

Mutational Analysis of Colistin-Resistant Pseudomonas aeruginosa Isolates: From Genomic Background to Antibiotic Resistance

 ,
,  ,
,  , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Pseudomonas aeruginosa Isolates
2.2. Evaluation of Antibacterial Activity for Colistin
2.3. Whole Genome Sequencing
2.4. Assembly and Annotation
2.5. Comparative Genomics Analysis
2.5.1. Phylogenomic Classification of Genomes
2.5.2. Identification of Antibiotic Resistance Genes and Virulence Genes
2.5.3. Genomic Characteristics of Colistin-Resistant P. aeruginosa and Mutation Analysis Based on Literature Reports
3. Results
3.1. P. aeruginosa Within-Strain Diversity
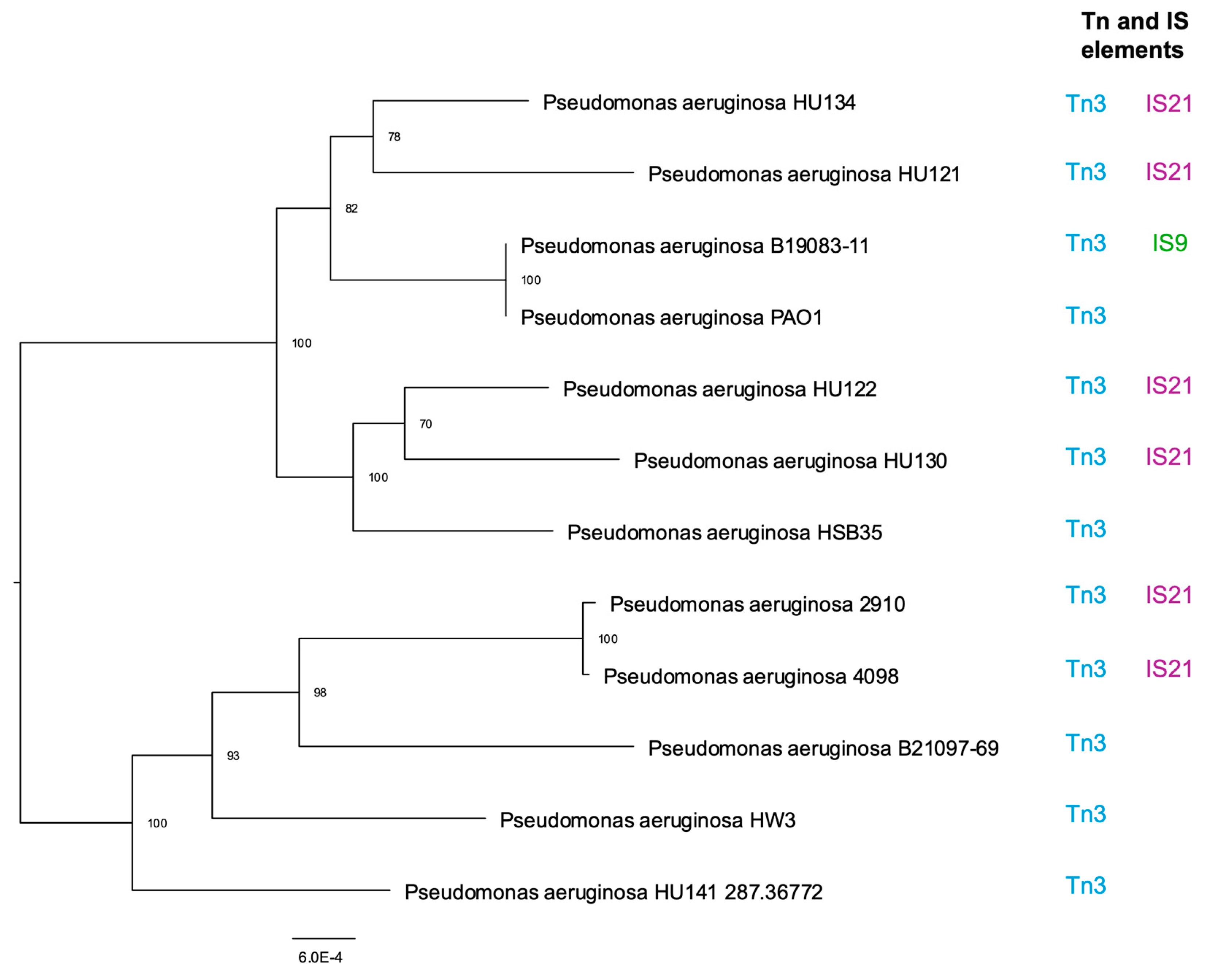
3.2. Tn and IS Elements
3.3. Geno-Phenotype in Antibiotic Resistance
3.4. Virulome
3.5. Genomic Profiling
3.6. Analysis of Mutations in Colistin-Resistant P. aeruginosa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laborda, P.; Sanz-García, F.; Hernando-Amado, S.; Martínez, J.L. Pseudomonas Aeruginosa: An Antibiotic Resilient Pathogen with Environmental Origin. Curr. Opin. Microbiol. 2021, 64, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Femenia, M.À.; Fernández-Muñoz, A.; Gomis-Font, M.A.; Taltavull, B.; López-Causapé, C.; Arca-Suárez, J.; Martínez-Martínez, L.; Cantón, R.; Larrosa, N.; Oteo-Iglesias, J.; et al. Pseudomonas aeruginosa Antibiotic Susceptibility Profiles, Genomic Epidemiology and Resistance Mechanisms: A Nation-Wide Five-Year Time Lapse Analysis. Lancet Reg. Health Eur. 2023, 34, 100736. [Google Scholar] [CrossRef] [PubMed]
- Avakh, A.; Grant, G.D.; Cheesman, M.J.; Kalkundri, T.; Hall, S. The Art of War with Pseudomonas aeruginosa: Targeting Mex Efflux Pumps Directly to Strategically Enhance Antipseudomonal Drug Efficacy. Antibiotics 2023, 12, 1304. [Google Scholar] [CrossRef] [PubMed]
- Jeannot, K.; Hagart, K.; Dortet, L.; Kostrzewa, M.; Filloux, A.; Plesiat, P.; Larrouy-Maumus, G. Detection of Colistin Resistance in Pseudomonas aeruginosa Using the MALDIxin Test on the Routine MALDI Biotyper Sirius Mass Spectrometer. Front. Microbiol. 2021, 12, 725383. [Google Scholar] [CrossRef]
- Biswas, S.; Brunel, J.M.; Dubus, J.C.; Reynaud-Gaubert, M.; Rolain, J.M. Colistin: An Update on the Antibiotic of the 21st Century. Expert Rev. Anti. Infect. Ther. 2012, 10, 917–934. [Google Scholar] [CrossRef]
- Fernández, L.; Álvarez-Ortega, C.; Wiegand, I.; Olivares, J.; Kocíncová, D.; Lam, J.S.; Martínez, J.L.; Hancock, R.E.W. Characterization of the Polymyxin B Resistome of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 110–119. [Google Scholar] [CrossRef]
- Lee, J.Y.; Park, Y.K.; Chung, E.S.; Na, I.Y.; Ko, K.S. Evolved Resistance to Colistin and Its Loss Due to Genetic Reversion in Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 25543. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chung, E.S.; Na, I.Y.; Kim, H.; Shin, D.; Ko, K.S. Development of Colistin Resistance in PmrA-, PhoP-, ParR- and CprR-Inactivated Mutants of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2014, 69, 2966–2971. [Google Scholar] [CrossRef]
- McPhee, J.B.; Bains, M.; Winsor, G.; Lewenza, S.; Kwasnicka, A.; Brazas, M.D.; Brinkman, F.S.L.; Hancock, R.E.W. Contribution of the PhoP-PhoQ and PmrA-PmrB Two-Component Regulatory Systems to Mg2+-Induced Gene Regulation in Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 3995–4006. [Google Scholar] [CrossRef]
- Beceiro, A.; Llobet, E.; Aranda, J.; Bengoechea, J.A.; Doumith, M.; Hornsey, M.; Dhanji, H.; Chart, H.; Bou, G.; Livermore, D.M.; et al. Phosphoethanolamine Modification of Lipid A in Colistin-Resistant Variants of Acinetobacter baumannii Mediated by the PmrAB Two-Component Regulatory System. Antimicrob. Agents Chemother. 2011, 55, 3370–3379. [Google Scholar] [CrossRef]
- Moskowitz, S.M.; Ernst, R.K.; Miller, S.I. PmrAB, a Two-Component Regulatory System of Pseudomonas aeruginosa That Modulates Resistance to Cationic Antimicrobial Peptides and Addition of Aminoarabinose to Lipid A. J. Bacteriol. 2004, 186, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Bolard, A.; Schniederjans, M.; Haüssler, S.; Triponney, P.; Valot, B.; Plésiat, P.; Jeannota, K. Production of Norspermidine Contributes to Aminoglycoside Resistance in Pmrab Mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, 10. [Google Scholar] [CrossRef]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Center for Genomic Epidemiology. Pseudomonas aeruginosa Serotyper. Available online: https://cge.food.dtu.dk/services/PAst/ (accessed on 25 May 2024).
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A Resource Combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella enterica Using a Newly Developed Web Tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Torsten Seemann Snippy. Available online: https://github.com/tseemann/snippy (accessed on 25 May 2024).
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- De Sousa, T.; Hébraud, M.; Dapkevicius, M.L.N.E.; Maltez, L.; Pereira, J.E.; Capita, R.; Alonso-Calleja, C.; Igrejas, G.; Poeta, P. Molecular Sciences Genomic and Metabolic Characteristics of the Pathogenicity in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2021, 22, 12892. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Dethlefsen, S.; Klockgether, J.; Tümmler, B. Phenotypic and Genomic Comparison of the Two Most Common ExoU-Positive Pseudomonas aeruginosa Clones, PA14 and ST235. mSystems 2020, 5, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Recio, R.; Sánchez-Diener, I.; Viedma, E.; Meléndez-Carmona, M.Á.; Villa, J.; Orellana, M.Á.; Mancheño, M.; Juan, C.; Zamorano, L.; Lora-Tamayo, J.; et al. Pathogenic Characteristics of Pseudomonas aeruginosa Bacteraemia Isolates in a High-Endemicity Setting for ST175 and ST235 High-Risk Clones. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 671–678. [Google Scholar] [CrossRef] [PubMed]
- del Barrio-Tofiño, E.; López-Causapé, C.; Oliver, A. Pseudomonas aeruginosa Epidemic High-Risk Clones and Their Association with Horizontally-Acquired β-Lactamases: 2020 Update. Int. J. Antimicrob. Agents 2020, 56, 106196. [Google Scholar] [CrossRef]
- Treepong, P.; Kos, V.N.; Guyeux, C.; Blanc, D.S.; Bertrand, X.; Valot, B.; Hocquet, D. Global Emergence of the Widespread Pseudomonas aeruginosa ST235 Clone. Clin. Microbiol. Infect. 2018, 24, 258–266. [Google Scholar] [CrossRef]
- Gómez-Zorrilla, S.; Juan, C.; Cabot, G.; Camoez, M.; Tubau, F.; Oliver, A.; Dominguez, M.A.; Ariza, J.; Peña, C. Impact of Multidrug Resistance on the Pathogenicity of Pseudomonas aeruginosa: In Vitro and in Vivo Studies. Int. J. Antimicrob. Agents 2016, 47, 368–374. [Google Scholar] [CrossRef]
- Sánchez-Diener, I.; Zamorano, L.; Peña, C.; Ocampo-Sosa, A.; Cabot, G.; Gómez-Zorrilla, S.; Almirante, B.; Aguilar, M.; Granados, A.; Calbo, E.; et al. Weighting the Impact of Virulence on the Outcome of Pseudomonas aeruginosa Bloodstream Infections. Clin. Microbiol. Infect. 2020, 26, 351–357. [Google Scholar] [CrossRef]
- Estepa, V.; Rojo-Bezares, B.; Torres, C.; Sáenz, Y. Genetic Lineages and Antimicrobial Resistance in Pseudomonas Spp. Isolates Recovered from Food Samples. Foodborne Pathog. Dis. 2015, 12, 486–491. [Google Scholar] [CrossRef]
- García-Castillo, M.; Del Campo, R.; Morosini, M.I.; Riera, E.; Cabot, G.; Willems, R.; Van Mansfeld, R.; Oliver, A.; Cantón, R. Wide Dispersion of ST175 Clone despite High Genetic Diversity of Carbapenem-Nonsusceptible Pseudomonas aeruginosa Clinical Strains in 16 Spanish Hospitals. J. Clin. Microbiol. 2011, 49, 2905–2910. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, D.; Ji, B.; Zhang, X.; Anbo, M.; Jelsbak, L. Whole-Genome Sequencing Reveals High-Risk Clones of Pseudomonas aeruginosa in Guangdong, China. Front. Microbiol. 2023, 14, 1134. [Google Scholar] [CrossRef]
- Subedi, D.; Vijay, A.K.; Kohli, G.S.; Rice, S.A.; Willcox, M. Comparative Genomics of Clinical Strains of Pseudomonas aeruginosa Strains Isolated from Different Geographic Sites. Sci. Rep. 2018, 8, 15668. [Google Scholar] [CrossRef]
- Dettman, J.R.; Kassen, R. Evolutionary Genomics of Niche-Specific Adaptation to the Cystic Fibrosis Lung in Pseudomonas aeruginosa. Mol. Biol. Evol. 2021, 38, 663–675. [Google Scholar] [CrossRef]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, 10-1128. [Google Scholar] [CrossRef]
- Benler, S.; Faure, G.; Altae-Tran, H.; Shmakov, S.; Zheng, F.; Koonin, E. Cargo Genes of Tn7-Like Transposons Comprise an Enormous Diversity of Defense Systems, Mobile Genetic Elements, and Antibiotic Resistance Genes. mBio 2021, 12, e02938-21. [Google Scholar] [CrossRef]
- Varani, A.; He, S.; Siguier, P.; Ross, K.; Chandler, M. The IS6 Family, a Clinically Important Group of Insertion Sequences Including IS26. Mob. DNA 2021, 12, 11. [Google Scholar] [CrossRef]
- Mahillon, J.; Léonard, C.; Chandler, M. IS Elements as Constituents of Bacterial Genomes. Res. Microbiol. 1999, 150, 675–687. [Google Scholar] [CrossRef]
- Mouton, J.W.; Den Hollander, J.G.; Horrevorts, A.M. Emergence of Antibiotic Resistance amongst Pseudomonas aeruginosa Isolates from Patients with Cystic Fibrosis. J. Antimicrob. Chemother. 1993, 31, 919–926. [Google Scholar] [CrossRef]
- Bai, Y.; Gong, Y.; Shen, F.; Li, H.; Cheng, Y.; Guo, J.; Liu, G.; Ji, A. fang Molecular Epidemiological Characteristics of Carbapenem-Resistant Pseudomonas aeruginosa Clinical Isolates in Southeast Shanxi, China. J. Glob. Antimicrob. Resist. 2024, 36, 301–306. [Google Scholar] [CrossRef]
- Pournaras, S.; Maniati, M.; Petinaki, E.; Tzouvelekis, L.S.; Tsakris, A.; Legakis, N.J.; Maniatis, A.N. Hospital Outbreak of Multiple Clones of Pseudomonas aeruginosa Carrying the Unrelated Metallo-β-Lactamase Gene Variants BlaVIM-2 and BlAVIM-4. J. Antimicrob. Chemother. 2003, 51, 1409–1414. [Google Scholar] [CrossRef]
- Tseng, S.P.; Hsueh, P.R.; Tsai, J.C.; Teng, L.J. Tn6001, a Transposon-like Element Containing the BlaVIM-3- Harboring Integron In450. Antimicrob. Agents Chemother. 2007, 51, 4187–4190. [Google Scholar] [CrossRef]
- Kazeminezhad, B.; Rad, A.B.; Gharib, A.; Zahedifard, S. BlaVIM and BlaIMP Genes Detection in Isolates of Carbapenem Resistant P. aeruginosa of Hospitalized Patients in Two Hospitals in Iran. Iran. J. Pathol. 2017, 12, 392. [Google Scholar] [CrossRef]
- Harris, P.N.A. Clinical Management of Infections Caused by Enterobacteriaceae That Express Extended-Spectrum β-Lactamase and AmpC Enzymes. Semin. Respir. Crit. Care Med. 2015, 36, 056–073. [Google Scholar] [CrossRef]
- Fraile-Ribot, P.A.; Cabot, G.; Mulet, X.; Periañez, L.; Luisa Martín-Pena, M.; Juan, C.; Pérez, J.L.; Oliver, A. Mechanisms Leading to in Vivo Ceftolozane/Tazobactam Resistance Development during the Treatment of Infections Caused by MDR Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2018, 73, 658–663. [Google Scholar] [CrossRef]
- Cabot, G.; Ocampo-Sosa, A.A.; Tubau, F.; Macia, M.D.; Rodríguez, C.; Moya, B.; Zamorano, L.; Suárez, C.; Peña, C.; Martínez-Martínez, L.; et al. Overexpression of AmpC and Efflux Pumps in Pseudomonas aeruginosa Isolates from Bloodstream Infections: Prevalence and Impact on Resistance in a Spanish Multicenter Study. Antimicrob. Agents Chemother. 2011, 55, 1906–1911. [Google Scholar] [CrossRef]
- Cabot, G.; Bruchmann, S.; Mulet, X.; Zamorano, L.; Moyá, B.; Juan, C.; Haussler, S.; Olivera, A. Pseudomonas aeruginosa Ceftolozane-Tazobactam Resistance Development Requires Multiple Mutations Leading to Overexpression and Structural Modification of Ampc. Antimicrob. Agents Chemother. 2014, 58, 3091–3099. [Google Scholar] [CrossRef]
- García-Betancur, J.C.; De La Cadena, E.; Mojica, M.F.; Hernández-Gómez, C.; Correa, A.; Radice, M.A.; Castañeda-Méndez, P.; Jaime-Villalon, D.A.; Gales, A.C.; Munita, J.M.; et al. Comparative In Vitro Activity of Ceftolozane/Tazobactam against Clinical Isolates of Pseudomonas aeruginosa and Enterobacterales from Five Latin American Countries. Antibiotics 2022, 11, 1101. [Google Scholar] [CrossRef]
- Ruedas-López, A.; Alonso-García, I.; Lasarte-Monterrubio, C.; Guijarro-Sánchez, P.; Gato, E.; Vázquez-Ucha, J.C.; Vallejo, J.A.; Fraile-Ribot, P.A.; Fernández-Pérez, B.; Velasco, D.; et al. Selection of AmpC β-Lactamase Variants and Metalloβ- Lactamases Leading to Ceftolozane/Tazobactam and Ceftazidime/Avibactam Resistance during Treatment of MDR/XDR Pseudomonas aeruginosa Infections. Antimicrob. Agents Chemother. 2022, 66, e02067-21. [Google Scholar] [CrossRef]
- Berrazeg, M.; Jeannot, K.; Ntsogo Enguéné, V.Y.; Broutin, I.; Loeffert, S.; Fournier, D.; Plésiat, P. Mutations in β-Lactamase AmpC Increase Resistance of Pseudomonas aeruginosa Isolates to Antipseudomonal Cephalosporins. Antimicrob. Agents Chemother. 2015, 59, 6248–6255. [Google Scholar] [CrossRef]
- Ding, Y.; Hao, J.; Xiao, W.; Ye, C.; Xiao, X.; Jian, C.; Tang, M.; Li, G.; Liu, J.; Zeng, Z. Role of Efflux Pumps, Their Inhibitors, and Regulators in Colistin Resistance. Front. Microbiol. 2023, 14, 1207441. [Google Scholar] [CrossRef]
- Rojo-Bezares, B.; Estepa, V.; Cebollada, R.; de Toro, M.; Somalo, S.; Seral, C.; Castillo, F.J.; Torres, C.; Sáenz, Y. Carbapenem-Resistant Pseudomonas aeruginosa Strains from a Spanish Hospital: Characterization of Metallo-Beta-Lactamases, Porin OprD and Integrons. Int. J. Med. Microbiol. 2014, 304, 405–414. [Google Scholar] [CrossRef]
- Adewoye, L.; Sutherland, A.; Srikumar, R.; Poole, K. The MexR Repressor of the MexAB-OprM Multidrug Efflux Operon in Pseudomonas aeruginosa: Characterization of Mutations Compromising Activity. J. Bacteriol. 2002, 184, 4308–4312. [Google Scholar] [CrossRef] [PubMed]
- Suresh, M.; Nithya, N.; Jayasree, P.R.; Vimal, K.P.; Manish Kumar, P.R. Mutational Analyses of Regulatory Genes, MexR, NalC, NalD and MexZ of MexAB-OprM and MexXY Operons, in Efflux Pump Hyperexpressing Multidrug-Resistant Clinical Isolates of Pseudomonas aeruginosa. World J. Microbiol. Biotechnol. 2018, 34, 83. [Google Scholar] [CrossRef]
- Vettoretti, L.; Plésiat, P.; Muller, C.; El Garch, F.; Phan, G.; Attrée, I.; Ducruix, A.; Llanes, C. Efflux Unbalance in Pseudomonas aeruginosa Isolates from Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2009, 53, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Galimand, M.; Lambert, T.; Gerbaud, G.; Courvalin, P. Characterization of the Aac(6’)-Ib Gene Encoding an Aminoglycoside 6’-N- Acetyltransferase in Pseudomonas aeruginosa BM2656. Antimicrob. Agents Chemother. 1993, 37, 1456–1462. [Google Scholar] [CrossRef] [PubMed]
- Gharbi, M.; Abbas, M.A.S.; Hamrouni, S.; Maaroufi, A. First Report of Aac(6′)-Ib and Aac(6′)-Ib-Cr Variant Genes Associated with Mutations in GyrA Encoded Fluoroquinolone Resistance in Avian Campylobacter coli Strains Collected in Tunisia. Int. J. Mol. Sci. 2023, 24, 16116. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Nikolaidis, N.; Tolmasky, M.E. Rise and Dissemination of Aminoglycoside Resistance: The Aac(6′)-Ib Paradigm. Front. Microbiol 2013, 4, 121. [Google Scholar] [CrossRef]
- Van Hoek, A.H.A.M.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J.M. Acquired Antibiotic Resistance Genes: An Overview. Front Microbiol 2011, 2, 203. [Google Scholar] [CrossRef]
- Zheng, D.; Bergen, P.J.; Landersdorfer, C.B.; Hirsch, E.B. Differences in Fosfomycin Resistance Mechanisms between Pseudomonas aeruginosa and Enterobacterales. Antimicrob. Agents Chemother. 2022, 66, e01446-21. [Google Scholar] [CrossRef]
- Ebrahim-Saraie, H.S.; Heidari, H.; Soltani, B.; Mardaneh, J.; Motamedifar, M. Prevalence of Antibiotic Resistance and Integrons, Sul and Smqnr Genes in Clinical Isolates of Stenotrophomonas maltophilia from a Tertiary Care Hospital in Southwest Iran. Iran J. Basic Med. Sci. 2019, 22, 872. [Google Scholar] [CrossRef]
- Venkatesan, M.; Fruci, M.; Verellen, L.A.; Skarina, T.; Mesa, N.; Flick, R.; Pham, C.; Mahadevan, R.; Stogios, P.J.; Savchenko, A. Molecular Mechanism of Plasmid-Borne Resistance to Sulfonamide Antibiotics. Nat. Commun. 2023, 14, 4031. [Google Scholar] [CrossRef]
- Pierson, L.S.; Pierson, E.A. Metabolism and Function of Phenazines in Bacteria: Impacts on the Behavior of Bacteria in the Environment and Biotechnological Processes. Appl. Microbiol. Biotechnol. 2010, 86, 1659–1670. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xiao, W.; Zhou, C.; Pu, Q.; Deng, X.; Lan, L.; Liang, H.; Song, X.; Wu, M. Pseudomonas aeruginosa: Pathogenesis, Virulence Factors, Antibiotic Resistance, Interaction with Host, Technology Advances and Emerging Therapeutics. Signal Transduct. Target Ther. 2022, 7, 199. [Google Scholar] [CrossRef]
- Trouillon, J.; Attrée, I.; Elsen, S. The Regulation of Bacterial Two-Partner Secretion Systems. Mol. Microbiol. 2023, 120, 159–177. [Google Scholar] [CrossRef]
- Linhartová, I.; Bumba, L.; Mašn, J.; Basler, M.; Osička, R.; Kamanová, J.; Procházková, K.; Adkins, I.; HejnováHolubová, J.; Sadílková, L.; et al. RTX Proteins: A Highly Diverse Family Secreted Bya Common Mechanism. FEMS Microbiol. Rev. 2010, 34, 1076–1112. [Google Scholar] [CrossRef]
- Elhosseiny, N.M.; El-Tayeb, O.M.; Yassin, A.S.; Lory, S.; Attia, A.S. The Secretome of Acinetobacter Baumannii ATCC 17978 Type II Secretion System Reveals a Novel Plasmid Encoded Phospholipase That Could Be Implicated in Lung Colonization. Int. J. Med. Microbiol. 2016, 306, 633–641. [Google Scholar] [CrossRef]
- Lo Sciuto, A.; Imperi, F. Aminoarabinosylation of Lipid a Is Critical for the Development of Colistin Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, 10-1128. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ko, K.S. Mutations and Expression of PmrAB and PhoPQ Related with Colistin Resistance in Pseudomonas aeruginosa Clinical Isolates. Diagn. Microbiol. Infect. Dis. 2014, 78, 271–276. [Google Scholar] [CrossRef]
- Nirwan, P.K.; Chatterjee, N.; Panwar, R.; Dudeja, M.; Jaggi, N. Mutations in Two Component System (PhoPQ and PmrAB) in Colistin Resistant Klebsiella pneumoniae from North Indian Tertiary Care Hospital. J. Antibiot. 2021, 74, 450–457. [Google Scholar] [CrossRef]
- McPhee, J.B.; Lewenza, S.; Hancock, R.E.W. Cationic Antimicrobial Peptides Activate a Two-Component Regulatory System, PmrA-PmrB, That Regulates Resistance to Polymyxin B and Cationic Antimicrobial Peptides in Pseudomonas aeruginosa. Mol. Microbiol. 2003, 50, 205–217. [Google Scholar] [CrossRef]
- Chen, H.D.; Groisman, E.A. The Biology of the PmrA/PmrB Two-Component System: The Major Regulator of Lipopolysaccharide Modifications. Annu. Rev. Microbiol. 2013, 67, 83–112. [Google Scholar] [CrossRef]
- Barrow, K.; Kwon, D.H. Alterations in Two-Component Regulatory Systems of PhoPQ and PmrAB Are Associated with Polymyxin B Resistance in Clinical Isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2009, 53, 5150–5154. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, S.M.; Brannon, M.K.; Dasgupta, N.; Pier, M.; Sgambati, N.; Miller, A.K.; Selgrade, S.E.; Miller, S.I.; Denton, M.; Conway, S.P.; et al. PmrB Mutations Promote Polymyxin Resistance of Pseudomonas aeruginosa Isolated from Colistin-Treated Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2012, 56, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Liu, C.; Pan, X.; Fu, W.; Fan, Z.; Jin, Y.; Bai, F.; Cheng, Z.; Wu, W. Identification of Novel PhoP-PhoQ Regulated Genes That Contribute to Polymyxin B Tolerance in Pseudomonas aeruginosa. Microorganisms 2021, 9, 344. [Google Scholar] [CrossRef]
- Erdmann, M.B.; Gardner, P.P.; Lamont, I.L. The PitA Protein Contributes to Colistin Susceptibility in Pseudomonas aeruginosa. PLoS ONE 2023, 18, e0292818. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.K.; Brannon, M.K.; Stevens, L.; Johansen, H.K.; Selgrade, S.E.; Miller, S.I.; Høiby, N.; Moskowitz, S.M. PhoQ Mutations Promote Lipid A Modification and Polymyxin Resistance of Pseudomonas aeruginosa Found in Colistin-Treated Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2011, 55, 5761–5769. [Google Scholar] [CrossRef]
- Gatzeva-Topalova, P.Z.; May, A.P.; Sousa, M.C. Structure and Mechanism of ArnA: Conformational Change Implies Ordered Dehydrogenase Mechanism in Key Enzyme for Polymyxin Resistance. Structure 2005, 13, 929–942. [Google Scholar] [CrossRef]
- Lee, J.Y.; Na, I.Y.; Park, Y.K.; Ko, K.S. Genomic Variations between Colistin-Susceptible and-Resistant Pseudomonas aeruginosa Clinical Isolates and Their Effects on Colistin Resistance. J. Antimicrob. Chemother. 2014, 69, 1248–1256. [Google Scholar] [CrossRef]
- López-Causapé, C.; Cabot, G.; del Barrio-Tofiño, E.; Oliver, A. The Versatile Mutational Resistome of Pseudomonas aeruginosa. Front. Microbiol. 2018, 9, 354671. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Isolate Reference No. | Biosample Accession | Host Disease | Isolation Source | Year | Geolocation |
|---|---|---|---|---|---|
| HU121 | SAMN40216622 | Urinary tract infection | Urine | 2022 | Portugal |
| HU122 | SAMN40216623 | Urinary tract infection | Urine | 2022 | Portugal |
| HU130 | SAMN40216624 | Urinary tract infection | Urine | 2022 | Portugal |
| HU134 | SAMN40216625 | Urinary tract infection | Urine | 2022 | Portugal |
| HU141 | SAMN40216632 | Urinary tract infection | Urine | 2022 | Portugal |
| HW3 | SAMN40216626 | Wound infection | Skin wound | 2022 | Portugal |
| HSB35 | SAMN40216631 | Pulmonary infection | Bronchial secretion | 2022 | Portugal |
| 4098 | SAMN40216628 | Urinary tract infection | Urine | 2012 | Taiwan |
| 2910 | SAMN40216627 | Urinary tract infection | Urine | 2010 | Taiwan |
| B19083-11 | SAMN40216629 | Bacteremia | Blood | 2019 | Taiwan |
| B21097-69 | SAMN40216630 | Bacteremia | Blood | 2021 | Taiwan |
| P. aeruginosa Reference No. | Genome Size (bp) | DNA GC (%) | Protein Coding Genes | rRNA Genes | tRNA Genes | ST | Serotype |
|---|---|---|---|---|---|---|---|
| HU121 | 6,474,782 | 66.39 | 6133 | 5 | 58 | 2438 | O6 |
| HU122 | 6,488,087 | 66.36 | 6163 | 5 | 58 | 270 | O2 |
| HU130 | 6,707,783 | 66.04 | 6450 | 7 | 60 | 348 | O12 |
| HU134 | 6,361,249 | 66.48 | 5997 | 6 | 56 | 4258 | O6 |
| HU141 | 6,983,523 | 65.85 | 6752 | 4 | 63 | 253 | O12 |
| HW3 | 6,774,228 | 65.97 | 6486 | 7 | 59 | 1601 | O11 |
| HSB35 | 6,307,730 | 66.48 | 5966 | 3 | 62 | 1053 | O2 |
| 4098 | 7,003,552 | 65.95 | 6880 | 12 | 63 | 4258 | O11 |
| 2910 | 6,962,389 | 66.00 | 6708 | 12 | 65 | 298 | O11 |
| B19083-11 | 6,713,745 | 66.10 | 6472 | 11 | 63 | 3002 | O5 |
| B21097-69 | 7,017,151 | 66.06 | 6977 | 12 | 64 | 235 | O11 |
| Class and⁄or Antimicrobial | Breakpoints (mm; S≥/R<) | MIC (mg/L; S≥/R<) | Number of Resistant Isolates |
|---|---|---|---|
| Ampicillin | - | - | 1 |
| Amoxicillin/Clavulanate | - | - | 2 |
| Piperacillin | 50/18 | 0.001/16 | 9 |
| Piperacillin/Tazobactam | 50/18 | 0.001/16 | 11 |
| Ceftazidime | 50/17 | 0.001/8 | 10 |
| Cefepime | 50/21 | 0.001/8 | 11 |
| Doripenem | 50/22 | 0.001/2 | 11 |
| Ertapenem | - | - | 2 |
| Imipenem | 50/20 | 0.001/4 | 9 |
| Meropenem | 20/14 | 02/8 | 6 |
| Aztreonam | 50/18 | 0.001/16 | 4 |
| Ciprofloxacin | 50/26 | 0.001/0.5 | 11 |
| Levofloxacin | 50/18 | 0.001/2 | 8 |
| Amikacin | 15/15 | 16/16 | 3 |
| Tobramicyn | 18/18 | 02/f2 | 1 |
| Gentamicin | 15/15 | - | 5 |
| Colistin * | - | 2 * | 11 |
| Isolates | Phenotype Resistance | Resistance Genes |
|---|---|---|
| HU121 | PIP/TAZ CAZ CIP FEP DOR IMP LVX PIP | aph(3′)-Iib, catB7, blaPDC-3, blaOXA-486, blaOXA-903, fosA |
| HU122 | ERT PIP/TAZ CAZ CIP IMP LVX FEP DOR PIP | aph(3′)-Iib, catB7, blaPDC-8, blaOXA-486, blaOXA-903, fosA |
| HU130 | PIP/TAZ CAZ CIP FEP DOR IMP MEM LVX PIP | aph(3′)-Iib, catB8, blaPDC-5, blaOXA-396, blaOXA-494, fosA, sul1 |
| HU134 | ERT PIP/TAZ CIP FEP DOR IMP PIP | aph(3′)-Iib, catB7, blaPDC-3, blaOXA-396, blaOXA-494, fosA |
| HU141 | PIP/TAZ CAZ CIP FEP DOR GM TOB IMP LVX MEM PIP | aph(3′)-Iib, aadA7, catB7, blaPDC-34, blaOXA-488, fosA, sul1 |
| HW3 | AMP ATM CAZ CIP FEP DOR IMP LVX MEM PIP TZP | aph(3′)-Iib, catB7, blaPDC-39, blaOXA-396, blaOXA-494, fosA, |
| HSB35 | ATM CAZ CIP FEP DOR LVX PIP TZP | aph(3′)-Iib, catB7, blaOXA-396, blaOXA-847, blaOXA-494, blaPCD-1, fosA, crpP |
| 4098 | AK AMC ATM CAZ CIP FEP DOR GM IPM MEM PIP TZP | aph(3′)-Iib, catB7, blaVIM-3, blaPDC-16, blaOXA-395, blaOXA-848, fosA, sul1, qacE |
| 2910 | AK ATM CAZ CIP FEP DOR GM IPM MEM PIP TZP | aph(3′)-IIb, aac(6′)-Ib3, aac(6′)-Ib-cr, aac(6′)-Ib-Hangzhou, ant(2″)-Ia, catB7, blaVIM-3, blaPDC-16, blaOXA-395, blaOXA-848, fosA, fosX, sul1, crpP, qacE |
| B19083-11 | AK AMC CAZ CIP FEP DOR GM IPM LVX MEM TZP | aph(3′)-IIb, aac(6′)-Ib3, aac(6′)-Ib-cr, ant(2″)-Ia, catB7, blaPDC-3, blaOXA-10, blaOXA-50, blaSHV-12, fosA, sul1, crpP |
| B21097-69 | CAZ CIP FEP DOR GM LVX TZP | aph(3′)-IIb, aac(6′)-IIa, ant(2″)-Ia, catB2, catB7, blaPDC-35, blaOXA-488, blaOXA-17, fosA, crpP, tet(G), floR, qacE |
| Isolates | Related Genes | Type of Mutation | Gene and Protein Change | Locus_Tag |
|---|---|---|---|---|
| HU134 | cprS | snp | missense_variant c.347C > T p.Ala116Val | PA3078 |
| parS | snp | missense_variant c.1033G > A p.Ala345Thr | PA1798 | |
| HU141 | parS | snp | missense_variant c.467C > T p.Pro156Leu | PA1798 |
| HW3 | cprS | snp | missense_variant c.322G > A p.Asp108Asn | PA3078 |
| missense_variant c.475G > A p.Val159Ile | ||||
| missense_variant c.1158G > C p.Glu386Asp | ||||
| 2910 | parR | snp | missense_variant c.695G > A p.Gly232Asp | PA1799 |
| pmrB | snp | missense_variant c.73T > G p.Trp25Gly | PA4777 | |
| ins | frameshift_variant c.1403dupG p.Gly469fs | |||
| rsmA | del | non_coding_transcript_variant | PA0905 | |
| 4098 | parR | snp | missense_variant c.695G > A p.Gly232Asp | PA1799 |
| pmrB | snp | missense_variant c.739G > A p.Ala247Thr | PA4777 | |
| rsmA | del | non_coding_transcript_variant | PA0905 | |
| B19083-11 | cprS | snp | missense_variant c.1282G > T p.Ala428Ser | PA3078 |
| parR | snp | missense_variant c.259G > A p.Glu87Lys | PA1799 | |
| phoQ | complex | frameshift_variant&missense_variant c p.Val152fs | PA1180 | |
| rsmA | del | non_coding_transcript_variant | PA0905 | |
| B21097-69 | cprS | snp | missense_variant c.47C > G p.Thr16Ser | PA3078 |
| ins | frameshift_variant c.209_210insT p.Phe71fs | |||
| del | disruptive_inframe_deletion c.273_275delGCC p.Pro92del | |||
| snp | missense_variant c.1158G > C p.Glu386Asp | |||
| parS | ins | frameshift_variant c.248_249insA p.Gln84fs | PA1798 | |
| parR | snp | missense_variant c.509G > A p.Ser170Asn | PA1799 | |
| snp | synonymous_variant c.465C > A p.Ile155Ile | |||
| pmrB | snp | missense_variant c.739G > A p.Ala247Thr | PA4777 | |
| phoQ | complex | missense_variant c.254_258delACGACinsTCGAT p.Tyr85Phe | PA1180 | |
| rsmA | ins | non_coding_transcript_variant | PA0905 | |
| del | non_coding_transcript_variant |
| Isolates | Related Genes | Type of Mutation | Protein Change | Locus_Tag |
|---|---|---|---|---|
| HU121 | arnB | snp | Val302Ala | PA3552 |
| arnA | complex | CysSer312SerGly | PA3554 | |
| arnT | snp | Val266Ile | PA3556 | |
| HU122 | arnB | snp | Val302Ala | PA3552 |
| arnC | snp | Arg198His | PA3553 | |
| arnA | complex | CysSer312SerGly | PA3554 | |
| arnT | del | Leu11del | PA3556 | |
| snp | Val266Ile | |||
| snp | Ala282Ser | |||
| snp | Arg502Gln | |||
| snp | Ile509Val | |||
| HU130 | arnB | snp | Val302Ala | PA3552 |
| arnC | del | Ala274_Phe277del | PA3553 | |
| arnA | complex | CysSer312SerGly | PA3554 | |
| arnT | snp | Ala267Ser | PA3556 | |
| snp | Ala330Val | |||
| snp | Leu439Ile | |||
| snp | Arg502Gln | |||
| snp | lle509Val | |||
| arnF | snp | Val14Met | PA3558 | |
| HU134 | arnB | snp | Lys286Glu | PA3552 |
| snp | Val302Ala | |||
| arnA | complex | CysSer312SerGly | PA3554 | |
| snp | Ile388Val | |||
| arnD | snp | Phe58Leu | PA3555 | |
| arnT | snp | Val88Phe | PA3556 | |
| snp | Thr443Ile | |||
| snp | Arg446His | |||
| snp | Arg502Gln | |||
| snp | Ile509Val | |||
| arnF | snp | Val14Met | PA3558 | |
| HU141 | arnB | snp | Val302Ala | PA3552 |
| snp | Glu376Asp | |||
| arnC | snp | Ala327Val | PA3553 | |
| arnA | complex | CysSer312SerGly | PA3554 | |
| snp | Ile388Val | |||
| arnD | snp | Glu25Asp | PA3555 | |
| complex | Ala118Val | |||
| snp | Val123Ala | |||
| arnT | snp | Cys7Trp | PA3556 | |
| complex | His151Tyr | |||
| snp | Thr166Ile | |||
| snp | Ala265Val | |||
| snp | Ile509Val | |||
| arnE | snp | Ala109Val | PA3557 | |
| arnF | snp | Val14Met | PA3558 | |
| complex | Ala55Thr | |||
| HW3 | arnB | snp | Val302Ala | PA3552 |
| snp | Glu376Asp | |||
| arnA | complex | Phe80Tyr | PA3554 | |
| snp | Thr297Ala | |||
| complex | CysSer312SerGly | |||
| snp | Ile388Val | |||
| arnD | snp | Phe58Leu | PA3555 | |
| arnT | snp | His151Tyr | PA3556 | |
| snp | Ala267Ser | |||
| snp | Leu337Gln | |||
| snp | Thr443Ala | |||
| snp | Ile509Val | |||
| arnF | snp | Val113Met | PA3558 | |
| HSB35 | arnB | snp | Lys286Glu | PA3552 |
| snp | Val302Ala | |||
| arnA | complex | CysSer312SerGly | PA3554 | |
| snp | Gln661Leu | |||
| arnD | snp | Ser272Asn | PA3555 | |
| arnT | snp | Cys7Trp | PA3556 | |
| snp | Ala267Ser | |||
| snp | Arg446His | |||
| snp | Val511Met | |||
| 2910 | arnB | snp | Thr143Ala | PA3552 |
| snp | Lys286Glu | |||
| snp | Val302Ala | |||
| snp | Arg340Cys | |||
| snp | Glu375Lys | |||
| snp | Glu376Asp | |||
| arnC | snp | Ala327Val | PA3553 | |
| arnA | complex | CysSer312SerGly | PA3554 | |
| snp | Ile388Val | |||
| arnD | snp | Glu25Asp | PA3555 | |
| complex | Ala118Val | |||
| snp | Val123Ala | |||
| arnT | complex | His151Tyr | PA3556 | |
| snp | Thr166Ile | |||
| snp | Ala265Val | |||
| snp | Thr443Ala | |||
| snp | Ile509Val | |||
| arnE | snp | Thr13Ser | PA3557 | |
| 4098 | arnB | snp | Thr143Ala | PA3552 |
| snp | Lys286Glu | |||
| snp | Val302Ala | |||
| snp | Arg340Cys | |||
| snp | Glu375Lys | |||
| snp | Glu376Asp | |||
| arnC | snp | Ala327Val | PA3553 | |
| arnA | complex | CysSer312SerGly | PA3554 | |
| snp | Ile388Val | |||
| arnD | snp | Glu25Asp | PA3555 | |
| complex | Ala118Val | |||
| snp | Val123Ala | |||
| arnT | complex | His151Tyr | PA3556 | |
| snp | Thr166Ile | |||
| snp | Ala265Val | |||
| snp | Asn346Asp | |||
| snp | Thr443Ala | |||
| complex | Leu493fs | |||
| snp | Ile509Val | |||
| arnE | snp | Thr13Ser | PA3557 | |
| B19083-11 | arnB | snp | Val302Ala | PA3552 |
| arnA | complex | CysSerProGln312SerGlyProLys | PA3554 | |
| arnT | snp | Gly156Arg | PA3556 | |
| snp | Asp232Asn | |||
| snp | Ala267Ser | |||
| snp | Arg446His | |||
| snp | Ile509Val | |||
| B21097-69 | arnB | snp | Arg105Ser | PA3552 |
| snp | Val302Ala | |||
| snp | Glu376Asp | |||
| arnC | snp | Ala327Val | PA3553 | |
| arnA | complex | CysSer312SerGly | PA3554 | |
| snp | Ile388Val | |||
| ins | Ala662fs | |||
| arnD | snp | Phe58Leu | PA3555 | |
| snp | Ser272Asn | |||
| arnT | snp | Cys7Trp | PA3556 | |
| complex | His151Tyr | |||
| snp | Thr166Ile | |||
| snp | Ala265Val | |||
| snp | Thr443Ala | |||
| snp | Val468Met | |||
| snp | Ile509Val | |||
| snp | 16Arg535Leu | |||
| arnE | snp | Arg28His | PA3557 | |
| snp | Ala109Val | |||
| arnF | snp | Val14Met | PA3558 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Sousa, T.; Wang, H.-Y.; Lin, T.-W.; Caniça, M.; Ramos, M.J.N.; Santos, D.; Silva, C.; Saraiva, S.; Beyrouthy, R.; Bonnet, R.; et al. Mutational Analysis of Colistin-Resistant Pseudomonas aeruginosa Isolates: From Genomic Background to Antibiotic Resistance. Pathogens 2025, 14, 387. https://doi.org/10.3390/pathogens14040387
De Sousa T, Wang H-Y, Lin T-W, Caniça M, Ramos MJN, Santos D, Silva C, Saraiva S, Beyrouthy R, Bonnet R, et al. Mutational Analysis of Colistin-Resistant Pseudomonas aeruginosa Isolates: From Genomic Background to Antibiotic Resistance. Pathogens. 2025; 14(4):387. https://doi.org/10.3390/pathogens14040387
Chicago/Turabian StyleDe Sousa, Telma, Hsin-Yao Wang, Ting-Wei Lin, Manuela Caniça, Miguel J. N. Ramos, Daniela Santos, Catarina Silva, Sónia Saraiva, Racha Beyrouthy, Richard Bonnet, and et al. 2025. "Mutational Analysis of Colistin-Resistant Pseudomonas aeruginosa Isolates: From Genomic Background to Antibiotic Resistance" Pathogens 14, no. 4: 387. https://doi.org/10.3390/pathogens14040387
APA StyleDe Sousa, T., Wang, H.-Y., Lin, T.-W., Caniça, M., Ramos, M. J. N., Santos, D., Silva, C., Saraiva, S., Beyrouthy, R., Bonnet, R., Hébraud, M., Igrejas, G., & Poeta, P. (2025). Mutational Analysis of Colistin-Resistant Pseudomonas aeruginosa Isolates: From Genomic Background to Antibiotic Resistance. Pathogens, 14(4), 387. https://doi.org/10.3390/pathogens14040387