---|---|
| Favipiravir (T-705) | Nucleoside Analog | Inhibits RNA polymerase; induces lethal mutagenesis | Oral |
|
| Licensed for influenza treatment in Japan; studied during 2014 EBOV outbreak |
| Galidesivir (BCX4430) | Nucleoside Analog | Targets RNA-dependent RNA polymerase (RdRp) | Intraperitoneally (IP); intravenously (IV) |
| Effective against other filoviruses like Ebola virus | N/A |
| Remdesivir (GS-5734) | Monophosphoramidate Prodrug | Inhibits viral RNA synthesis via vRNA-dependent RNA polymerase | Intravenously (IV) |
| SUDV COVID-19 Hepatitis C Respiratory Syncytial Virus (RSV) Human Immunodeficiency Virus (HIV) | Authorized for treating COVID-19 in several countries |
| Obeldesivir (GS-5245) | Nucleoside Prodrug | Targets RNA-dependent RNA polymerase (RdRp) | Oral |
| SUDV COVID-19 | N/A |
| Treatment Modality | Virus | Antibody Type | Timing of Administration | Survival Rate/Outcome |
|---|---|---|---|---|
| Polyclonal IgG treatment [43] | MARV | Polyclonal IgG | 15–30 min after exposure, followed by additional doses at day 4 and day 8 | 100% protection (3/3 NHPs), with no clinical features and no viremia. MARV IgM response observed by day 4 and day 6 after challenge. IgG response increased and remained |
| Polyclonal IgG treatment [43] | MARV | Polyclonal IgG | 48 h, followed by additional doses at day 4, day 8, and day 12 | 2/3 NHPs showed no clinical signs and completely cured; 1/3 NHPs showed mild clinical signs associated with high liver enzyme and low viremia, however fully recovered by day 16 |
| Human monoclonal antibodies against MARV GP (MR-78-N; MR82-N; MR191-N) [44] | MARV, Ravn | Monoclonal antibody MR-78-N; MR82-N; MR191-N | 2 days post-inoculation in guinea pigs | 100% survival using MR-78-N and MR191-N, with virus not detected within plasma at 7 days post-infection; MR-82-N provided less protection and virus was detected within plasma |
| Monoclonal antibody MR191-N [44] | MARV | Monoclonal antibody MR191-N | 4 days post-inoculation | 100% survival, decline in viral load by day 7 post-inoculation with no evidence of virus by day 10 |
| MR186-YTE [31] | MARV | Monoclonal antibody MR186-YTE | 5 days post-inoculation single dose | 100% survival (4/4 NHPs), all animals developed clinical features of disease; however, by day 16, no evidence of virus had been detected |
| Combination therapy of MR186-YTE and remdesivir [31] | MARV | MR186-YTE + remdesivir | 6 days post-infection | 80% efficacy |
| Trial ID. | Phase | Location(s) | Date | Estimated Completion Date | Purpose | Trial Name |
|---|---|---|---|---|---|---|
| NCT05817422 | Phase 2 | Uganda and Kenya | 19 October 2023 | 1 May 2025 | Vaccine | Monovalent chimpanzee adenoviral-vectored Marburg virus vaccine in healthy adults |
| NCT06620003 | Phase 2 | USA | January 2025 | 7/2026 | Vaccine | A phase 2, randomized, double-blind, placebo-controlled trial to evaluate safety, tolerability, and immune responses of an investigational monovalent chimpanzee adenoviral-vectored Marburg virus vaccine in healthy adults |
| NCT03475056 | Phase 1 | USA | 9 October 2018 | 19 December 2019 | Vaccine | A phase 1, open-label study to examine the safety, tolerability, and immunogenicity of an investigational Marburg vaccine given by intramuscular (IM) injection to healthy adults. The study was a dose escalation of VRC-MARADC087-00-VP, a chimpanzee adenovirus serotype 3 (cAd3) vector vaccine, which encodes wild-type (WT) glycoprotein (GP) from Marburg virus |
| NCT06265012 | Phase 1 | USA | 5 February 2024 | 16 September 2024 | Vaccine | A phase 1 randomized, single-blind, placebo-controlled, ascending-dose study to evaluate the safety and immunogenicity of rVSV∆G-MARV-GP [Angola] (PHV01, Marburg virus glycoprotein [MARV GP] vaccine) in healthy adults. PHV01 is a live, attenuated rVSV vaccine expressing the MARV GP |
| NCT03800173 | Phase 1 | USA | 10 December 2018 | 30 April 2019 | Antiviral | A phase 1 double-blind, placebo-controlled, dose-ranging study to evaluate the safety, tolerability, and pharmacokinetics of galidesivir (BCX4430) administered as single doses via intravenous infusion in healthy subjects |
| NCT04723602 | Phase 1 | USA | 6 January 2021 | 14 December 2021 | Vaccine | A phase 1b trial to evaluate safety, tolerability, and immune responses of 2 monovalent chimpanzee adenoviral-vectored filovirus (Ebola-S and Marburg) vaccines to healthy adults, collection of plasma/serum for the purposes of assay development |
| NCT00997607 | Phase 1 | Uganda | February 2010 | April 2012 | Vaccine | A phase 1B study to evaluate the safety and immunogenicity of an Ebola DNA plasmid vaccine, VRC-EBODNA023-00-VP, and a Marburg DNA plasmid vaccine, VRC-MARDNA025-00-VP, in healthy adults in Kampala, Uganda |
| NCT00605514 | Phase 1 | USA | 25 January 2008 | 21 June 2010 | Vaccine | A phase 1 study to evaluate the safety and immunogenicity of an Ebola DNA plasmid vaccine, VRC-EBODNA023-00-VP, and a Marburg DNA plasmid vaccine, VRC-MARDNA025-00-VP, in healthy adults |
| NCT01353040 | USA | May 2011 | December 2011 | Treatment | A randomized, double-blind, placebo-controlled, single-dose, dose-escalation study to assess the safety, tolerability, and pharmacokinetics of AVI-6003 in healthy adult volunteers | |
| NCT02891980 | Phase 1 | USA | 24 March 2017 | 21 March 2019 | Vaccine | A phase 1 trial to utilize systems biology approaches to examine the safety, immunogenicity, and -omics response to MVA-BN(R)-Filo and Ad26.ZEBOV vaccines in healthy volunteers |
| NCT01566877 | USA | May 2013 | January 2014 | Treatment | A randomized, double-blind, placebo-controlled, multiple-dose, dose-escalation study to assess the safety, tolerability, and pharmacokinetics of AVI-7288 in healthy adult volunteers |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musafiri, S.; Siddig, E.E.; Nkuranga, J.B.; Rukundo, A.; Mpunga, T.; Sendegeya, A.; Twagirumugabe, T.; Ahmed, A.; Muvunyi, C.M. Emerging Strategies and Progress in the Medical Management of Marburg Virus Disease. Pathogens 2025, 14, 322. https://doi.org/10.3390/pathogens14040322
Musafiri S, Siddig EE, Nkuranga JB, Rukundo A, Mpunga T, Sendegeya A, Twagirumugabe T, Ahmed A, Muvunyi CM. Emerging Strategies and Progress in the Medical Management of Marburg Virus Disease. Pathogens. 2025; 14(4):322. https://doi.org/10.3390/pathogens14040322
Chicago/Turabian StyleMusafiri, Sanctus, Emmanuel Edwar Siddig, John Baptist Nkuranga, Athanase Rukundo, Tharcisse Mpunga, Augustin Sendegeya, Theogene Twagirumugabe, Ayman Ahmed, and Claude Mambo Muvunyi. 2025. "Emerging Strategies and Progress in the Medical Management of Marburg Virus Disease" Pathogens 14, no. 4: 322. https://doi.org/10.3390/pathogens14040322
APA StyleMusafiri, S., Siddig, E. E., Nkuranga, J. B., Rukundo, A., Mpunga, T., Sendegeya, A., Twagirumugabe, T., Ahmed, A., & Muvunyi, C. M. (2025). Emerging Strategies and Progress in the Medical Management of Marburg Virus Disease. Pathogens, 14(4), 322. https://doi.org/10.3390/pathogens14040322