
Animal Models of Chronic Hepatitis Delta Virus Infection Host–Virus Immunologic Interactions

Abstract
:1. Introduction
1.1. The Disease, Chronic HDV Infection
1.2. Main Characteristics of HDV
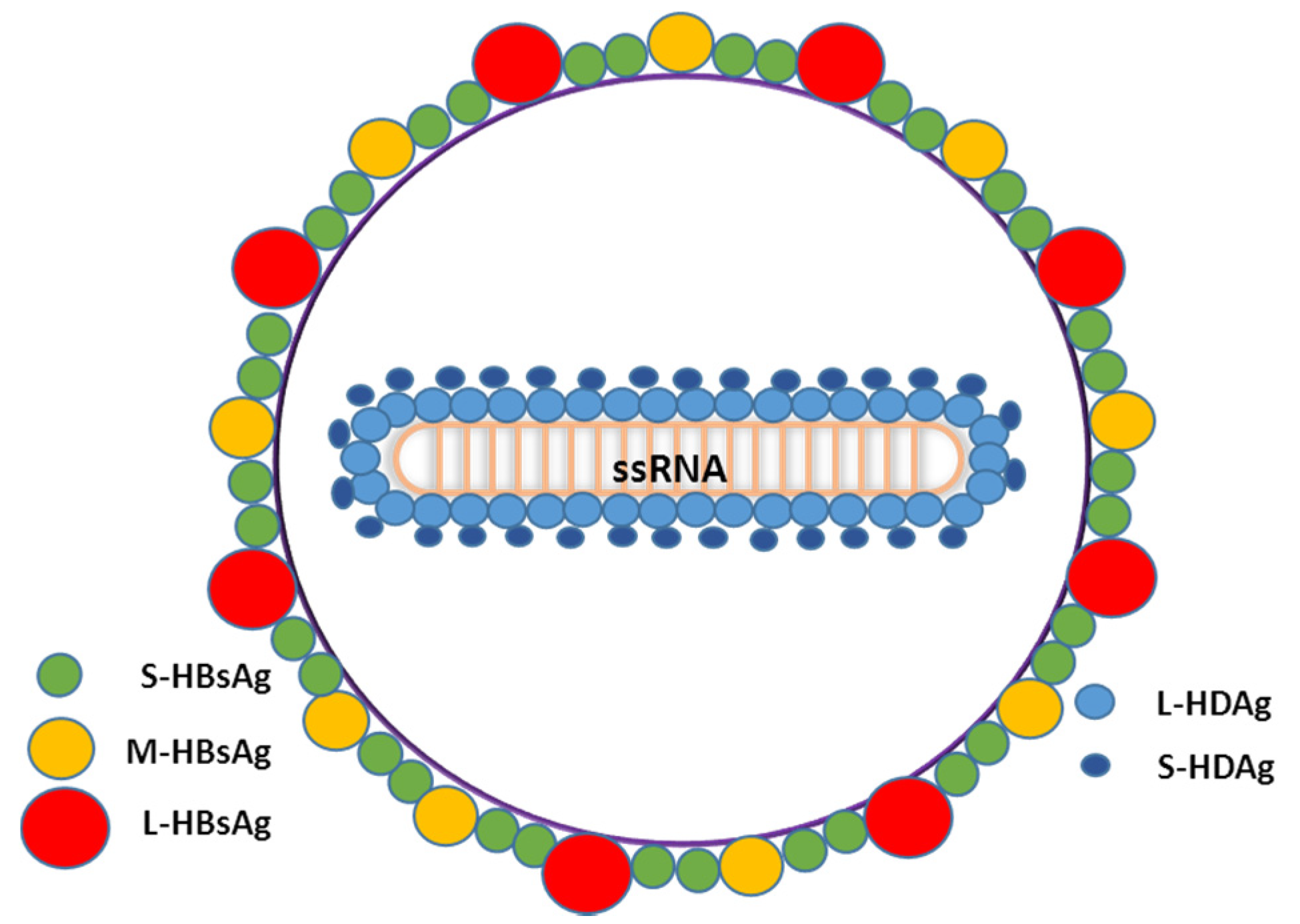
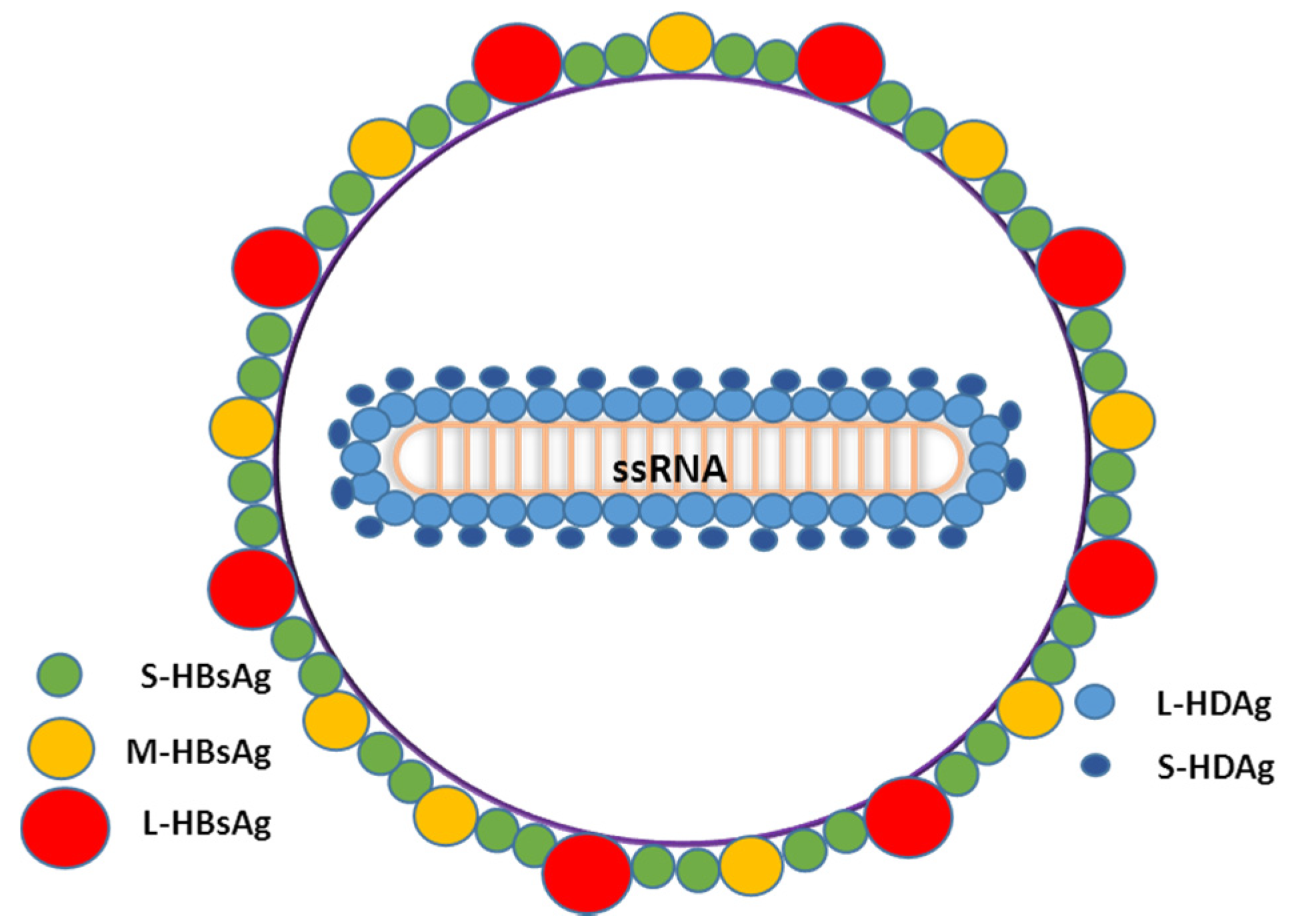
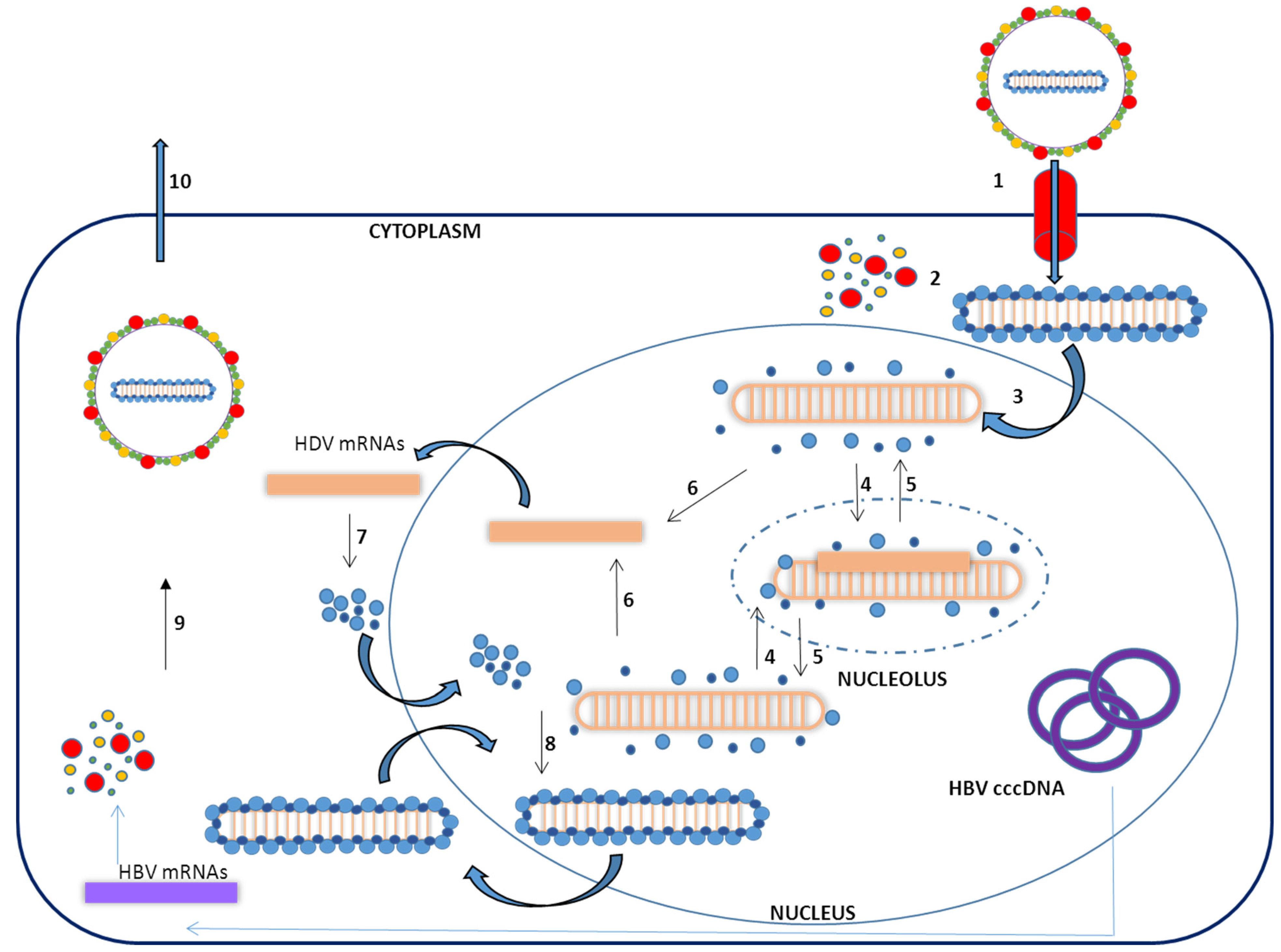
1.3. Virus Life Cycle
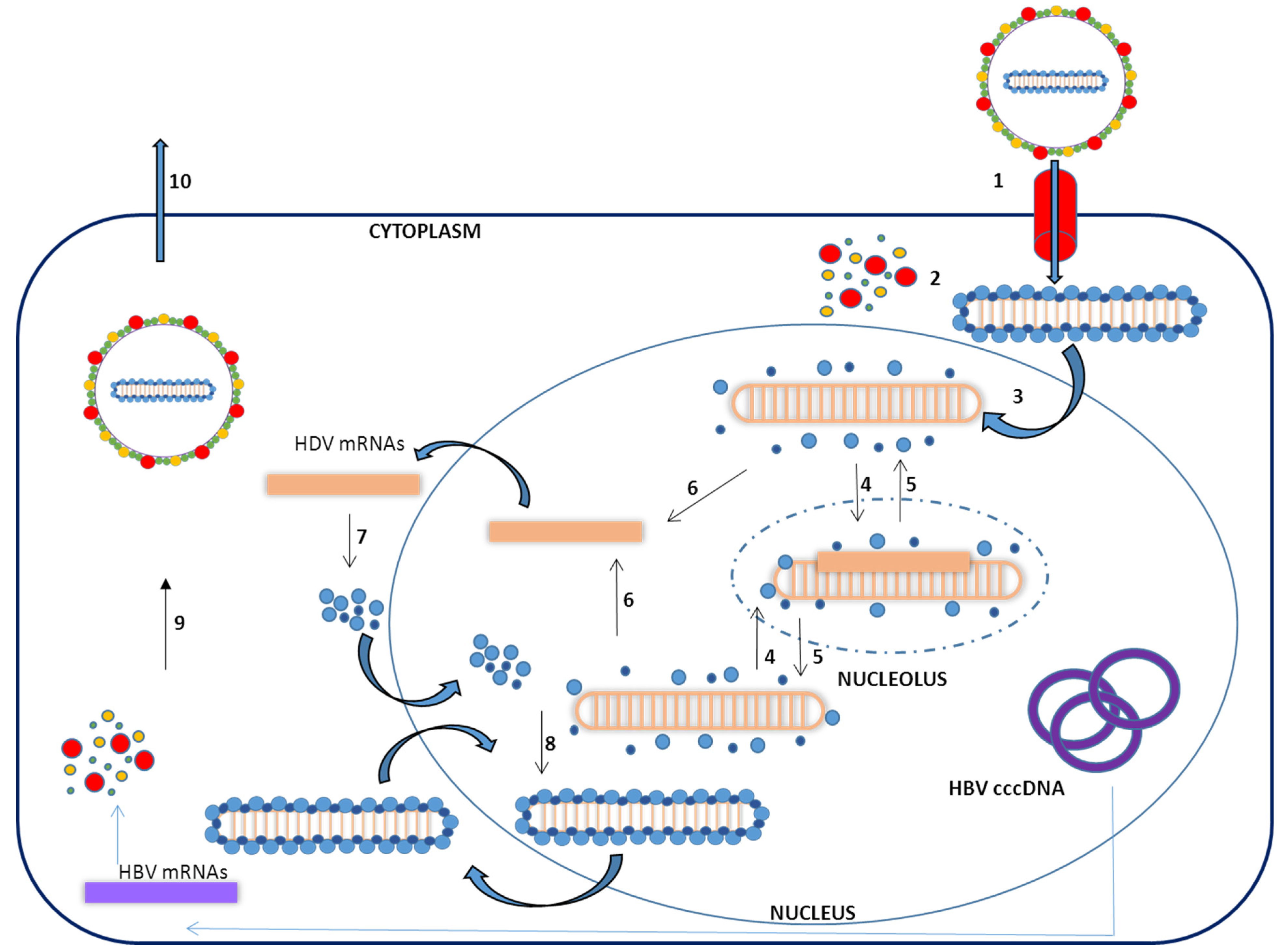
2. In Vitro HDV–Host Cell Interaction
3. Chronic HDV Infection Animal Models
3.1. “Natural” Animal Models
3.2. Mouse Models of HDV Replication
3.2.1. Injection of Mice with HDV Infectious Particles
3.2.2. HDV Transgenic Mice
3.2.3. Mice with “Humanized” Livers
- The availability of primary human hepatocytes is limited and they cannot be propagated in vitro, which limits the number of available animals.
- The susceptibility of primary human hepatocytes to HBV infection is highly variable and generally low, since these cells rapidly dedifferentiate, losing the expression of the NTCP receptor.
- The engraftment of human hepatocytes in the liver of mice requires the use of immunodeficient mice, thus the interaction of the immune system and the virus is lost and consequently experiments are restricted to studies analyzing virus host–cell interactions.
3.2.4. Development of Gene Delivery Vectors to Deliver HDV Genomes into Mice
4. Conclusions

{kind=link}
{kind=link}
| Animal model | Main characteristics | References | |
|---|---|---|---|
| Natural animal model | chimpanzees | Chimpanzees can be efficiently infected by HDV, coinfection and superinfection experiments have been performed resulting in moderately severe and severe liver damage, respectively. | 79–81 |
| woodchucks | Several laboratories have shown that woodchucks chronically infected with WHV can be infected by HDV and produce new HDV virions using WHV surface antigen to form the envelope. This animal model recapitulate many of the characterists of HDV infection in humans. | 76–78 | |
| bats | HDV pseudotyped with surface proteins of bat hepadnavirus TBHBV is able to infect both primary hepatocytes, a pattern similar to the pattern observed with HDV-HBV | 83 | |
| WT mice | wild-type & inmunodeficient mice | Mice injected with HDV virus obtained from woodchucks transiently infect and replicate in the liver | 44 |
| HDV-transgenic mice | S-HDAg transgenic mice | S-HDAg is expressed in hepatocytes and localized in the nucleus. No liver injury was observed, thus S-HDAg protein is not responsible for HDV inducing liver damage. | 97 |
| L-HDAg transgenic mice | L-HDAg is expressed in hepatocytes an localized in the nucleus. No liver injury was observed, thus L-HDAg protein is not responsible for HDV inducing liver damage. | 97 | |
| HDV Ag x HBV sAg | The phenotype of these mice does not differ from the phenotype of the parents | 97 | |
| HDV genome under the control of the Albumin promoter | HDV replication and S-HDAg expression was detected in several organs including the liver, muscle, and brain. However, no large HDV large antigen were detected in any tissue, indicating that there was no efficient HDV RNA editing. Furthermore, no liver pathology was observed. | 98 | |
| Humanized mouse models | Fah-/-RAG-/-IL-2Rg-/- + human hepatocytes | Establishment of HDV infection was highly efficient in both HBV-infected and naïve chimeric mice, representing an interesting animal model to study HDV-human hepatocyte interaction and new antivirals. | 20,55,99 |
| HDV-transfected mice | HDV plasmid hydrodynamic injection | In this animal model there is an increase of HDV genome and antigenome in the liver but only during the first two weeks after hydrodinamic injetion. There is no associated damage. Edition is observed in the liver. | 107 |
| HDV-transfected mice | HDV plasmid intramuscular injection | Sustained RNA accumulation at least for 7 weeks. HDAg antibody generation | 106 |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rizzetto, M.; Ciancio, A. Epidemiology of hepatitis D. Semin. Liver Dis. 2012, 32, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Canese, M.G.; Arico, S.; Crivelli, O.; Trepo, C.; Bonino, F.; Verme, G. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut 1977, 18, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Hoyer, B.; Canese, M.G.; Shih, J.W.; Purcell, R.H.; Gerin, J.L. Delta agent: Association of delta antigen with hepatitis B surface antigen and RNA in serum of delta-infected chimpanzees. Proc. Natl. Acad. Sci. USA 1980, 77, 6124–6128. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D.; Wang, J.Y.; Wu, J.C.; Tsai, Y.T.; Lo, K.J.; Lai, K.H.; Tsay, S.H.; Govindarajan, S. Hepatitis D virus (delta agent) superinfection in an endemic area of hepatitis B infection: Immunopathologic and serologic findings. Scand. J. Infect. Dis. 1987, 19, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.H.; Liaw, Y.F.; Chen, T.J.; Chu, C.M.; Huang, M.J. Natural course of patients with chronic type B hepatitis following acute hepatitis delta virus superinfection. Liver 1989, 9, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Wedemeyer, H. Hepatitis D revival. Liver Int. 2011, 31 (Suppl. 1), 140–144. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Alavian, S.M. Hepatitis delta: The rediscovery. Clin. Liver Dis. 2013, 17, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Ciancio, A.; Rizzetto, M. Chronic hepatitis D at a standstill: Where do we go from here? Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 68–71. [Google Scholar] [CrossRef] [PubMed]
- Gaeta, G.B.; Stroffolini, T.; Smedile, A.; Niro, G.; Mele, A. Hepatitis delta in Europe: Vanishing or refreshing? Hepatology 2007, 46, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Heidrich, B.; Deterding, K.; Tillmann, H.L.; Raupach, R.; Manns, M.P.; Wedemeyer, H. Virological and clinical characteristics of delta hepatitis in central Europe. J. Viral Hepat. 2009, 16, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Servant-Delmas, A.; Le Gal, F.; Gallian, P.; Gordien, E.; Laperche, S. Increasing prevalence of HDV/HBV infection over 15 years in France. J. Clin. Virol. 2014, 59, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z.; Khan, M.A.; Salih, M.; Jafri, W. Interferon alpha for chronic hepatitis D. Cochrane Database Syst. Rev. 2011, 12. [Google Scholar] [CrossRef]
- Abbas, Z.; Memon, M.S.; Mithani, H.; Jafri, W.; Hamid, S. Treatment of chronic hepatitis D patients with pegylated interferon: A real-world experience. Antivir. Ther. 2014, 19, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Alvarado-Mora, M.V.; Locarnini, S.; Rizzetto, M.; Pinho, J.R. An update on HDV: Virology, pathogenesis and treatment. Antivir. Ther. 2013, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Taylor, J.M. Susceptibility of human hepatitis delta virus RNAs to small interfering RNA action. J. Virol. 2003, 77, 9728–9731. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Gupta, S.K.; Nischal, A.; Khattri, S.; Nath, R.; Pant, K.K.; Seth, P.K. Design of potential siRNA molecules for hepatitis delta virus gene silencing. Bioinformation 2012, 8, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Abou-Jaoude, G.; Molina, S.; Maurel, P.; Sureau, C. Myristoylation signal transfer from the large to the middle or the small HBV envelope protein leads to a loss of HDV particles infectivity. Virology 2007, 365, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Abou-Jaoude, G.; Sureau, C. Entry of hepatitis delta virus requires the conserved cysteine residues of the hepatitis B virus envelope protein antigenic loop and is blocked by inhibitors of thiol-disulfide exchange. J. Virol. 2007, 81, 13057–13066. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, M.; Sureau, C.; Labonte, P. Use of FDA approved therapeutics with hNTCP metabolic inhibitory properties to impair the HDV lifecycle. Antivir. Res. 2014, 106, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Lütgehetmann, M.; Mancke, L.V.; Volz, T.; Helbig, M.; Allweiss, L.; Bornscheuer, T.; Pollok, J.M.; Lohse, A.W.; Petersen, J.; Urban, S.; et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology 2012, 55, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Bartenschlager, R.; Kubitz, R.; Zoulim, F. Strategies to inhibit entry of HBV and HDV into hepatocytes. Gastroenterology 2014, 147, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Bordier, B.B.; Marion, P.L.; Ohashi, K.; Kay, M.A.; Greenberg, H.B.; Casey, J.L.; Glenn, J.S. A prenylation inhibitor prevents production of infectious hepatitis delta virus particles. J. Virol. 2002, 76, 10465–10472. [Google Scholar] [CrossRef] [PubMed]
- Bordier, B.B.; Ohkanda, J.; Liu, P.; Lee, S.Y.; Salazar, F.H.; Marion, P.L.; Ohashi, K.; Meuse, L.; Kay, M.A.; Cas, J.L.; et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J. Clin. Investig. 2003, 112, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M. Hepatitis delta virus. Virology 2006, 344, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Kos, A.; Dijkema, R.; Arnberg, A.C.; van der Meide, P.H.; Schellekens, H. The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986, 323, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M. Structure and replication of hepatitis delta virus RNA. Curr. Top. Microbiol. Immunol. 2006, 307, 1–23. [Google Scholar] [PubMed]
- Makino, S.; Chang, M.F.; Shieh, C.K.; Kamahora, T.; Vannier, D.M.; Govindarajan, S.; Lai, M.M. Molecular cloning and sequencing of a human hepatitis delta (delta) virus RNA. Nature 1987, 329, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Grubb, D.; Elleuch, A.; Nohales, M.Á.; Delgado, S.; Gago, S. Rolling-circle replication of viroids, viroid-like satellite RNAs and hepatitis delta virus: Variations on a theme. RNA Biol. 2011, 8, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, R.; Ruiz-Ruiz, S.; Serra, P. Viroids and hepatitis delta virus. Semin. Liver Dis. 2012, 32, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Ke, A.; Zhou, K.; Ding, F.; Cate, J.H.; Doudna, J.A. A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 2004, 429, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Deny, P. Hepatitis delta virus genetic variability: From genotypes I, II, III to eight major clades? Curr. Top. Microbiol. Immunol. 2006, 307, 151–171. [Google Scholar] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Peng, B.; He, W.; Zhong, G.; Qi, Y.; Ren, B.; Gao, Z.; Jing, Z.; Song, M.; et al. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J. Virol. 2013, 87, 7977–7991. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Pellicoro, A.; Faber, K.N. Review article: The function and regulation of proteins involved in bile salt biosynthesis and transport. Aliment. Pharmacol. Ther. 2007, 26 (Suppl. 2), 149–160. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.P.; Yeh, C.T.; Ou, J.H.; Lai, M.M. Characterization of nuclear targeting signal of hepatitis delta antigen: Nuclear transport as a protein complex. J. Virol. 1992, 66, 914–921. [Google Scholar] [PubMed]
- Chang, J.; Nie, X.; Chang, H.E.; Han, Z.; Taylor, J. Transcription of hepatitis delta virus RNA by RNA polymerase II. J. Virol. 2008, 82, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H.; Lai, M.M. Hepatitis delta virus RNA replication. Viruses 2009, 1, 818–831. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M. Replication of the hepatitis delta virus RNA genome. Adv. Virus Res. 2009, 74, 103–121. [Google Scholar] [PubMed]
- Lehmann, E.; Brueckner, F.; Cramer, P. Molecular basis of RNA-dependent RNA polymerase II activity. Nature 2007, 450, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Greco-Stewart, V.S.; Schissel, E.; Pelchat, M. The hepatitis delta virus RNA genome interacts with the human RNA polymerases I and III. Virology 2009, 386, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Haussecker, D.; Huang, Y.; Kay, M.A. Combined proteomic-RNAi screen for host factors involved in human hepatitis delta virus replication. RNA 2009, 15, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- MacNaughton, T.B.; Gowans, E.J.; McNamara, S.P.; Burrell, C.J. Hepatitis delta antigen is necessary for access of hepatitis delta virus RNA to the cell transcriptional machinery but is not part of the transcriptional complex. Virology 1991, 184, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Netter, H.J.; Kajino, K.; Taylor, J.M. Experimental transmission of human hepatitis delta virus to the laboratory mouse. J. Virol. 1993, 67, 3357–3362. [Google Scholar] [PubMed]
- Lai, M.M. The molecular biology of hepatitis delta virus. Annu. Rev. Biochem. 1995, 64, 259–286. [Google Scholar] [CrossRef] [PubMed]
- Reid, C.E.; Lazinski, D.W. A host-specific function is required for ligation of a wide variety of ribozyme-processed RNAs. Proc. Natl. Acad. Sci. USA 2000, 97, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Weiner, A.J.; Choo, Q.L.; Wang, K.S.; Govindarajan, S.; Redeker, A.G.; Gerin, J.L.; Houghton, M. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta antigen polypeptides p24 delta and p27 delta. J. Virol. 1988, 62, 594–599. [Google Scholar] [PubMed]
- Chen, R.; Linnstaedt, S.D.; Casey, J.L. RNA editing and its control in hepatitis delta virus replication. Viruses 2010, 2, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, D.; Schütte, C.; Warnecke, J.; Dorn, I.; Hennig, H.; Kirchner, H.; Schlenke, P. The large form of ADAR 1 is responsible for enhanced hepatitis delta virus RNA editing in interferon-alpha-stimulated host cells. J. Viral Hepat. 2006, 13, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Polson, A.G.; Bass, B.L.; Casey, J.L. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature 1996, 380, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef] [PubMed]
- Macnaughton, T.B.; Lai, M.M. Genomic but not antigenomic hepatitis delta virus RNA is preferentially exported from the nucleus immediately after synthesis and processing. J. Virol. 2002, 76, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.J.; Chen, P.J.; Wu, J.C.; Patel, D.; Chen, D.S. Small-form hepatitis B surface antigen is sufficient to help in the assembly of hepatitis delta virus-like particles. J. Virol. 1991, 65, 6630–6636. [Google Scholar] [PubMed]
- Freitas, N.; Salisse, J.; Cunha, C.; Toshkov, I.; Menne, S.; Gudima, S.O. Hepatitis delta virus infects the cells of hepadnavirus-induced hepatocellular carcinoma in woodchucks. Hepatology 2012, 56, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Helbig, M.; Volz, T.; Allweiss, L.; Mancke, L.V.; Lohse, A.W.; Polywka, S.; Pollok, J.M.; Petersen, J.; Taylor, J.; et al. Persistent hepatitis D virus mono-infection in humanized mice is efficiently converted by hepatitis B virus to a productive co-infection. J. Hepatol. 2014, 60, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Davies, S.E.; Lau, J.Y.; O’Grady, J.G.; Portmann, B.C.; Alexander, G.J.; Williams, R. Evidence that hepatitis D virus needs hepatitis B virus to cause hepatocellular damage. Am. J. Clin. Pathol. 1992, 98, 554–558. [Google Scholar] [PubMed]
- Sheldon, J.; Ramos, B.; Toro, C.; Rios, P.; Martinez-Alarcon, J.; Bottecchia, M.; Romero, M.; Garcia-Samaniego, J.; Soriano, V.; et al. Does treatment of hepatitis B virus (HBV) infection reduce hepatitis delta virus (HDV) replication in HIV-HBV-HDV-coinfected patients? Antivir. Ther. 2008, 13, 97–102. [Google Scholar] [PubMed]
- Babiker, Z.O.; Hogan, C.; Ustianowski, A.; Wilkins, E. Does interferon-sparing tenofovir disoproxil fumarate-based therapy have a role in the management of severe acute hepatitis delta superinfection? J. Med. Microbiol. 2012, 61, 1780–1783. [Google Scholar] [CrossRef] [PubMed]
- Sureau, C. The use of hepatocytes to investigate HDV infection: The HDV/HepaRG model. Methods Mol. Biol. 2010, 640, 463–473. [Google Scholar] [PubMed]
- Brazas, R.; Ganem, D. A cellular homolog of hepatitis delta antigen: Implications for viral replication and evolution. Science 1996, 274, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Chang, S.C.; Huang, C.; Li, Y.P.; Lee, C.H.; Chang, M.F. Novel nuclear export signal-interacting protein, NESI, critical for the assembly of hepatitis delta virus. J. Virol. 2005, 79, 8113–8120. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.; Mendes, M.; Penque, D.; Coelho, A.V.; Cunha, C. Changes in the proteome of Huh7 cells induced by transient expression of hepatitis D virus RNA and antigens. J. Proteomics 2008, 71, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.; Mendes, M.; Freitas, N.; Penque, D.; Coelho, A.V.; Cunha, C. Proteome analysis of a human liver carcinoma cell line stably expressing hepatitis delta virus ribonucleoproteins. J. Proteomics 2009, 72, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Greco-Stewart, V.S.; Thibault, C.S.; Pelchat, M. Binding of the polypyrimidine tract-binding protein-associated splicing factor (PSF) to the hepatitis delta virus RNA. Virology 2006, 356, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Sikora, D.; Greco-Stewart, V.S.; Miron, P.; Pelchat, M. The hepatitis delta virus RNA genome interacts with eEF1A1, p54(nrb), hnRNP-L, GAPDH and ASF/SF2. Virology 2009, 390, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.; Brichler, S.; Khan, E.; Chami, M.; Dény, P.; Kremsdorf, D.; Gordien, E. Large hepatitis delta antigen activates STAT-3 and NF-kappaB via oxidative stress. J. Viral Hepat. 2012, 19, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Jeong, S.H.; Hwang, S.B. Large hepatitis delta antigen modulates transforming growth factor-beta signaling cascades: Implication of hepatitis delta virus-induced liver fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Pugnale, P.; Pazienza, V.; Guilloux, K.; Negro, F. Hepatitis delta virus inhibits alpha interferon signaling. Hepatology 2009, 49, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Kato, N.; Yoshida, H.; Otsuka, M.; Moriyama, M.; Shiratori, Y.; Koike, K.; Matsumura, M.; Omata, M. Synergistic activation of the serum response element-dependent pathway by hepatitis B virus x protein and large-isoform hepatitis delta antigen. J. Infect. Dis. 2003, 187, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.; Sheu, G.T.; Lai, M.M. Inhibition of Cellular RNA polymerase II transcription by delta antigen of hepatitis delta virus. Virology 1998, 247, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Filipovska, J.; Yano, K.; Furuya, A.; Inukai, N.; Narita, T.; Wada, T.; Sugimoto, S.; Konarska, M.M.; Handa, H. Stimulation of RNA polymerase II elongation by hepatitis delta antigen. Science 2001, 293, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.T.; Lee, Y.J.; Ko, J.L.; Tsai, C.C.; Tseng, C.J.; Sheu, G.T. Hepatitis delta virus epigenetically enhances clusterin expression via histone acetylation in human hepatocellular carcinoma cells. J. Gen. Virol. 2009, 90, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.; Perez-Hernandez, D.; Vazquez, J.; Coelho, A.V.; Cunha, C. Proteomic changes in HEK-293 cells induced by hepatitis delta virus replication. J. Proteomics 2013, 89, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Pearlberg, J.; Liu, Y.T.; Ganem, D. Deleterious effects of hepatitis delta virus replication on host cell proliferation. J. Virol. 2001, 75, 3600–3604. [Google Scholar] [CrossRef] [PubMed]
- Heinicke, L.A.; Bevilacqua, P.C. Activation of PKR by RNA misfolding: HDV ribozyme dimers activate PKR. RNA 2012, 18, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Korba, B.E.; Forzani, B.; Baroudy, B.M.; Brown, T.L.; Gerin, J.L.; Ponzetto, A. Hepatitis delta virus (HDV) and woodchuck hepatitis virus (WHV) nucleic acids in tissues of HDV-infected chronic WHV carrier woodchucks. J. Virol. 1989, 63, 1612–1618. [Google Scholar] [PubMed]
- Casey, J.L.; Gerin, J.L. The woodchuck model of HDV infection. Curr. Top. Microbiol. Immunol. 2006, 307, 211–225. [Google Scholar] [PubMed]
- Caselmann, W.H. HBV and HDV replication in experimental models: Effect of interferon. Antivir. Res. 1994, 24, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Kos, T.; Molijn, A.; van Doorn, L.J.; van Belkum, A.; Dubbeld, M.; Schellekens, H. Hepatitis delta virus cDNA sequence from an acutely HBV-infected chimpanzee: Sequence conservation in experimental animals. J. Med. Virol. 1991, 34, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Bergmann, K.F.; Baroudy, B.M.; Satterfield, W.C.; Popper, H.; Purcell, R.H.; Gerin, J.L. Chronic hepatitis D virus (HDV) infection in hepatitis B virus carrier chimpanzees experimentally superinfected with HDV. J. Infect. Dis. 1988, 158, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Gerin, J.L. Animal models of hepatitis delta virus infection and disease. ILAR J. 2001, 42, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Engelke, M.; Mills, K.; Seitz, S.; Simon, P.; Gripon, P.; Schnölzer, M.; Urban, S. Characterization of a hepatitis B and hepatitis delta virus receptor binding site. Hepatology 2006, 43, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Geipel, A.; König, A.; Corman, V.M.; van Riel, D.; Leijten, L.M.; Bremer, C.M.; Rasche, A.; Cottontail, V.M.; Maganga, G.D.; et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 16151–16156. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Yan, H.; Wang, H.; He, W.; Jing, Z.; Qi, Y.; Fu, L.; Gao, Z.; Huang, Y.; Xu, G.; et al. Sodium taurocholate cotransporting polypeptide mediates woolly monkey hepatitis B virus infection of Tupaia hepatocytes. J. Virol. 2013, 87, 7176–7184. [Google Scholar] [CrossRef] [PubMed]
- Walter, E.; Keist, R.; Niederost, B.; Pult, I.; Blum, H.E. Hepatitis B virus infection of tupaia hepatocytes in vitro and in vivo. Hepatology 1996, 24, 1–5. [Google Scholar] [PubMed]
- Li, Q.; Ding, M.; Wang, H. The infection of hepatitis D virus in adult tupaia. Zhonghua Yi Xue Za Zhi 1995, 75, 611–613, 639–640. [Google Scholar] [PubMed]
- Ponzetto, A.; Hoyer, B.H.; Popper, H.; Engle, R.; Purcell, R.H.; Gerin, J.L. Titration of the infectivity of hepatitis D virus in chimpanzees. J. Infect. Dis. 1987, 155, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Yang, P.M.; Chen, C.R.; Chen, D.S. Characterization of the transcripts of hepatitis D and B viruses in infected human livers. J. Infect. Dis. 1989, 160, 944–947. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, P.; Goldin, R.; Luther, S.; Carman, W.F.; Monjardino, J.; Thomas, H.C. Effect of cyclosporin-A in woodchucks with chronic hepatitis delta virus infection. J. Med. Virol. 1992, 36, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, P.; Saldanha, J.; Monjardino, J.; Jackson, A.; Luther, S.; Thomas, H.C. Immunisation of woodchucks with hepatitis delta antigen expressed by recombinant vaccinia and baculoviruses, controls HDV superinfection. Prog. Clin. Biol. Res. 1993, 382, 193–199. [Google Scholar] [PubMed]
- Fiedler, M.; Lu, M.; Siegel, F.; Whipple, J.; Roggendorf, M. Immunization of woodchucks (Marmota monax) with hepatitis delta virus DNA vaccine. Vaccine 2001, 19, 4618–4626. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, M.; Roggendorf, M. Vaccination against hepatitis delta virus infection: Studies in the woodchuck (Marmota monax) model. Intervirology 2001, 44, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, M.; Kosinska, A.; Schumann, A.; Brovko, O.; Walker, A.; Lu, M.; Johrden, L.; Mayer, A.; Wildner, O.; Roggendorf, M. Prime/boost immunization with DNA and adenoviral vectors protects from hepatitis D virus (HDV) infection after simultaneous infection with HDV and woodchuck hepatitis virus. J. Virol. 2013, 87, 7708–7716. [Google Scholar] [CrossRef] [PubMed]
- D’Ugo, E.; Paroli, M.; Palmieri, G.; Giuseppetti, R.; Argentini, C.; Tritarelli, E.; Bruni, R.; Barnaba, V.; Houghton, M.; Rapicetta, M. Immunization of woodchucks with adjuvanted sHDAg (p24): Immune response and outcome following challenge. Vaccine 2004, 22, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, S.; Fields, H.A.; Humphrey, C.D.; Margolis, H.S. Pathologic and ultrastructural changes of acute and chronic delta hepatitis in an experimentally infected chimpanzee. Am. J. Pathol. 1986, 122, 315–322. [Google Scholar] [PubMed]
- Cole, S.M.; Macnaughton, T.B.; Gowans, E.J. Differential roles for HDAg-p24 and -p27 in HDV pathogenesis. Prog. Clin. Biol. Res. 1993, 382, 131–138. [Google Scholar] [PubMed]
- Guilhot, S.; Huang, S.N.; Xia, Y.P.; La Monica, N.; Lai, M.M.; Chisari, F.V. Expression of the hepatitis delta virus large and small antigens in transgenic mice. J. Virol. 1994, 68, 1052–1058. [Google Scholar] [PubMed]
- Polo, J.M.; Jeng, K.S.; Lim, B.; Govindarajan, S.; Hofman, F.; Sangiorgi, F.; Lai, M.M. Transgenic mice support replication of hepatitis delta virus RNA in multiple tissues, particularly in skeletal muscle. J. Virol. 1995, 69, 4880–4887. [Google Scholar] [PubMed]
- Grompe, M.; Strom, S. Mice with human livers. Gastroenterology 2013, 145, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Navarro, B.; Gisel, A.; Rodio, M.E.; Delgado, S.; Flores, R.; Di Serio, F. Viroids: How to infect a host and cause disease without encoding proteins. Biochimie 2012, 94, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Kuo, M.Y.; Chen, M.L.; Tu, S.J.; Chiu, M.N.; Wu, H.L.; Hsu, H.C.; Chen, D.S. Continuous expression and replication of the hepatitis delta virus genome in Hep G2 hepatoblastoma cells transfected with cloned viral DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 5253–5257. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [PubMed]
- Sureau, C.; Taylor, J.; Chao, M.; Eichberg, J.W.; Lanford, R.E. Cloned hepatitis delta virus cDNA is infectious in the chimpanzee. J. Virol. 1989, 63, 4292–4297. [Google Scholar] [PubMed]
- Wu, J.C.; Chen, T.Z.; Huang, Y.S.; Yen, F.S.; Ting, L.T.; Sheng, W.Y.; Tsay, S.H.; Lee, S.D. Natural history of hepatitis D viral superinfection: Significance of viremia detected by polymerase chain reaction. Gastroenterology 1995, 108, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Dienes, H.P.; Purcell, R.H.; Popper, H.; Ponzetto, A. The significance of infections with two types of viral hepatitis demonstrated by histologic features in chimpanzees. J. Hepatol. 1990, 10, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Polo, J.M.; Lim, B.; Govindarajan, S.; Lai, M.M. Replication of hepatitis delta virus RNA in mice after intramuscular injection of plasmid DNA. J. Virol. 1995, 69, 5203–5207. [Google Scholar] [PubMed]
- Chang, J.; Sigal, L.J.; Lerro, A.; Taylor, J. Replication of the human hepatitis delta virus genome is initiated in mouse hepatocytes following intravenous injection of naked DNA or RNA sequences. J. Virol. 2001, 75, 3469–3473. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Huang, L.R.; Yang, H.C.; Tzeng, H.T.; Hsu, P.N.; Wu, H.L.; Chen, P.J.; Chen, D.S. Hepatitis B virus core antigen determines viral persistence in a C57BL/6 mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 9340–9345. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.R.; Gäbel, Y.A.; Graf, S.; Arzberger, S.; Kurts, C.; Heikenwalder, M.; Knolle, P.A.; Protzer, U. Transfer of HBV genomes using low doses of adenovirus vectors leads to persistent infection in immune competent mice. Gastroenterology 2012, 142, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zeng, D.; Yu, F.; He, J.; Zhou, Z.; Tu, W.; Deng, H.; Tian, D.A.; Liu, M. Role of autophagy in monokine induced by interferon gamma (Mig) production during adenovirus-hepatitis B virus infection. Hepatogastroenterology 2012, 59, 1245–1250. [Google Scholar] [PubMed]
- Dion, S.; Bourgine, M.; Godon, O.; Levillayer, F.; Michel, M.L. Adeno-associated virus-mediated gene transfer leads to persistent hepatitis B virus replication in mice expressing HLA-A2 and HLA-DR1 molecules. J. Virol. 2013, 87, 5554–5563. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Fang, C.C.; Tsuneyama, K.; Chou, H.Y.; Pan, W.Y.; Shih, Y.M.; Wu, P.Y.; Chen, Y.; Leung, P.S.; Gershwin, M.E.; et al. A murine model of hepatitis B-associated hepatocellular carcinoma generated by adeno-associated virus-mediated gene delivery. Int. J. Oncol. 2011, 39, 1511–1519. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldabe, R.; Suárez-Amarán, L.; Usai, C.; González-Aseguinolaza, G. Animal Models of Chronic Hepatitis Delta Virus Infection Host–Virus Immunologic Interactions. Pathogens 2015, 4, 46-65. https://doi.org/10.3390/pathogens4010046
Aldabe R, Suárez-Amarán L, Usai C, González-Aseguinolaza G. Animal Models of Chronic Hepatitis Delta Virus Infection Host–Virus Immunologic Interactions. Pathogens. 2015; 4(1):46-65. https://doi.org/10.3390/pathogens4010046
Chicago/Turabian StyleAldabe, Rafael, Lester Suárez-Amarán, Carla Usai, and Gloria González-Aseguinolaza. 2015. "Animal Models of Chronic Hepatitis Delta Virus Infection Host–Virus Immunologic Interactions" Pathogens 4, no. 1: 46-65. https://doi.org/10.3390/pathogens4010046