Targeting Plasmodium falciparum Hsp90: Towards Reversing Antimalarial Resistance

Abstract
:
{kind=link}
{kind=link}
{kind=link}
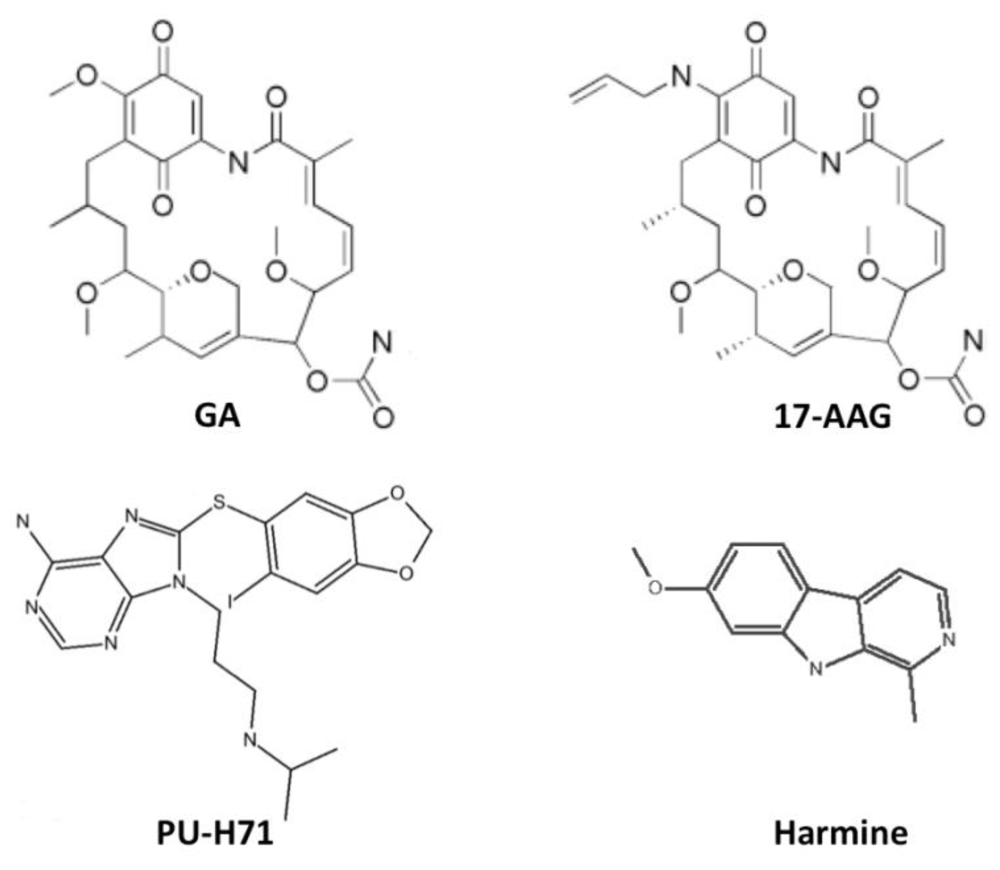{kind=link}
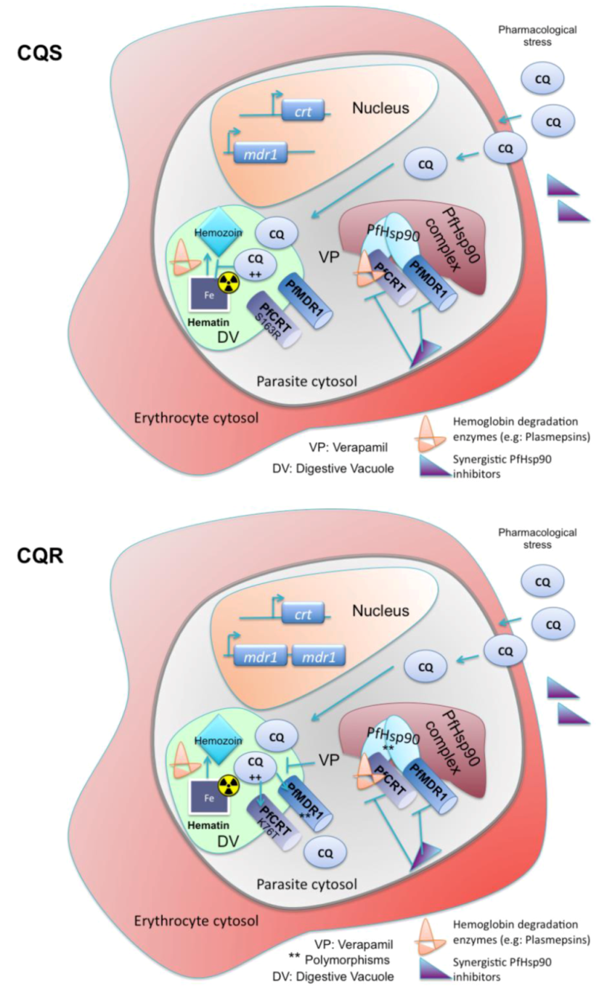{kind=link}
1. Introduction
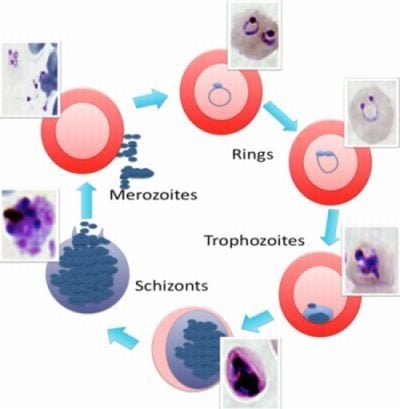
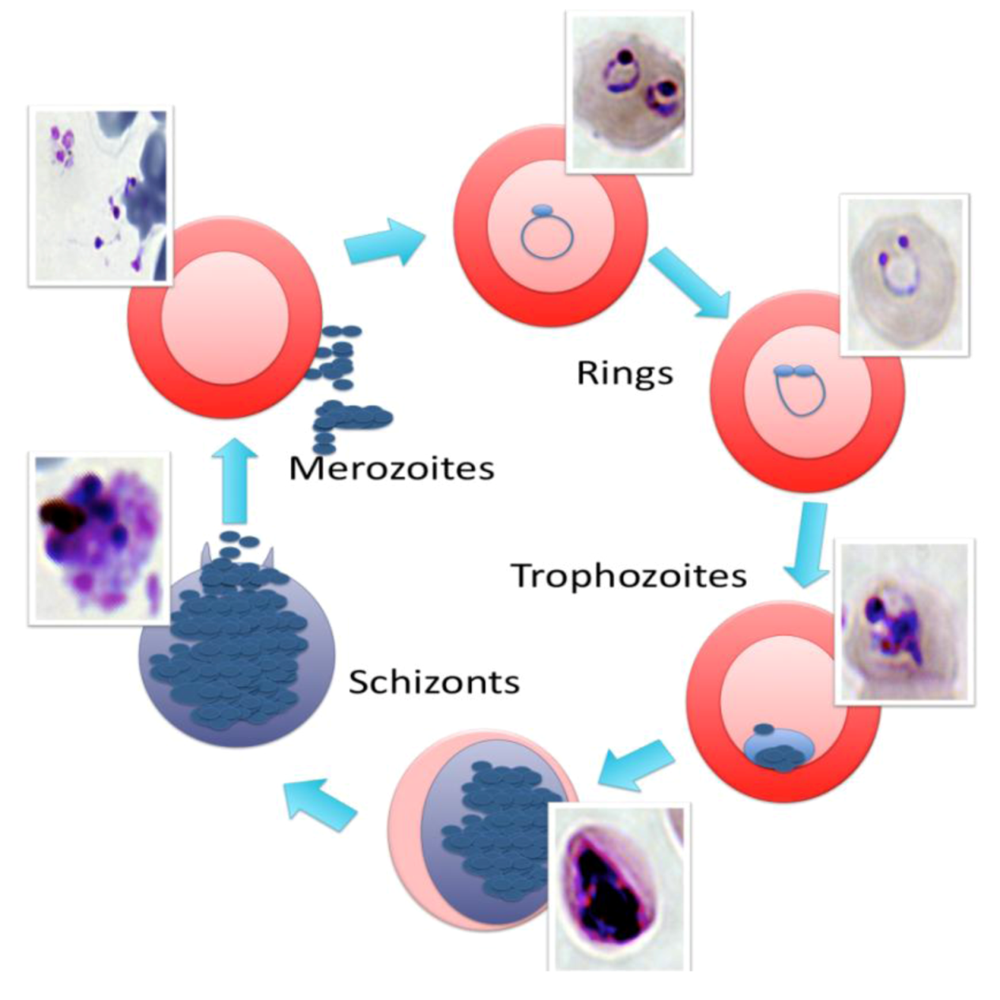
2. The Malaria Life Cycle

3. The Emergence of Antimalarial Resistance
5. Heat Shock Protein 90

6. Inhibitors of Heat Shock Protein 90

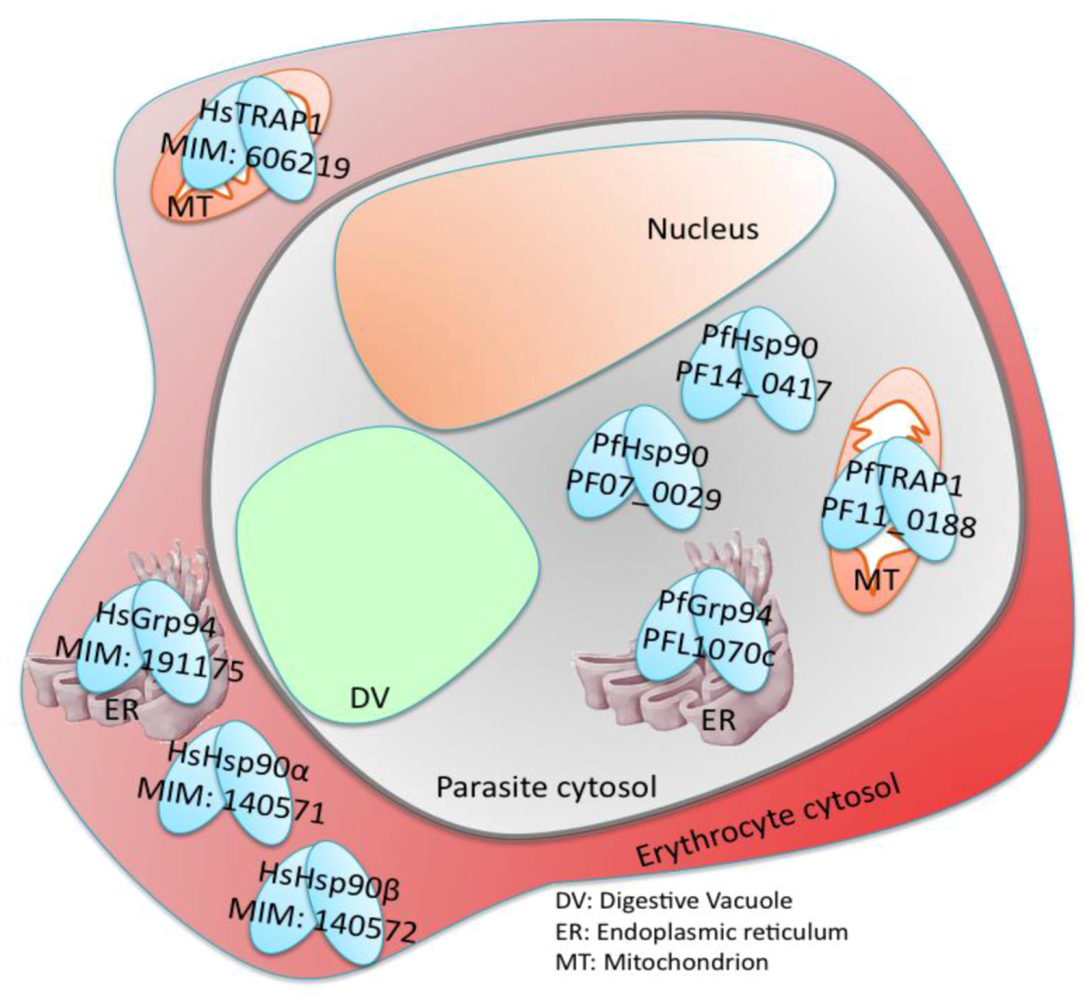
7. Plasmodium falciparum Hsp90
8. The PfHsp90 Drug Pipeline
9. Conclusions

Acknowledgement
References
- Sachs, J.; Malaney, P. The economic and social burden of malaria. Nature 2002, 415, 680–685. [Google Scholar] [CrossRef]
- Snow, R.W.; Guerra, C.A.; Noor, A.M.; Myint, H.Y.; Hay, S.I. The global distribution of clinical episodes of plasmodium falciparum malaria. Nature 2005, 434, 214–217. [Google Scholar]
- Hay, S.I.; Guerra, C.A.; Tatem, A.J.; Noor, A.M.; Snow, R.W. The global distribution and population at risk of malaria: Past, present, and future. Lancet Infect. Dis. 2004, 4, 327–336. [Google Scholar]
- Acharya, P.; Kumar, R.; Tatu, U. Chaperoning a cellular upheaval in malaria: Heat shock proteins in plasmodium falciparum. Mol. Biochem. Parasitol. 2007, 153, 85–94. [Google Scholar] [CrossRef]
- Sherman, I.W. The life of plasmodium: An overview. In Molecular approaches to malaria; Sherman, I.W., Ed.; ASM Press: Washington, D.C., USA, 2005; pp. 3–23. [Google Scholar]
- Zak, O.; Carbon, C.; Fantin, B.; Kaminsky, R. Malaria. In Handbook of Animal Models of Infection: Experimental Models in Antimicrobial Chemotherapy; Sande, M.A., O'Reilly, T., Eds.; Academic Press: London, UK, 1999; pp. 757–773. [Google Scholar]
- Ashley, E.; McGready, R.; Singhasivanon, P.; Nosten, F.; Carrara, V.; Price, R. In vivo sensitivity monitoring of mefloquine monotherapy and artesunate-mefloquine combinations for the treatment of uncomplicated falciparum malaria in thailand in 2003. Trop. Med. Int. Health 2006, 11, 1898–1899. [Google Scholar] [CrossRef]
- Ndam, N.T.; Deloron, P. Molecular aspects of plasmodium falciparum infection during pregnancy. J. Biomed. Biotech. 2007, 2007, 43785. [Google Scholar]
- Douradinha, B.; Doolan, D.L. Harnessing immune responses against plasmodium for rational vaccine design. Trends Para. 2011, 27, 274–283. [Google Scholar] [CrossRef]
- Doolan, D.L.; Dobano, C.; Baird, J.K. Acquired immunity to malaria. Clin. Micro. Rev. 2009, 22, 13–36. [Google Scholar]
- Langhorne, J.; Ndungu, F.M.; Sponaas, A.M.; Marsh, K. Immunity to malaria: More questions than answers. Nat. Immunol. 2008, 9, 725–732. [Google Scholar]
- Casares, S.; Brumeanu, T.D.; Richie, T.L. The rts,s malaria vaccine. Vaccine 2010, 28, 4880–4894. [Google Scholar] [CrossRef]
- Alonso, P.L.; Sacarlal, J.; Aponte, J.J.; Leach, A.; Macete, E.; Milman, J.; Mandomando, I.; Spiessens, B.; Guinovart, C.; Espasa, M.; et al. Efficacy of the rts,s/as02a vaccine against plasmodium falciparum infection and disease in young african children: Randomised controlled trial. Lancet 2004, 364, 1411–1420. [Google Scholar]
- Alonso, P.L.; Sacarlal, J.; Aponte, J.J.; Leach, A.; Macete, E.; Aide, P.; Sigauque, B.; Milman, J.; Mandomando, I.; Bassat, Q.; et al. Duration of protection with rts,s/as02a malaria vaccine in prevention of plasmodium falciparum disease in mozambican children: Single-blind extended follow-up of a randomised controlled trial. Lancet 2005, 366, 2012–2018. [Google Scholar]
- Bejon, P.; Lusingu, J.; Olotu, A.; Leach, A.; Lievens, M.; Vekemans, J.; Mshamu, S.; Lang, T.; Gould, J.; Dubois, M.C.; et al. Efficacy of rts,s/as01e vaccine against malaria in children 5 to 17 months of age. New Eng. J. Med. 2008, 359, 2521–2532. [Google Scholar]
- Fidock, D.A.; Rosenthal, P.J.; Croft, S.L.; Brun, R.; Nwaka, S. Antimalarial drug discovery: Efficacy models for compound screening. Nat Rev. Drug Disc. 2004, 3, 509–520. [Google Scholar]
- Peters, W.; Robinson, B.L.; Ellis, D.S. The chemotherapy of rodent malaria. Xlii. Halofantrine and halofantrine resistance. Ann. Trop. Med. Para. 1987, 81, 639–646. [Google Scholar]
- Tuteja, R. Malaria - an overview. FEBS 2007, 274, 4670–4679. [Google Scholar]
- Cowman, A.F.; Crabb, B.S. Invasion of red blood cells by malaria parasites. Cell 2006, 124, 755–766. [Google Scholar] [CrossRef]
- Bray, R.S.; Garnham, P.C. The life-cycle of primate malaria parasites. Brit. Med. Bull. 1982, 38, 117–122. [Google Scholar]
- Chin, W.; Contacos, P.G.; Coatney, G.R.; Kimball, H.R. A naturally acquited quotidian-type malaria in man transferable to monkeys. Science 1965, 149, 865. [Google Scholar]
- Bannister, L.; Mitchell, G. The ins, outs and roundabouts of malaria. Trends Para. 2003, 19, 209–213. [Google Scholar]
- Baumeister, S.; Winterberg, M.; Przyborski, J.M.; Lingelbach, K. The malaria parasite plasmodium falciparum: Cell biological peculiarities and nutritional consequences. Protoplasma 2010, 240, 3–12. [Google Scholar] [CrossRef]
- Francis, S.E.; Sullivan, D.J., Jr.; Goldberg, D.E. Hemoglobin metabolism in the malaria parasite plasmodium falciparum. Annu. Rev. Micro. 1997, 51, 97–123. [Google Scholar]
- Przyborski, J.M.; Wickert, H.; Krohne, G.; Lanzer, M. Maurer's clefts--a novel secretory organelle? Mol. Biochem. Para. 2003, 132, 17–26. [Google Scholar] [CrossRef]
- Cooke, B.M.; Mohandas, N.; Coppel, R.L. Malaria and the red blood cell membrane. Sem. Hematol. 2004, 41, 173–188. [Google Scholar]
- Ben Mamoun, C.; Gluzman, I.Y.; Hott, C.; MacMillan, S.K.; Amarakone, A.S.; Anderson, D.L.; Carlton, J.M.; Dame, J.B.; Chakrabarti, D.; Martin, R.K.; et al. Co-ordinated programme of gene expression during asexual intraerythrocytic development of the human malaria parasite plasmodium falciparum revealed by microarray analysis. Mol. Micro. 2001, 39, 26–36. [Google Scholar] [CrossRef]
- Koussis, K.; Withers-Martinez, C.; Yeoh, S.; Child, M.; Hackett, F.; Knuepfer, E.; Juliano, L.; Woehlbier, U.; Bujard, H.; Blackman, M.J. A multifunctional serine protease primes the malaria parasite for red blood cell invasion. EMBO 2009, 28, 725–735. [Google Scholar] [CrossRef]
- Fidock, D.A. Drug discovery: Priming the antimalarial pipeline. Nature 2010, 465, 297–298. [Google Scholar] [CrossRef]
- Gregson, A.; Plowe, C.V. Mechanisms of resistance of malaria parasites to antifolates. Pharm. Rev. 2005, 57, 117–145. [Google Scholar] [CrossRef]
- Taylor, D.K.; Avery, T.D.; Greatrex, B.W.; Tiekink, E.R.; Macreadie, I.G.; Macreadie, P.I.; Humphries, A.D.; Kalkanidis, M.; Fox, E.N.; Klonis, N.; et al. Novel endoperoxide antimalarials: Synthesis, heme binding, and antimalarial activity. J. Med. Chem. 2004, 47, 1833–1839. [Google Scholar] [CrossRef]
- Imwong, M.; Pukrittakayamee, S.; Looareesuwan, S.; Pasvol, G.; Poirreiz, J.; White, N.J.; Snounou, G. Association of genetic mutations in plasmodium vivax dhfr with resistance to sulfadoxine-pyrimethamine: Geographical and clinical correlates. Antimicrob. Agents Chemother. 2001, 45, 3122–3127. [Google Scholar] [CrossRef]
- White, N.J.; Nosten, F.; Looareesuwan, S.; Watkins, W.M.; Marsh, K.; Snow, R.W.; Kokwaro, G.; Ouma, J.; Hien, T.T.; Molyneux, M.E.; et al. Averting a malaria disaster. Lancet 1999, 353, 1965–1967. [Google Scholar]
- Krogstad, D.J.; Gluzman, I.Y.; Kyle, D.E.; Oduola, A.M.; Martin, S.K.; Milhous, W.K.; Schlesinger, P.H. Efflux of chloroquine from plasmodium falciparum: Mechanism of chloroquine resistance. Science 1987, 238, 1283–1285. [Google Scholar]
- Wellems, T.E.; Walker-Jonah, A.; Panton, L.J. Genetic mapping of the chloroquine-resistance locus on plasmodium falciparum chromosome 7. Proc. Nat. Acad. Sci. USA 1991, 88, 3382–3386. [Google Scholar] [CrossRef]
- Djimde, A.; Doumbo, O.K.; Steketee, R.W.; Plowe, C.V. Application of a molecular marker for surveillance of chloroquine-resistant falciparum malaria. Lancet 2001, 358, 890–891. [Google Scholar]
- Djimde, A.; Doumbo, O.K.; Cortese, J.F.; Kayentao, K.; Doumbo, S.; Diourte, Y.; Dicko, A.; Su, X.Z.; Nomura, T.; Fidock, D.A.; et al. A molecular marker for chloroquine-resistant falciparum malaria. New Eng. J. Med. 2001, 344, 257–263. [Google Scholar]
- Eckstein-Ludwig, U.; Webb, R.J.; Van Goethem, I.D.; East, J.M.; Lee, A.G.; Kimura, M.; O'Neill, P.M.; Bray, P.G.; Ward, S.A.; Krishna, S. Artemisinins target the serca of plasmodium falciparum. Nature 2003, 424, 957–961. [Google Scholar]
- Uhlemann, A.C.; Krishna, S. Antimalarial multi-drug resistance in asia: Mechanisms and assessment. Curr. Topics Micro. Immunol. 2005, 295, 39–53. [Google Scholar] [CrossRef]
- White, N.J. Preventing antimalarial drug resistance through combinations. Drug Res. Updates 1998, 1, 3–9. [Google Scholar] [CrossRef]
- Crandall, I.; Charuk, J.; Kain, K.C. Nonylphenolethoxylates as malarial chloroquine resistance reversal agents. Antimicrob. Agents Chemother. 2000, 44, 2431–2434. [Google Scholar] [CrossRef]
- Pereira, M.R.; Henrich, P.P.; Sidhu, A.B.; Johnson, D.; Hardink, J.; Van Deusen, J.; Lin, J.; Gore, K.; O'Brien, C.; Wele, M.; et al. In vivo and in vitro antimalarial properties of azithromycin-chloroquine combinations that include the resistance reversal agent amlodipine. Antimicrob. Agents Chemother. 2011, 55, 3115–3124. [Google Scholar]
- Pesce, E.R.; Cockburn, I.L.; Goble, J.L.; Stephens, L.L.; Blatch, G.L. Malaria heat shock proteins: Drug targets that chaperone other drug targets. Infect. Dis. Drug Tartgets 2010, 10, 147–157. [Google Scholar]
- Shonhai, A. Plasmodial heat shock proteins: Targets for chemotherapy. FEMS Immunol. Med. Micro. 2010, 58, 61–74. [Google Scholar] [CrossRef]
- Wegele, H.; Muller, L.; Buchner, J. Hsp70 and hsp90--a relay team for protein folding. Rev. Physiol. Biochem. Pharmacol. 2004, 151, 1–44. [Google Scholar] [CrossRef]
- Smith, D.F.; Sullivan, W.P.; Marion, T.N.; Zaitsu, K.; Madden, B.; McCormick, D.J.; Toft, D.O. Identification of a 60-kilodalton stress-related protein, p60, which interacts with hsp90 and hsp70. Mol. Cell. Biol. 1993, 13, 869–876. [Google Scholar]
- Cowen, L.E.; Lindquist, S. Hsp90 potentiates the rapid evolution of new traits: Drug resistance in diverse fungi. Science 2005, 309, 2185–2189. [Google Scholar]
- Cowen, L.E.; Singh, S.D.; Kohler, J.R.; Collins, C.; Zaas, A.K.; Schell, W.A.; Aziz, H.; Mylonakis, E.; Perfect, J.R.; Whitesell, L.; et al. Harnessing hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc. Nat. Acad. Sci. USA 2009, 106, 2818–2823. [Google Scholar]
- Marubayashi, S.; Koppikar, P.; Taldone, T.; Abdel-Wahab, O.; West, N.; Bhagwat, N.; Caldas-Lopes, E.; Ross, K.N.; Gonen, M.; Gozman, A.; et al. Hsp90 is a therapeutic target in jak2-dependent myeloproliferative neoplasms in mice and humans. J. Clin. Invest. 2010, 120, 3578–3593. [Google Scholar]
- Mollapour, M.; Neckers, L. Post-translational modifications of hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 2011, 1823, 648–655. [Google Scholar] [CrossRef]
- Moulick, K.; Ahn, J.H.; Zong, H.; Rodina, A.; Cerchietti, L.; Gomes DaGama, E.M.; Caldas-Lopes, E.; Beebe, K.; Perna, F.; Hatzi, K.; et al. Affinity-based proteomics reveal cancer-specific networks coordinated by hsp90. Nat. Chem. Bio. 2011, 7, 818–826. [Google Scholar] [CrossRef]
- Tatokoro, M.; Koga, F.; Yoshida, S.; Kawakami, S.; Fujii, Y.; Neckers, L.; Kihara, K. Potential role of hsp90 inhibitors in overcoming cisplatin resistance of bladder cancer-initiating cells. Int. J. Cancer 2011, 131, 987–996. [Google Scholar]
- Wang, Y.; Trepel, J.B.; Neckers, L.M.; Giaccone, G. Sta-9090, a small-molecule hsp90 inhibitor for the potential treatment of cancer. Curr. Opin. Invest. Drugs 2010, 11, 1466–1476. [Google Scholar]
- Cowen, L.E.; Carpenter, A.E.; Matangkasombut, O.; Fink, G.R.; Lindquist, S. Genetic architecture of hsp90-dependent drug resistance. Euk. Cell 2006, 5, 2184–2188. [Google Scholar] [CrossRef]
- Pavithra, S.R.; Kumar, R.; Tatu, U. Systems analysis of chaperone networks in the malarial parasite plasmodium falciparum. PLoS Comp. Bio. 2007, 3, 1701–1715. [Google Scholar]
- Cerchietti, L.C.; Hatzi, K.; Caldas-Lopes, E.; Yang, S.N.; Figueroa, M.E.; Morin, R.D.; Hirst, M.; Mendez, L.; Shaknovich, R.; Cole, P.A.; et al. Bcl6 repression of ep300 in human diffuse large b cell lymphoma cells provides a basis for rational combinatorial therapy. J. Clin. Invest. 2010. [Google Scholar] [CrossRef]
- Taldone, T.; Gillan, V.; Sun, W.; Rodina, A.; Patel, P.; Maitland, K.; O'Neill, K.; Chiosis, G.; Devaney, E. Assay strategies for the discovery and validation of therapeutics targeting brugia pahangi hsp90. PLoS Neg. Trop. Dis. 2010, 4, e714. [Google Scholar]
- Taldone, T.; Gomes-DaGama, E.M.; Zong, H.; Sen, S.; Alpaugh, M.L.; Zatorska, D.; Alonso-Sabadell, R.; Guzman, M.L.; Chiosis, G. Synthesis of purine-scaffold fluorescent probes for heat shock protein 90 with use in flow cytometry and fluorescence microscopy. Bio. Med. Chem. Let. 2011, 21, 5347–5352. [Google Scholar] [CrossRef]
- Taldone, T.; Zatorska, D.; Patel, P.D.; Zong, H.; Rodina, A.; Ahn, J.H.; Moulick, K.; Guzman, M.L.; Chiosis, G. Design, synthesis, and evaluation of small molecule hsp90 probes. Bioorg. Med. Chem. 2011, 19, 2603–2614. [Google Scholar] [CrossRef]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2011, 1823, 742–755. [Google Scholar]
- Usmani, S.Z.; Chiosis, G. Hsp90 inhibitors as therapy for multiple myeloma. Clin. Lymp. Myel. Leuk. 2011, 11 (Suppl. 1), S77–S81. [Google Scholar] [CrossRef]
- Banumathy, G.; Singh, V.; Pavithra, S.R.; Tatu, U. Heat shock protein 90 function is essential for plasmodium falciparum growth in human erythrocytes. J. Biol. Chem. 2003, 278, 18336–18345. [Google Scholar]
- Kumar, R.; Pavithra, S.R.; Tatu, U. Three-dimensional structure of heat shock protein 90 from plasmodium falciparum: Molecular modelling approach to rational drug design against malaria. J. Biosc. 2007, 32, 531–536. [Google Scholar]
- Pallavi, R.; Roy, N.; Nageshan, R.K.; Talukdar, P.; Pavithra, S.R.; Reddy, R.; Venketesh, S.; Kumar, R.; Gupta, A.K.; Singh, R.K.; et al. Heat shock protein 90 as a drug target against protozoan infections: Biochemical characterization of hsp90 from plasmodium falciparum and trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J. Biol. Chem. 2010, 285, 37964–37975. [Google Scholar]
- Pavithra, S.R.; Banumathy, G.; Joy, O.; Singh, V.; Tatu, U. Recurrent fever promotes plasmodium falciparum development in human erythrocytes. J. Biol. Chem. 2004, 279, 46692–46699. [Google Scholar]
- Dutta, R.; Inouye, M. Ghkl, an emergent atpase/kinase superfamily. Trends Biochem. Sci. 2000, 25, 24–28. [Google Scholar]
- Singh, S.D.; Robbins, N.; Zaas, A.K.; Schell, W.A.; Perfect, J.R.; Cowen, L.E. Hsp90 governs echinocandin resistance in the pathogenic yeast candida albicans via calcineurin. PLoS Path. 2009, 5, e1000532. [Google Scholar] [CrossRef]
- Dollins, D.E.; Immormino, R.M.; Gewirth, D.T. Structure of unliganded grp94, the endoplasmic reticulum hsp90. Basis for nucleotide-induced conformational change. J. Biol. Chem. 2005, 280, 30438–30447. [Google Scholar] [CrossRef]
- Dollins, D.E.; Warren, J.J.; Immormino, R.M.; Gewirth, D.T. Structures of grp94-nucleotide complexes reveal mechanistic differences between the hsp90 chaperones. Mol. Cell 2007, 28, 41–56. [Google Scholar]
- Immormino, R.M.; Dollins, D.E.; Shaffer, P.L.; Soldano, K.L.; Walker, M.A.; Gewirth, D.T. Ligand-induced conformational shift in the n-terminal domain of grp94, an hsp90 chaperone. J. Bio. Chem. 2004, 279, 46162–46171. [Google Scholar]
- Immormino, R.M.; Metzger, L.E.t.; Reardon, P.N.; Dollins, D.E.; Blagg, B.S.; Gewirth, D.T. Different poses for ligand and chaperone in inhibitor-bound hsp90 and grp94: Implications for paralog-specific drug design. J. Mol. Bio. 2009, 388, 1033–1042. [Google Scholar]
- Johnson, J.L.; Brown, C. Plasticity of the hsp90 chaperone machine in divergent eukaryotic organisms. Cell Stress Chap. 2009, 14, 83–94. [Google Scholar] [CrossRef]
- Shonhai, A.; Maier, A.G.; Przyborski, J.M.; Blatch, G.L. Intracellular protozoan parasites of humans: The role of molecular chaperones in development and pathogenesis. Pro. Pep. Lett. 2011, 18, 143–157. [Google Scholar] [CrossRef]
- Gitau, G.W.; Mandal, P.; Blatch, G.L.; Przyborski, J.; Shonhai, A. Characterisation of the plasmodium falciparum hsp70-hsp90 organising protein (pfhop). Cell Stress Chap. 2012, 17, 191–202. [Google Scholar]
- Chiosis, G.; Caldas Lopes, E.; Solit, D. Heat shock protein-90 inhibitors: A chronicle from geldanamycin to today's agents. Curr. Opin. Invest. Drugs 2006, 7, 534–541. [Google Scholar]
- Whitesell, L.; Mimnaugh, E.G.; De Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein hsp90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Nat. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef]
- Chiosis, G.; Vilenchik, M.; Kim, J.; Solit, D. Hsp90: The vulnerable chaperone. Drug Disc. Today 2004, 9, 881–888. [Google Scholar] [CrossRef]
- Taldone, T.; Gillan, V.; Sun, W.; Rodina, A.; Patel, P.; Maitland, K.; O'Neill, K.; Chiosis, G.; Devaney, E. Assay strategies for the discovery and validation of therapeutics targeting brugia pahangi hsp90. PLoS Negl. Trop. Dis. 2010, 4, e714. [Google Scholar]
- Elo, M.A.; Kaarniranta, K.; Helminen, H.J.; Lammi, M.J. Hsp90 inhibitor geldanamycin increases hsp70 mrna stabilisation but fails to activate hsf1 in cells exposed to hydrostatic pressure. Biochim. Biophys. Acta 2005, 1743, 115–119. [Google Scholar] [CrossRef]
- Taldone, T.; Chiosis, G. Purine-scaffold hsp90 inhibitors. Curr. Topics Med. Chem. 2009, 9, 1436–1446. [Google Scholar]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef]
- Caldas-Lopes, E.; Cerchietti, L.; Ahn, J.H.; Clement, C.C.; Robles, A.I.; Rodina, A.; Moulick, K.; Taldone, T.; Gozman, A.; Guo, Y.; et al. Hsp90 inhibitor pu-h71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc. Natl. Acad. Sci. USA 2009, 106, 8368–8373. [Google Scholar]
- Krause, S.; Willighagen, E.; Steinbeck, C. Jchempaint: Using the collaborative forces of the internet to develop a free editor for 2d chemical structures. Molecules 2000, 5, 93–98. [Google Scholar] [CrossRef]
- Su, X.Z.; Wellems, T.E. Sequence, transcript characterization and polymorphisms of a plasmodium falciparum gene belonging to the heat-shock protein (hsp) 90 family. Gene 1994, 151, 225–230. [Google Scholar] [CrossRef]
- Banumathy, G. Functional insights into heat shock protein 90 multichaperone complex in plasmodium falciparum. A Thesis submitted for the Degree of Doctor, Philosophy in the Faculty of Science, Department of Biochemistry, Bangalore University, 2004. [Google Scholar]
- Immormino, R.M.; Kang, Y.; Chiosis, G.; Gewirth, D.T. Structural and quantum chemical studies of 8-aryl-sulfanyl adenine class hsp90 inhibitors. J. Med. Chem. 2006, 49, 4953–4960. [Google Scholar] [CrossRef]
- Corbett, K.D.; Berger, J.M. Structure of the atp-binding domain of plasmodium falciparum hsp90. Proteins 2010, 78, 2738–2744. [Google Scholar] [CrossRef]
- Wider, D.; Peli-Gulli, M.P.; Briand, P.A.; Tatu, U.; Picard, D. The complementation of yeast with human or plasmodium falciparum hsp90 confers differential inhibitor sensitivities. Mol. Biochem. Parasitol. 2009, 164, 147–152. [Google Scholar] [CrossRef]
- Shahinas, D.; Liang, M.; Datti, A.; Pillai, D.R. A repurposing strategy identifies novel synergistic inhibitors of plasmodium falciparum heat shock protein 90. J. Med. Chem. 2010, 53, 3552–3557. [Google Scholar]
- Ancolio, C.; Azas, N.; Mahiou, V.; Ollivier, E.; Di Giorgio, C.; Keita, A.; Timon-David, P.; Balansard, G. Antimalarial activity of extracts and alkaloids isolated from six plants used in traditional medicine in mali and sao tome. Phyto. Res. 2002, 16, 646–649. [Google Scholar] [CrossRef]
- Azas, N.; Laurencin, N.; Delmas, F.; Di, G.C.; Gasquet, M.; Laget, M.; Timon-David, P. Synergistic in vitro antimalarial activity of plant extracts used as traditional herbal remedies in Mali. Par. Res. 2002, 88, 165–171. [Google Scholar]
- Fiot, J.; Sanon, S.; Azas, N.; Mahiou, V.; Jansen, O.; Angenot, L.; Balansard, G.; Ollivier, E. Phytochemical and pharmacological study of roots and leaves of guiera senegalensis j.F. Gmel (combretaceae). J. Ethnopharmacol. 2006, 106, 173–178. [Google Scholar] [CrossRef]
- Traore-Keita, F.; Gasquet, M.; Di Giorgio, C.; Ollivier, E.; Delmas, F.; Keita, A.; Doumbo, O.; Balansard, G.; Timon-David, P. Antimalarial activity of four plants used in traditional medicine in mali. Phyto. Res. 2000, 14, 45–47. [Google Scholar] [CrossRef]
- Shahinas, D.; Macmullin, G.; Benedict, C.; Crandall, I.; Pillai, D.R. Harmine is a potent antimalarial targeting hsp90 and synergizes with chloroquine and artemisinin. Antimicrob. Agents Chemother. 2012, 56, 4207–4213. [Google Scholar] [CrossRef]
- Biamonte, M.A.; Van de Water, R.; Arndt, J.W.; Scannevin, R.H.; Perret, D.; Lee, W.C. Heat shock protein 90: Inhibitors in clinical trials. J. Med. Chem. 2010, 53, 3–17. [Google Scholar] [CrossRef]
- Berenbaum, M.C. What is synergy? Pharmaco. Rev. 1989, 41, 93–141. [Google Scholar]
- Cooper, R.A.; Hartwig, C.L.; Ferdig, M.T. Pfcrt is more than the plasmodium falciparum chloroquine resistance gene: A functional and evolutionary perspective. Acta Trop. 2005, 94, 170–180. [Google Scholar] [CrossRef]
- Cooper, R.A.; Ferdig, M.T.; Su, X.Z.; Ursos, L.M.; Mu, J.; Nomura, T.; Fujioka, H.; Fidock, D.A.; Roepe, P.D.; Wellems, T.E. Alternative mutations at position 76 of the vacuolar transmembrane protein pfcrt are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in plasmodium falciparum. Mol. Pharmacol. 2002, 61, 35–42. [Google Scholar]
- Chinappi, M.; Via, A.; Marcatili, P.; Tramontano, A. On the mechanism of chloroquine resistance in plasmodium falciparum. PloS One 2010, 5, e14064. [Google Scholar]
- Johnson, D.J.; Fidock, D.A.; Mungthin, M.; Lakshmanan, V.; Sidhu, A.B.; Bray, P.G.; Ward, S.A. Evidence for a central role for pfcrt in conferring plasmodium falciparum resistance to diverse antimalarial agents. Mol. Cell 2004, 15, 867–877. [Google Scholar] [CrossRef]
- Wellems, T.E. Transporter of a malaria catastrophe. Nat. Med. 2004, 10, 1169–1171. [Google Scholar]
- Mu, J.; Myers, R.A.; Jiang, H.; Liu, S.; Ricklefs, S.; Waisberg, M.; Chotivanich, K.; Wilairatana, P.; Krudsood, S.; White, N.J.; et al. Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat. Genet. 2010, 42, 268–271. [Google Scholar] [CrossRef]
- Leeson, P.D.; St-Gallay, S.A. The influence of the 'organizational factor' on compound quality in drug discovery. Nat. Rev. Drug Discovery 2011, 10, 749–765. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shahinas, D.; Folefoc, A.; Pillai, D.R. Targeting Plasmodium falciparum Hsp90: Towards Reversing Antimalarial Resistance. Pathogens 2013, 2, 33-54. https://doi.org/10.3390/pathogens2010033
Shahinas D, Folefoc A, Pillai DR. Targeting Plasmodium falciparum Hsp90: Towards Reversing Antimalarial Resistance. Pathogens. 2013; 2(1):33-54. https://doi.org/10.3390/pathogens2010033
Chicago/Turabian StyleShahinas, Dea, Asongna Folefoc, and Dylan R. Pillai. 2013. "Targeting Plasmodium falciparum Hsp90: Towards Reversing Antimalarial Resistance" Pathogens 2, no. 1: 33-54. https://doi.org/10.3390/pathogens2010033
APA StyleShahinas, D., Folefoc, A., & Pillai, D. R. (2013). Targeting Plasmodium falciparum Hsp90: Towards Reversing Antimalarial Resistance. Pathogens, 2(1), 33-54. https://doi.org/10.3390/pathogens2010033