KSHV and the Role of Notch Receptor Dysregulation in Disease Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Kaposi’s Sarcoma-Associated Herpesvirus
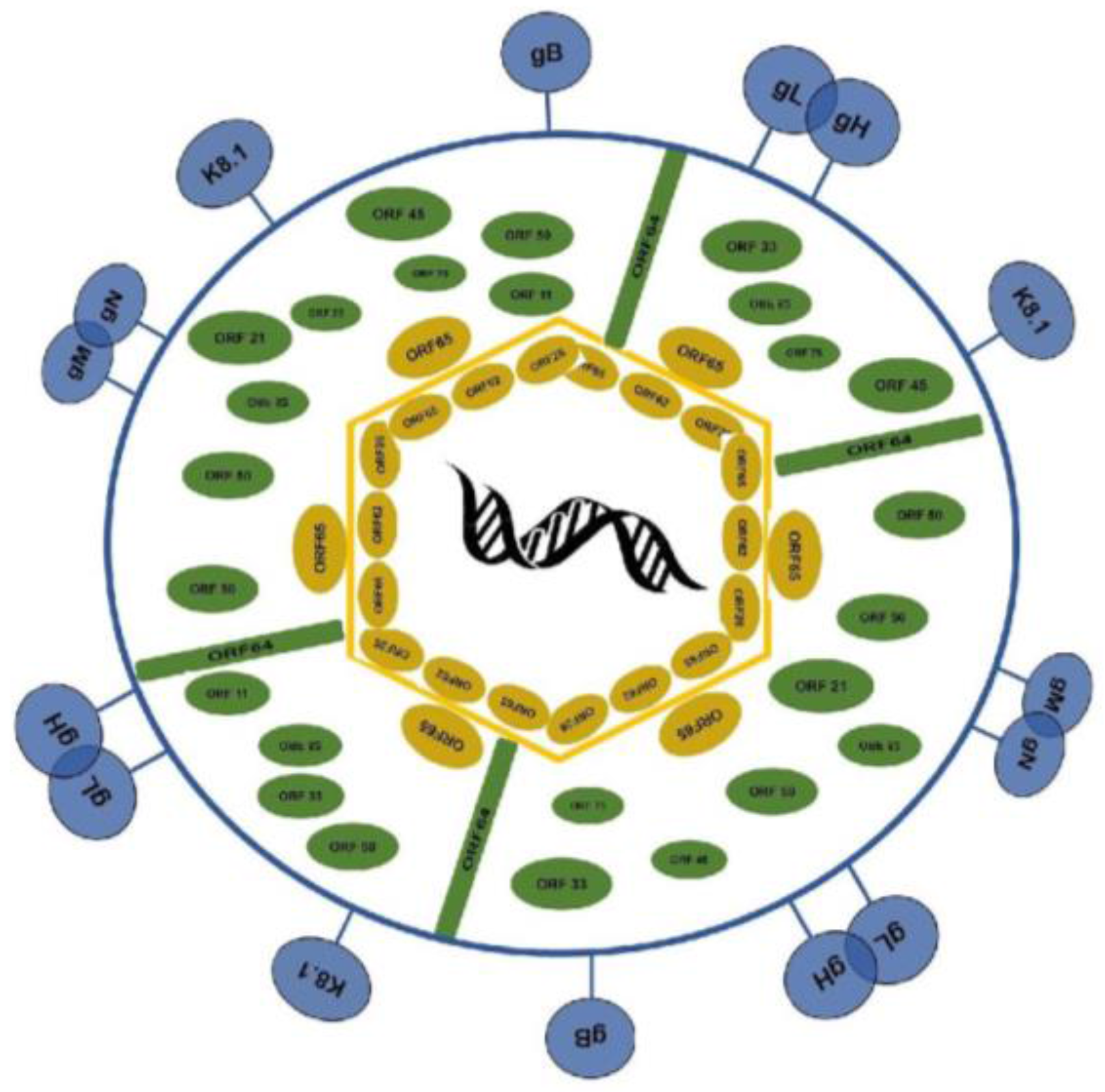
1.1. Virion Structure
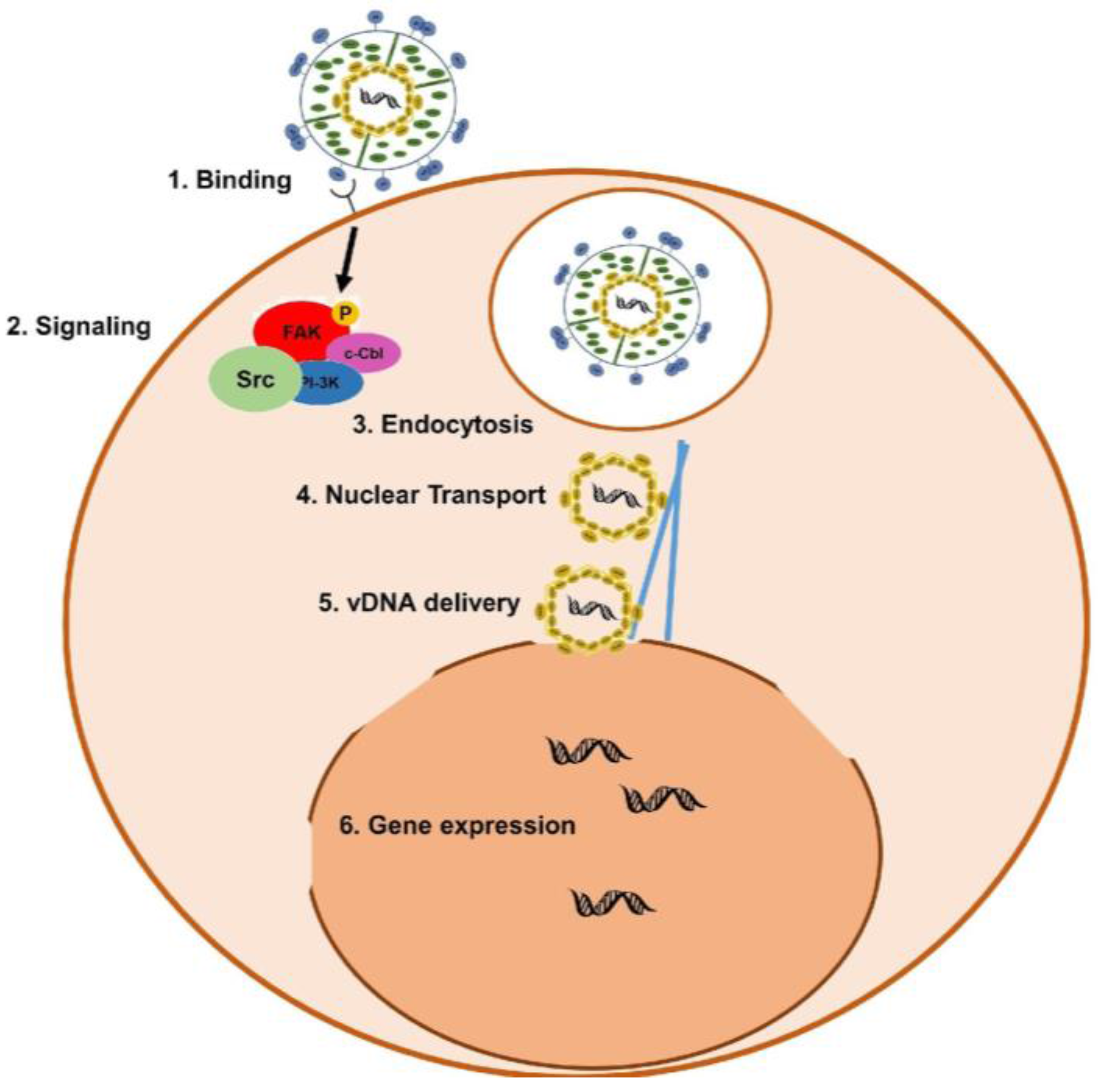
1.2. KSHV Infection of Target Cells and Egress
1.3. Pathogenesis
1.4. Transmission
1.5. Disease
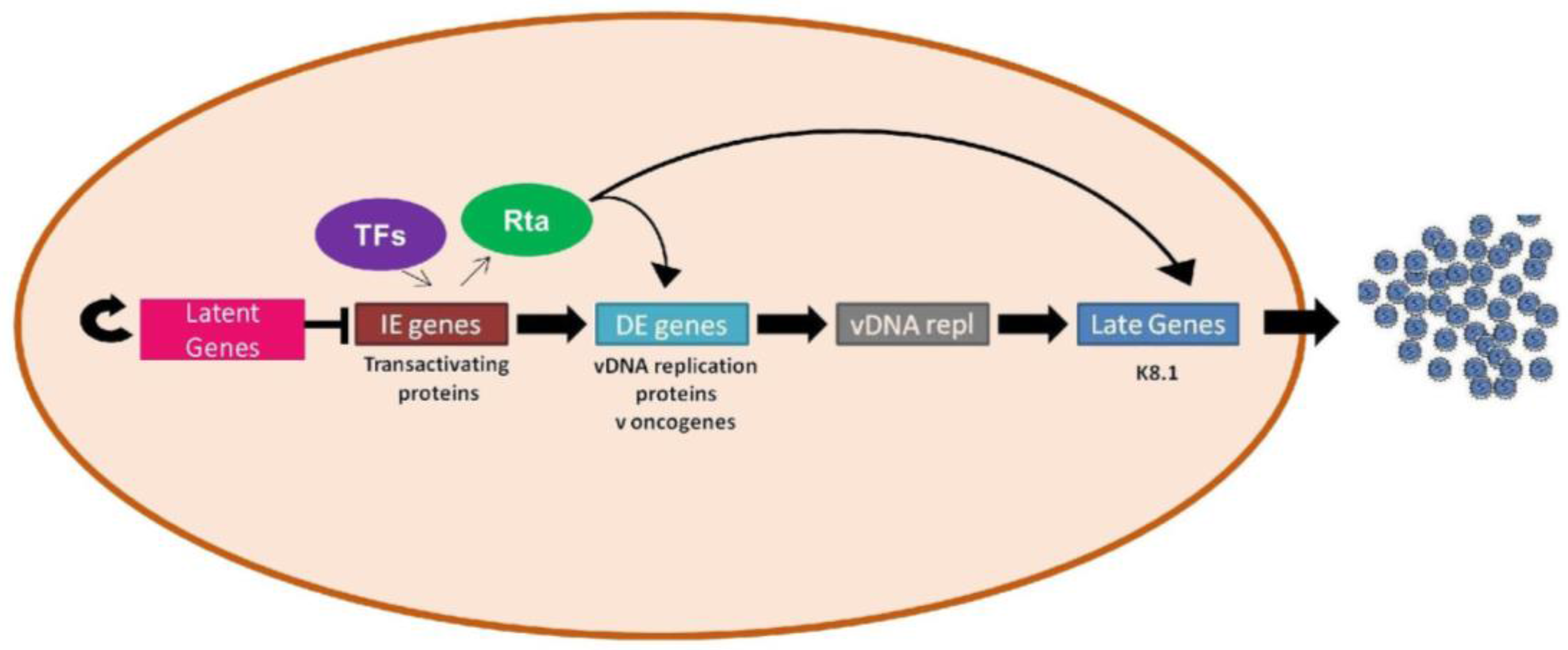
1.6. Lytic vs. Latent Infection
2. KSHV Gene Regulation by Replication and Transcriptional Activator (Rta) Partially Mimics the Cellular Notch Protein
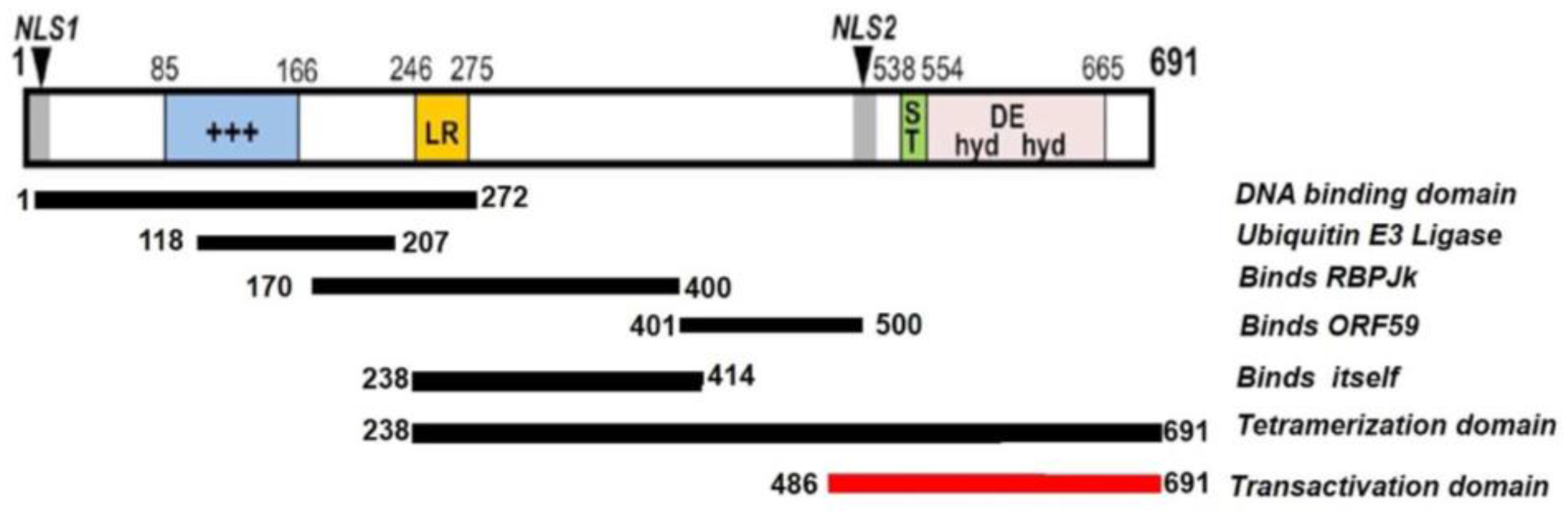
2.1. Structure
2.2. Functions
2.2.1. Viral DNA Replication
2.2.2. Transactivation of Viral Promoters
2.2.3. Degradation of Cellular Proteins
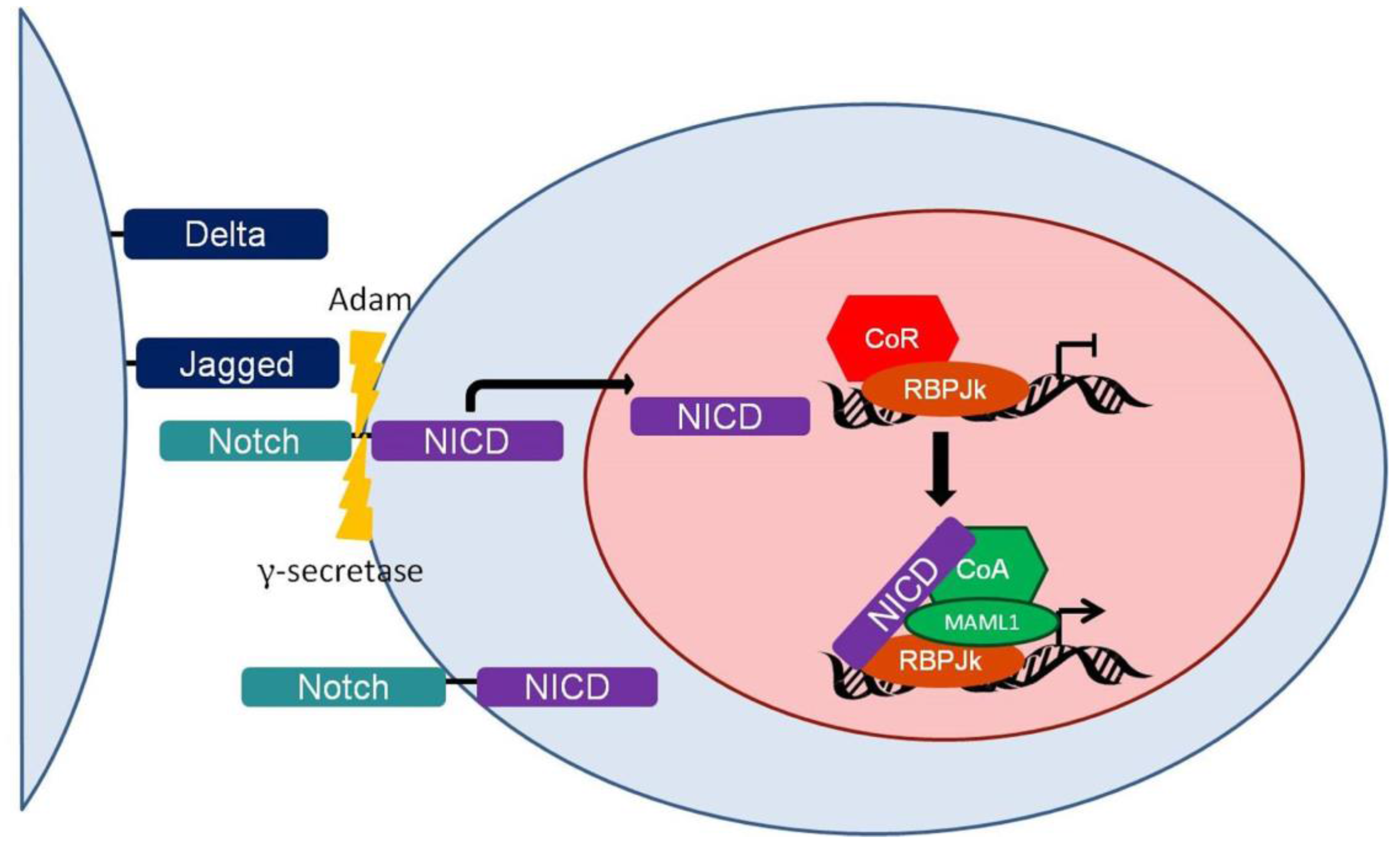
3. Notch
3.1. Function
3.2. Structure
3.3. Dysregulation in KSHV Infection
3.3.1. Notch in KSHV Disease Progression and Oncogenesis
3.3.2. Notch in KSHV Reactivation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.; Lee, F.; Culpepper, J.; Knowles, D.; Moore, P. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.; Clauvel, J.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Dittmer, D.P.; Richards, K.L.; Damania, B. Treatment of Kaposi sarcoma-associated herpesvirus (KSHV)-associated cancers. Front. Microbiol. 2012, 3, 141. [Google Scholar] [CrossRef] [PubMed]
- Fatahzadeh, M. Kaposi sarcoma: Review and medical management update. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 113, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Curry, C.L.; Reed, L.L.; Golde, T.E.; Miele, L.; Nickoloff, B.J.; Foreman, K.E. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi’s sarcoma tumor cells. Oncogene 2005, 24, 6333–6344. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Pekkonen, P.; Laurinavicius, S.; Sugiyama, N.; Henderson, S.; Günther, T.; Rantanen, V.; Kaivanto, E.; Aavikko, M.; Sarek, G.; et al. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell Host Microbe 2011, 10, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Madonna, R.; Melino, G.; Caruso, C. The emerging role of notch pathway in ageing: Focus on the related mechanisms in age-related diseases. Ageing Res. Rev. 2016, 29, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Gridley, T. Notch signaling and inherited disease syndromes. Hum. Mol. Genet. 2003, 12, R9–R13. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Lagos, D.; Pizzey, A.; Gratrix, F.; Henderson, S.R.; Boshoff, C. KSHV manipulates notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Choudhuri, T.; Murakami, M.; Kuppers, D.A.; Robertson, E.S. Intracellular activated Notch1 is critical for proliferation of Kaposi’s sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J. Virol. 2006, 80, 6411–6419. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Murakami, M.; Bajaj, B.; Kaul, R.; He, Z.; Gan, R.; Feldman, M.; Robertson, E.S. Inhibition of KSHV-infected primary effusion lymphomas in NOD/SCID mice by gamma-secretase inhibitor. Cancer Biol. Ther. 2009, 8, 2136–2143. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Bu, W.; Palmeri, D.; Spadavecchia, S.; Lynch, S.J.; Marras, S.A.E.; Tyagi, S.; Lukac, D.M. Kaposi’s sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the notch pathway. J. Virol. 2006, 80, 9697–9709. [Google Scholar] [CrossRef] [PubMed]
- DeCotiis, J.L.; Ortiz, N.C.; Vega, B.A.; Lukac, D.M. An easily transfectable cell line that produces an infectious reporter virus for routine and robust quantitation of Kaposi’s sarcoma-associated herpesvirus reactivation. J. Virol. Methods 2017, 247, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Murakami, M.; Choudhuri, T.; Kuppers, D.A.; Robertson, E.S. Intracellular-activated Notch1 can reactivate Kaposi’s sarcoma-associated herpesvirus from latency. Virology 2006, 351, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hu, H.; He, Z.; Liang, D.; Sun, R.; Lan, K. Fine-tuning of the Kaposi’s sarcoma-associated herpesvirus life cycle in neighboring cells through the RTA-JAG1-Notch pathway. PLoS Pathog. 2016, 12, e1005900. [Google Scholar] [CrossRef] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.-C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Cullen, B.R. Transcriptional origin of Kaposi’s sarcoma-associated herpesvirus MicroRNAs. J. Virol. 2006, 80, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Pramod, N.P.; Wang, F.-Z.; Chandran, B. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 2001, 284, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.-Z.; Akula, S.M.; Pramod, N.P.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 2001, 75, 7517–7527. [Google Scholar] [CrossRef] [PubMed]
- Rozen, R.; Sathish, N.; Li, Y.; Yuan, Y. Virion-wide protein interactions of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 4742–4750. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C. Herpesvirus assembly and egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Wang, X.; Yuan, Y. Tegument proteins of Kaposi’s sarcoma-associated herpesvirus and related gamma-herpesviruses. Front. Microbiol. 2012, 3, 98. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Liang, D.; Lan, K. MicroRNAs and unusual small RNAs discovered in Kaposi’s sarcoma-associated herpesvirus virions. J. Virol. 2012, 86, 12717–12730. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; Sathish, N.; Yuan, Y. Antagonism of host antiviral responses by Kaposi’s sarcoma-associated herpesvirus tegument protein ORF45. PLoS ONE 2010, 5, e10573. [Google Scholar] [CrossRef] [PubMed]
- Inn, K.-S.; Lee, S.-H.; Rathbun, J.Y.; Wong, L.-Y.; Toth, Z.; Machida, K.; Ou, J.-H.J.; Jung, J.U. Inhibition of RIG-i-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol. 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.-i.; Reed, J.C.; Ting, J.P.Y.; Damania, B. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Wies, E.; Thurau, M.; Marquardt, G.; Naschberger, E.; Hentschel, S.; Jochmann, R.; Schulz, T.F.; Erfle, H.; Brors, B.; et al. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-κB activation. J. Virol. 2009, 83, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Lo, P.; Yu, X.; Stoops, J.K.; Forghani, B.; Zhou, Z.H. Three-dimensional structure of the human herpesvirus 8 capsid. J. Virol. 2000, 74, 9646–9654. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Lagunoff, M.; Zhong, W.; Ganem, D. The size and conformation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 1996, 70, 8151–8154. [Google Scholar] [PubMed]
- Persson, L.M.; Wilson, A.C. Wide-scale use of notch signaling factor CSL/RBP-Jκ in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J. Virol. 2010, 84, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Birkmann, A.; Mahr, K.; Ensser, A.; Yağuboğlu, S.; Titgemeyer, F.; Fleckenstein, B.; Neipel, F. Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J. Virol. 2001, 75, 11583–11593. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Birkmann, A.; Wies, E.; Dorer, D.; Mahr, K.; Stürzl, M.; Titgemeyer, F.; Neipel, F. Kaposi’s sarcoma-associated herpesvirus gH/gL: Glycoprotein export and interaction with cellular receptors. J. Virol. 2009, 83, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Pramod, N.P.; Wang, F.-Z.; Chandran, B. Integrin α3β1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002, 108, 407–419. [Google Scholar] [CrossRef]
- Hahn, A.S.; Desrosiers, R.C. Binding of the Kaposi’s sarcoma-associated herpesvirus to the ephrin binding surface of the EphA2 receptor and its inhibition by a small molecule. J. Virol. 2014, 88, 8724–8734. [Google Scholar] [CrossRef] [PubMed]
- Rappocciolo, G.; Hensler, H.R.; Jais, M.; Reinhart, T.A.; Pegu, A.; Jenkins, F.J.; Rinaldo, C.R. Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J. Virol. 2008, 82, 4793–4806. [Google Scholar] [CrossRef] [PubMed]
- Chandran, B. Early events in Kaposi’s sarcoma-associated herpesvirus infection of target cells. J. Virol. 2010, 84, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.; Bandyopadhyay, C.; Dutta, D.; Chandran, B. Interaction of KSHV with host cell surface receptors and cell entry. Viruses 2014, 6, 4024. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Veettil, M.V.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. USA 2012, 109, E1163–E1172. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Sharma-Walia, N.; Sadagopan, S.; Raghu, H.; Sivakumar, R.; Naranatt, P.P.; Chandran, B. RhoA-GTPase facilitates entry of Kaposi’s sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 2006, 80, 11432–11446. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Akula, S.M.; Zien, C.A.; Krishnan, H.H.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the Phosphatidylinositol 3-Kinase-PKC-ζ-MEK-ERK signaling pathway in target cells early during infection: Implications for infectivity. J. Virol. 2003, 77, 1524–1539. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; Naranatt, P.P.; Krishnan, H.H.; Zeng, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-Phosphatidylinositol 3-Kinase-Rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 2004, 78, 4207–4223. [Google Scholar] [CrossRef] [PubMed]
- Raghu, H.; Sharma-Walia, N.; Veettil, M.V.; Sadagopan, S.; Caballero, A.; Sivakumar, R.; Varga, L.; Bottero, V.; Chandran, B. Lipid rafts of primary endothelial cells are essential for Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8-induced Phosphatidylinositol 3-Kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J. Virol. 2007, 81, 7941–7959. [Google Scholar] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Smith, M.S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J. Virol. 2005, 79, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Svojanovsky, S.R.; Bloomer, C.; Mathur, S.; Chandran, B. Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: Insights into Modulation Events Early during Infection. Cancer Res. 2004, 64, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Sharma, N.; Murakami, M.; Robertson, E.S. Induction of Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: A novel mechanism for establishment of latency. J. Virol. 2005, 79, 7453–7465. [Google Scholar] [CrossRef] [PubMed]
- Shamay, M.; Krithivas, A.; Zhang, J.; Hayward, S.D. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. USA 2006, 103, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Cai, Q.; Robertson, E.S. The single RBP-Jκ site within the LANA promoter is crucial for establishing Kaposi’s sarcoma-associated herpesvirus latency during primary infection. J. Virol. 2011, 85, 6148–6161. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.-R.; Izumiya, Y.; Tepper, C.; Kung, H.-J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Uppal, T.; Jha, H.; Verma, S.; Robertson, E. Chromatinization of the KSHV genome during the KSHV life cycle. Cancers 2015, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Skepper, J.N.; Whiteley, A.; Browne, H.; Minson, A. Herpes simplex virus nucleocapsids mature to progeny virions by an envelopment → deenvelopment → reenvelopment pathway. J. Virol. 2001, 75, 5697–5702. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Forest, T.; Barnard, S.; Baines, J.D. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 2005, 7, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Granzow, H.; Klupp, B.G.; Fuchs, W.; Veits, J.; Osterrieder, N.; Mettenleiter, T.C. Egress of alphaherpesviruses: Comparative ultrastructural study. J. Virol. 2001, 75, 3675–3684. [Google Scholar] [CrossRef] [PubMed]
- Gershon, A.A.; Sherman, D.L.; Zhu, Z.; Gabel, C.A.; Ambron, R.T.; Gershon, M.D. Intracellular transport of newly synthesized varicella-zoster virus: Final envelopment in the trans-Golgi network. J. Virol. 1994, 68, 6372–6390. [Google Scholar] [PubMed]
- Mettenleiter, T.C. Intriguing interplay between viral proteins during herpesvirus assembly or: The herpesvirus assembly puzzle. Vet. Microbiol. 2006, 113, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Corbellino, M.; Poirel, L.; Bestetti, G.; Pizzuto, M.; Aubin, J.T.; Capra, M.; Bifulco, C.; Berti, E.; Agut, H.; Rizzardini, G.; et al. Restricted tissue distribution of extralesional Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS patients with Kaposi’s sarcoma. AIDS Res. Hum. Retrovir. 1996, 12, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Luppi, M.; Barozzi, P.; Schulz, T.F.; Setti, G.; Staskus, K.; Trovato, R.; Narni, F.; Donelli, A.; Maiorana, A.; Marasca, R.; et al. Bone marrow failure associated with human herpesvirus 8 infection after transplantation. N. Engl. J. Med. 2000, 343, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Ambroziak, J.A.; Blackbourn, D.J.; Herndier, B.G.; Glogau, R.G.; Gullett, J.H.; McDonald, A.R.; Lennette, E.T.; Levy, J.A. Herpes-like sequences in HIV-infected and uninfected Kaposi’s sarcoma patients. Science 1995, 268, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Blackbourn, D.J.; Ambroziak, J.; Lennette, E.; Adams, M.; Ramachandran, B.; Levy, J.A. Infectious human herpesvirus 8 in a healthy north american blood donor. Lancet 1997, 349, 609–611. [Google Scholar] [CrossRef]
- Münz, C. Epstein barr virus—A tumor virus that needs cytotoxic lymphocytes to persist asymptomatically. Curr. Opin. Virol. 2016, 20, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Usherwood, E.J. Immune escape of γ-herpesviruses from adaptive immunity. Rev. Med. Virol. 2014, 24, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Whitby, D.; Boshoff, C.; Hatzioannou, T.; Weiss, R.A.; Schulz, T.F.; Howard, M.R.; Brink, N.S.; Tedder, R.S.; Tenant-Flowers, M.; Copas, A.; et al. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi’s sarcoma. Lancet 1995, 346, 799–802. [Google Scholar] [CrossRef]
- Martin, D.F.; Kuppermann, B.D.; Wolitz, R.A.; Palestine, A.G.; Li, H.; Robinson, C.A.; Group, The Roche Ganciclovir Study Group. Oral ganciclovir for patients with Cytomegalovirus Retinitis treated with a ganciclovir implant. N. Engl. J. Med. 1999, 340, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Diem, M.D.; Chan, C.C.; Younis, I.; Dreyfuss, G. PYM binds the cytoplasmic exon-junction complex and ribosomes to enhance translation of spliced mRNAs. Nat. Struct. Mol. Biol. 2007, 14, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Alkharsah, K.R.; Dedicoat, M.; Blasczyk, R.; Newton, R.; Schulz, T.F. Influence of HLA alleles on shedding of Kaposi sarcoma-associated herpesvirus in saliva in an African population. J. Infect. Dis. 2007, 195, 809–816. [Google Scholar] [CrossRef] [PubMed]
- de França, T.R.T.; de Araújo, R.A.; Ribeiro, C.M.B.; Leao, J.C. Salivary shedding of HHV-8 in people infected or not by human immunodeficiency virus 1. J. Oral Pathol. Med. 2011, 40, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Hladik, W.; Dollard, S.C.; Mermin, J.; Fowlkes, A.L.; Downing, R.; Amin, M.M.; Banage, F.; Nzaro, E.; Kataaha, P.; Dondero, T.J.; et al. Transmission of human herpesvirus 8 by blood transfusion. N. Engl. J. Med. 2006, 355, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.R.; Whitby, D.; Bahadur, G.; Suggett, F.; Boshoff, C.; Tenant-Flowers, M.; Schulz, T.F.; Kirk, S.; Matthews, S.; Weller, I.V.D.; et al. Detection of human herpesvirus 8 DNA in semen from HIV-infected individuals but not healthy semen donors. AIDS 1997, 11, F15–F19. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Wachtman, L.M.; Pearson, C.B.; Lee, J.-S.; Lee, H.-R.; Lee, S.H.; Vieira, J.; Mansfield, K.G.; Jung, J.U. Non-human primate model of Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 2009, 5, e1000606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munshi, N.; Mehra, M.; van de Velde, H.; Desai, A.; Potluri, R.; Vermeulen, J. Use of a claims database to characterize and estimate the incidence rate for castleman disease. Leuk. Lymphoma 2015, 56, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Bonekamp, D.; Horton, K.M.; Hruban, R.H.; Fishman, E.K. Castleman disease: The great mimic. Radiographics 2011, 31, 1793–1807. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.; Mitsuyasu, R. HIV-associated multicentric Castleman disease. Curr. Opin. Oncol. 2011, 23, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Uldrick, T.S.; Wang, V.; O’Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Giffin, L.; Damania, B. KSHV: Pathways to tumorigenesis and persistent infection. Adv. Virus Res. 2014, 88, 111–159. [Google Scholar] [PubMed]
- Polizzotto, M.N.; Uldrick, T.S.; Hu, D.; Yarchoan, R. Clinical manifestations of kaposi sarcoma herpesvirus (kshv) lytic activation: Multicentric castleman disease (KSHV-MCD) and the KSHV inflammatory cytokine syndrome (KICS). Front. Microbiol. 2012, 3, 73. [Google Scholar] [CrossRef] [PubMed]
- Uphoff, C.C.; Carbone, A.; Gaidano, G.; Drexler, H.G. HHV-8 infection is specific for cell lines derived from primary effusion (body cavity-based) lymphomas. Leukemia 1998, 12, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-B.; Rahemtullah, A.; Hochberg, E. Primary effusion lymphoma. Oncologist 2007, 12, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Goedert, J.J.; Vitale, F.; Lauria, C.; Serraino, D.; Tamburini, M.; Montella, M.; Messina, A.; Brown, E.E.; Rezza, G.; Gafà, L.; et al. Risk factors for classical Kaposi’s sarcoma. JNCI J. Natl. Cancer Inst. 2002, 94, 1712–1718. [Google Scholar] [CrossRef] [PubMed]
- Chokunonga, E.; Levy, L.M.; Bassett, M.T.; Mauchaza, B.G.; Thomas, D.B.; Parkin, D.M. Cancer Incidence in the African Population of Harare, Zimbabwe: Second Results from the Cancer Registry 1993–1995. Int. J. Cancer 2000, 85, 54–59. [Google Scholar] [CrossRef]
- Wood, N.H.; Feller, L. The malignant potential of HIV-associated Kaposi sarcoma. Cancer Cell Int. 2008, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Penn, I. Kaposi’s sarcoma in organ transplant recipients: Report of 20 cases. Transplantation 1979, 27, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Andreoni, M.; Goletti, D.; Pezzotti, P.; Pozzetto, A.; Monini, P.; Sarmati, L.; Farchi, F.; Tisone, G.; Piazza, A.; Pisani, F.; et al. Prevalence, incidence and correlates of HHV-8/KSHV infection and Kaposi’s sarcoma in renal and liver transplant recipients. J. Infect. 2001, 43, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Louthrenoo, W.; Kasitanon, N.; Mahanuphab, P.; Bhoopat, L.; Thongprasert, S. Kaposi’s sarcoma in rheumatic diseases. Semin. Arthritis Rheum. 2003, 32, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Bottler, T.; Kuttenberger, J.; Hardt, N.; Oehen, H.P.; Baltensperger, M. Non-HIV-associated Kaposi’s sarcoma of the tongue. Int. J. Oral Maxillofac. Surg. 2007, 36, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Ferdinando, M.; Massimiliano, B.; Emanuela, V.; Ornella, S.; Ernesto, Z.; Umberto, T. AIDS-related Kaposi’s sarcoma: State of the art and therapeutic strategies. Curr. HIV Res. 2009, 7, 634–638. [Google Scholar]
- Krown, S.E. Highly active antiretroviral therapy in AIDS-associated Kaposi’s sarcoma: Implications for the design of therapeutic trials in patients with advanced, symptomatic Kaposi’s sarcoma. J. Clin. Oncol. 2004, 22, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lagunoff, M. Establishment and maintenance of Kaposi’s sarcoma-associated herpesvirus latency in B cells. J. Virol. 2005, 79, 14383–14391. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, A.D.A.; Cavallin, L.E.; Vincent, L.; Chiozzini, C.; Eroles, P.; Duran, E.M.; Asgari, Z.; Hooper, A.T.; La Perle, K.M.D.; Hilsher, C.; et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: A cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell 2007, 11, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S.; Sodhi, A.; Molinolo, A.; Bugge, T.H.; Sawai, E.T.; He, Y.; Li, Y.; Ray, P.E.; Gutkind, J.S. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell 2003, 3, 23–36. [Google Scholar] [CrossRef]
- Liang, Y.; Ganem, D. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi’s sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 2004, 78, 6818–6826. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Garber, A.C.; Renne, R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 2002, 76, 11677–11687. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [PubMed]
- Ye, F.; Lei, X.; Gao, S.-J. Mechanisms of Kaposi’s sarcoma-associated herpesvirus latency and reactivation. Adv. Virol. 2011, 2011, 19. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Verma, S.C.; Lampson, M.A.; Cai, Q.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol. 2008, 82, 6734–6746. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: A potential mechanism for virus-mediated control of latency. J. Virol. 2004, 78, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jκ, the major downstream effector of the Notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Cai, Q.; Saha, A.; Dzeng, R.K.; Robertson, E.S. The RBP-Jκ binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 2012, 8, e1002479. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; He, Z.; Liang, D.; Zhang, Q.; Zhang, H.; Deng, Q.; Robertson, E.S.; Lan, K. Carboxyl-terminal amino acids 1052 to 1082 of the latency-associated nuclear antigen (LANA) interact with RBP-Jκ and are responsible for LANA-mediated RTA repression. J. Virol. 2012, 86, 4956–4969. [Google Scholar] [CrossRef] [PubMed]
- Heinzelmann, K.; Scholz, B.A.; Nowak, A.; Fossum, E.; Kremmer, E.; Haas, J.; Frank, R.; Kempkes, B. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4/K10) is a novel interaction partner of CSL/CBF1, the major downstream effector of notch signaling. J. Virol. 2010, 84, 12255–12264. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641. [Google Scholar] [CrossRef] [PubMed]
- Fejér, G.; Medveczky, M.M.; Horvath, E.; Lane, B.; Chang, Y.; Medveczky, P.G. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts preferentially with the terminal repeats of the genome in vivo and this complex is sufficient for episomal DNA replication. J. Gen. Virol. 2003, 84, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [PubMed]
- Cai, Q.-L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Martin, H.J.; Liao, G.; Hayward, S.D. The Kaposi’s sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J. Virol. 2007, 81, 10451–10459. [Google Scholar] [CrossRef] [PubMed]
- Van Dross, R.; Yao, S.; Asad, S.; Westlake, G.; Mays, D.J.; Barquero, L.; Duell, S.; Pietenpol, J.A.; Browning, P.J. Constitutively active K-cyclin/cdk6 kinase in Kaposi sarcoma-associated herpesvirus-infected cells. JNCI J. Natl. Cancer Inst. 2005, 97, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Direkze, S.; Laman, H. Regulation of growth signalling and cell cycle by Kaposi’s sarcoma-associated herpesvirus genes. Int. J. Exp. Pathol. 2004, 85, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Field, N.; Low, W.; Daniels, M.; Howell, S.; Daviet, L.; Boshoff, C.; Collins, M. KSHV vFLIP binds to IKK-γ to activate IKK. J. Cell Sci. 2003, 116, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Li, Q.; Lee, J.-Y.; Lee, S.-H.; Jeong, J.H.; Lee, H.-R.; Chang, H.; Zhou, F.-C.; Gao, S.-J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Leidal, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.K.; McCormick, C. Subversion of autophagy by Kaposi’s sarcoma-associated herpesvirus impairs oncogene-induced senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [PubMed]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 2005, 307, 739. [Google Scholar] [CrossRef] [PubMed]
- Plaisance-Bonstaff, K.; Choi, H.; Beals, T.; Krueger, B.; Boss, I.; Gay, L.; Haecker, I.; Hu, J.; Renne, R. KSHV miRNAs decrease expression of lytic genes in latently infected PEL and endothelial cells by targeting host transcription factors. Viruses 2014, 6, 4005. [Google Scholar] [CrossRef] [PubMed]
- Moody, R.; Zhu, Y.; Huang, Y.; Cui, X.; Jones, T.; Bedolla, R.; Lei, X.; Bai, Z.; Gao, S.-J. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Renne, R.; Dittmer, D.; Ganem, D. Inflammatory cytokines and the reactivation of Kaposi’s sarcoma-associated herpesvirus lytic replication. Virology 2000, 266, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.-J. Reactive oxygen species hydrogen peroxide mediates Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, C.; Guo, Y.; Wei, F.; Lu, J.; Qin, J.; Banerjee, S.; Wang, J.; Shang, H.; Verma, S.C.; et al. Inhibition of KAP1 enhances hypoxia-induced Kaposi’s sarcoma-associated herpesvirus reactivation through RBP-Jκ. J. Virol. 2014, 88, 6873–6884. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.N.; Arbiser, J.L.; Offermann, M.K. Valproic acid induces human herpesvirus 8 lytic gene expression in BCBL-1 cells. AIDS 2000, 14, 899. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zhou, J.; Wiedmer, A.; Madden, K.; Yuan, Y.; Lieberman, P.M. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003, 77, 11425–11435. [Google Scholar] [CrossRef] [PubMed]
- Daigle, D.; Gradoville, L.; Tuck, D.; Schulz, V.; Wang’ondu, R.; Ye, J.; Gorres, K.; Miller, G. Valproic acid antagonizes the capacity of other histone deacetylase inhibitors to activate the Epstein-Barr virus lytic cycle. J. Virol. 2011, 85, 5628–5643. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Brodie, C.; Sarid, R. An essential role of ERK signalling in TPA-induced reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2006, 87, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Kati, S.; Hage, E.; Mynarek, M.; Ganzenmueller, T.; Indenbirken, D.; Grundhoff, A.; Schulz, T.F. Generation of high-titre virus stocks using BrK.219, a B-cell line infected stably with recombinant Kaposi’s sarcoma-associated herpesvirus. J. Virol. Methods 2015, 217, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Dollery, S.J.; Santiago-Crespo, R.J.; Kardava, L.; Moir, S.; Berger, E.A. Efficient infection of a human B cell line with cell-free Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014, 88, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Kati, S.; Tsao, E.H.; Günther, T.; Weidner-Glunde, M.; Rothämel, T.; Grundhoff, A.; Kellam, P.; Schulz, T.F. Activation of the B cell antigen receptor triggers reactivation of latent Kaposi’s sarcoma-associated herpesvirus in B cells. J. Virol. 2013, 87, 8004–8016. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 1999, 73, 9348–9361. [Google Scholar] [PubMed]
- Sun, R.; Lin, S.-F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [PubMed]
- Purushothaman, P.; Uppal, T.; Verma, S.C. Molecular biology of KSHV lytic reactivation. Viruses 2015, 7, 116–153. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Chen, J.; Tang, W.; Liu, C.; Chen, X. Kaposi’s sarcoma-associated herpesvirus ORF6 gene is essential in viral lytic replication. PLoS ONE 2014, 9, e99542. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Susilarini, N.K.; Pari, G.S. Interaction of Kaposi’s sarcoma-associated herpesvirus ORF59 with oriLyt is dependent on binding with K-Rta. J. Virol. 2011, 85, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, D.; Spadavecchia, S.; Carroll, K.D.; Lukac, D.M. Promoter- and cell-specific transcriptional transactivation by the Kaposi’s sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 2007, 81, 13299–13314. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Pripuzova, N.; McCoy, J.P.; Gao, S.-J.; Zheng, Z.-M. Targeted disruption of Kaposi’s sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, k8α, and k8.1 and the production of infectious virus. J. Virol. 2007, 81, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Massimelli, M.J.; Kang, J.-G.; Majerciak, V.; Le, S.-Y.; Liewehr, D.J.; Steinberg, S.M.; Zheng, Z.-M. Stability of a long noncoding viral RNA depends on a 9-nt core element at the RNA 5′ end to interact with viral ORF57 and cellular PABPC1. Int. J. Biol. Sci. 2011, 7, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-G.; Pripuzova, N.; Majerciak, V.; Kruhlak, M.; Le, S.-Y.; Zheng, Z.-M. Kaposi’s sarcoma-associated herpesvirus ORF57 promotes escape of viral and human interleukin-6 from microRNA-mediated suppression. J. Virol. 2011, 85, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Yamanegi, K.; Allemand, E.; Kruhlak, M.; Krainer, A.R.; Zheng, Z.-M. Kaposi’s sarcoma-associated herpesvirus ORF57 functions as a viral splicing factor and promotes expression of intron-containing viral lytic genes in spliceosome-mediated RNA splicing. J. Virol. 2008, 82, 2792–2801. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Ueda, K.; Guwanan, E.; Sakakibara, S.; Do, E.; Osaki, E.; Yada, K.; Okuno, T.; Yamanishi, K. A posttranscriptional regulator of Kaposi’s sarcoma-associated herpesvirus interacts with RNA-binding protein PCBP1 and controls gene expression through the IRES. Virology 2004, 325, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Boyne, J.R.; Jackson, B.R.; Taylor, A.; Macnab, S.A.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus ORF57 protein interacts with PYM to enhance translation of viral intronless mRNAs. EMBO J. 2010, 29, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 2012, 8, e1002680. [Google Scholar] [CrossRef] [PubMed]
- Ellison, T.J.; Izumiya, Y.; Izumiya, C.; Luciw, P.A.; Kung, H.-J. A comprehensive analysis of recruitment and transactivation potential of K-rta and K-bZIP during reactivation of Kaposi’s sarcoma-associated herpesvirus. Virology 2009, 387, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Lin, S.-F.; Ellison, T.; Chen, L.-Y.; Izumiya, C.; Luciw, P.; Kung, H.-J. Kaposi’s sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: Physical association and promoter-dependent transcriptional repression. J. Virol. 2003, 77, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.; Yamboliev, I.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 K-bZIP modulates latency-associated nuclear protein-mediated suppression of lytic origin-dependent DNA synthesis. J. Virol. 2009, 83, 8492–8501. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ganem, D. Lytic but not latent infection by Kaposi’s sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Nat. Acad. Sci. USA 2003, 100, 8490–8495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; Cusano, T.; Yuan, Y. Identification of the immediate-early transcripts of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5556–5567. [Google Scholar] [PubMed]
- Bu, W.; Palmeri, D.; Krishnan, R.; Marin, R.; Aris, V.M.; Soteropoulos, P.; Lukac, D.M. Identification of direct transcriptional targets of the Kaposi’s sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J. Virol. 2008, 82, 10709–10723. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Garibyan, L.; Kirshner, J.R.; Palmeri, D.; Ganem, D. DNA binding by Kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 2001, 75, 6786–6799. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Carroll, K.D.; Palmeri, D.; Lukac, D.M. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 2007, 81, 5788–5806. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chang, J.; Lynch, S.J.; Lukac, D.M.; Ganem, D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jκ (CSL), the target of the Notch signaling pathway. Genes Dev. 2002, 16, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Oswald, F. The notch signaling pathway: Transcriptional regulation at notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, D.; Carroll, K.D.; Gonzalez-Lopez, O.; Lukac, D.M. Kaposi’s sarcoma-associated herpesvirus Rta tetramers make high-affinity interactions with repetitive DNA elements in the Mta promoter to stimulate DNA binding of RBP-Jk/CSL. J. Virol. 2011, 85, 11901–11915. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, S.E.; Hayward, G.S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 2005, 22, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Guito, J.; Lukac, D. KSHV reactivation and novel implications of protein isomerization on lytic switch control. Viruses 2015, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Cei, S.A.; Papousková, I.; Tarrant, M.; Pari, G.S. Amplification of the Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting at-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 2004, 318, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ciustea, M.; Ricciardi, R.P. Processivity factor of KSHV contains a nuclear localization signal and binding domains for transporting viral DNA polymerase into the nucleus. Virology 2005, 340, 183–191. [Google Scholar] [CrossRef] [PubMed]
- McDowell, M.E.; Purushothaman, P.; Rossetto, C.C.; Pari, G.S.; Verma, S.C. Phosphorylation of Kaposi’s sarcoma-associated herpesvirus processivity factor ORF59 by a viral kinase modulates its ability to associate with RTA and oriLyt. J. Virol. 2013, 87, 8038–8052. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, Y. Essential role of RBP-Jκ in activation of the K8 delayed-early promoter of Kaposi’s sarcoma-associated herpesvirus by ORF50/RTA. Virology 2007, 359, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Scholz, B.A.; Harth-Hertle, M.L.; Malterer, G.; Haas, J.; Ellwart, J.; Schulz, T.F.; Kempkes, B. Abortive lytic reactivation of KSHV in CBF1/CSL deficient human B cell lines. PLoS Pathog. 2013, 9, e1003336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.J.; Li, X.; Brown, H.J.; Sun, R. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 2002, 76, 5000–5013. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Khadim, F.; Spadavecchia, S.; Palmeri, D.; Lukac, D.M. Direct interactions of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 2007, 81, 8451–8467. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.-J.; Boonsiri, J.; Wang, S.-S.; Chen, L.-Y.; Miller, G. Binding of RBP-Jκ (CSL) protein to the promoter of the Kaposi’s sarcoma-associated herpesvirus ORF47 (gL) gene is a critical but not sufficient determinant of transactivation by ORF50 protein. Virology 2010, 398, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Papugani, A.; Coleman, T.; Jones, C.; Zhang, L. The interaction between KSHV RTA and cellular RBP-Jκ and their subsequent DNA binding are not sufficient for activation of RBP-Jκ. Virus Res. 2008, 131, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, Z.; Wood, C. Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 2008, 82, 3590–3603. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, E.S.; Chmura, J.C.; Smith, J.C.; Kalu, N.N.; Hayward, G.S. KSHV RTA abolishes NFκB responsive gene expression during lytic reactivation by targeting vFLIP for degradation via the proteasome. PLoS ONE 2014, 9, e91359. [Google Scholar] [CrossRef] [PubMed]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Yada, K.; Do, E.; Sakakibara, S.; Ohsaki, E.; Ito, E.; Watanabe, S.; Ueda, K. KSHV RTA induces a transcriptional repressor, HEY1 that represses rta promoter. Biochem. Biophys. Res. Commun. 2006, 345, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.H. The theory of the gene. Am. Nat. 1917, 51, 513–544. [Google Scholar] [CrossRef]
- Weng, A.P.; Nam, Y.; Wolfe, M.S.; Pear, W.S.; Griffin, J.D.; Blacklow, S.C.; Aster, J.C. Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol. Cell. Biol. 2003, 23, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhasz, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.-J. Activation of Notch1 signaling is required for β-catenin-mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [PubMed]
- Park, J.T.; Li, M.; Nakayama, K.; Mao, T.-L.; Davidson, B.; Zhang, Z.; Kurman, R.J.; Eberhart, C.G.; Shih, I.-M.; Wang, T.-L. Notch3 gene amplification in ovarian cancer. Cancer Res. 2006, 66, 6312–6318. [Google Scholar] [CrossRef] [PubMed]
- Rebay, I.; Fleming, R.J.; Fehon, R.G.; Cherbas, L.; Cherbas, P.; Artavanis-Tsakonas, S. Specific EGF repeats of Notch mediate interactions with delta and serrate: Implications for Notch as a multifunctional receptor. Cell 1991, 67, 687–699. [Google Scholar] [CrossRef]
- D’Souza, B.; Meloty-Kapella, L.; Weinmaster, G. Canonical and non-canonical notch ligands. Curr. Top. Dev. Biol. 2010, 92, 73–129. [Google Scholar] [PubMed]
- Kopan, R.; Ilagan, M.X.G. The canonical notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Mumm, J.S.; Kopan, R. Notch signaling: From the outside in. Dev. Biol. 2000, 228, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.J.; Yuan, Z.; Kovall, R.A. Structure and function of the CSL-KyoT2 corepressor complex: A negative regulator of notch signaling. Structure 2014, 22, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Kovall, R.A. Crystal structure of the CSL-Notch-mastermind ternary complex bound to DNA. Cell 2006, 124, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.-T.; Cheng, H.-T.; Chang, L.-W.; Ohtsuka, T.; Kageyama, R.; Stormo, G.D.; Kopan, R. Target selectivity of vertebrate notch proteins: Collaboration between discrete domains and CSL-binding site architecture determines activation probability. J. Biol. Chem. 2006, 281, 5106–5119. [Google Scholar] [CrossRef] [PubMed]
- Wharton, K.A.; Johansen, K.M.; Xu, T.; Artavanis-Tsakonas, S. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 1985, 43, 567–581. [Google Scholar] [CrossRef]
- Cordle, J.; RedfieldZ, C.; Stacey, M.; van der Merwe, P.A.; Willis, A.C.; Champion, B.R.; Hambleton, S.; Handford, P.A. Localization of the delta-like-1-binding site in human Notch-1 and its modulation by calcium affinity. J. Biol. Chem. 2008, 283, 11785–11793. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Choi, S.H.; Hu, T.; Tiyanont, K.; Habets, R.; Groot, A.J.; Vooijs, M.; Aster, J.C.; Chopra, R.; Fryer, C.; et al. Insights into autoregulation of Notch3 from structural and functional studies of its negative regulatory region. Structure 2015, 23, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Luca, V.C.; Kim, B.C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger, R.S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [CrossRef] [PubMed]
- Deatherage, C.L.; Lu, Z.; Kim, J.-H.; Sanders, C.R. Notch transmembrane domain: Secondary structure and topology. Biochemistry 2015, 54, 3565–3568. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Taniguchi, Y.; Minoguchi, S.; Sakai, T.; Tun, T.; Furukawa, T.; Honjo, T. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-Jκ/Su(H). Curr. Biol. 1995, 5, 1416–1423. [Google Scholar] [CrossRef]
- Kovall, R.A.; Hendrickson, W.A. Crystal structure of the nuclear effector of Notch signaling, CSL, bound to DNA. EMBO J. 2004, 23, 3441–3451. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.E.; Ilagan, M.X.G.; Kopan, R.; Barrick, D. Thermodynamic analysis of the CSL·x notch interaction: Distribution of binding energy of the Notch RAM region to the CSL β-trefoil domain and the mode of competition with the viral transactivator EBNA2. J. Biol. Chem. 2010, 285, 6681–6692. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, Q.; Li, D.; Ching, K.; Zhang, C.; Zheng, X.; Ozeck, M.; Shi, S.; Li, X.; Wang, H.; et al. PEST domain mutations in Notch receptors comprise an oncogenic driver segment in triple-negative breast cancer sensitive to a γ-secretase inhibitor. Clin. Cancer Res. 2015, 21, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Lyapina, S.; Das, I.; Li, J.; Gurney, M.; Pauley, A.; Chui, I.; Deshaies, R.J.; Kitajewski, J. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol. Cell. Biol. 2001, 21, 7403–7415. [Google Scholar] [CrossRef] [PubMed]
- Bigas, A.; Martin, D.I.K.; Milner, L.A. Notch1 and Notch2 inhibit myeloid differentiation in response to different cytokines. Mol. Cell. Biol. 1998, 18, 2324–2333. [Google Scholar] [CrossRef] [PubMed]
- Foltz, D.R.; Santiago, M.C.; Berechid, B.E.; Nye, J.S. Glycogen synthase kinase-3β modulates notch signaling and stability. Curr. Biol. 2002, 12, 1006–1011. [Google Scholar] [CrossRef]
- Pekkonen, P.; Järviluoma, A.; Zinovkina, N.; Cvrljevic, A.; Prakash, S.; Westermarck, J.; Evan, G.I.; Cesarman, E.; Verschuren, E.W.; Ojala, P.M. KSHV viral cyclin interferes with T-cell development and induces lymphoma through Cdk6 and Notch activation in vivo. Cell Cycle 2014, 13, 3670–3684. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Tulpule, A.; Zhou, Y.; Scehnet, J.S.; Zhang, S.; Lee, J.-S.; Chaudhary, P.M.; Jung, J.; Gill, P.S. KSHV-induced notch components render endothelial and mural cell characteristics and cell survival. Blood 2010, 115, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Verma, S.C.; Murakami, M.; Bajaj, B.; Kaul, R.; Robertson, E.S. Kaposi’s sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein. Proc. Natl. Acad. Sci. USA 2007, 104, 16287–16292. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, Z.; Xia, T.; Li, X.; Liang, D.; Lin, X.; Wen, H.; Lan, K. Latency-associated nuclear antigen of kaposi sarcoma–associated herpesvirus promotes angiogenesis through targeting notch signaling effector Hey1. Cancer Res. 2014, 74, 2026. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, P.; Espigol-Frigole, G.; McCormick, P.J.; Salvucci, O.; Maric, D.; Uldrick, T.S.; Polizzotto, M.N.; Yarchoan, R.; Tosato, G. Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through Notch-dependent signaling. Cancer Res. 2012, 72, 1157. [Google Scholar] [CrossRef] [PubMed]
- Chang, H. Notch signal transduction induces a novel profile of Kaposi’s sarcoma-associated herpesvirus gene expression. J. Microbiol. 2006, 44, 217–225. [Google Scholar] [PubMed]
- Chang, H.; Dittmer, D.P.; Chul, S.-Y.; Hong, Y.; Jung, J.U. Role of notch signal transduction in Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 2005, 79, 14371–14382. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Henkel, T.; Salmon, P.; Robey, E.; Peterson, M.G.; Hayward, S.D. Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol. Cell. Biol. 1996, 16, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.D. Viral interactions with the Notch pathway. Semin. Cancer Biol. 2004, 14, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Gordadze, A.V.; Peng, R.; Tan, J.; Liu, G.; Sutton, R.; Kempkes, B.; Bornkamm, G.W.; Ling, P.D. Notch1IC partially replaces EBNA2 function in B cells immortalized by Epstein-Barr virus. J. Virol. 2001, 75, 5899–5912. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Kobberup, S.; Jørgensen, M.C.; Kalisz, M.; Klein, T.; Kageyama, R.; Gegg, M.; Lickert, H.; Lindner, J.; Magnuson, M.A.; et al. Mind bomb 1 is required for pancreatic β-cell formation. Proc. Natl. Acad. Sci. USA 2012, 109, 7356–7361. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.X.; Xu, A.; Zhang, C.C.; Olson, P.; Chen, L.; Lee, T.K.; Cheung, T.T.; Lo, C.M.; Wang, X.Q. Notch inhibitor PF-03084014 inhibits hepatocellular carcinoma growth and metastasis via suppression of cancer stemness due to reduced activation of Notch1-Stat3. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef] [PubMed]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeCotiis, J.L.; Lukac, D.M. KSHV and the Role of Notch Receptor Dysregulation in Disease Progression. Pathogens 2017, 6, 34. https://doi.org/10.3390/pathogens6030034
DeCotiis JL, Lukac DM. KSHV and the Role of Notch Receptor Dysregulation in Disease Progression. Pathogens. 2017; 6(3):34. https://doi.org/10.3390/pathogens6030034
Chicago/Turabian StyleDeCotiis, Jennifer L., and David M. Lukac. 2017. "KSHV and the Role of Notch Receptor Dysregulation in Disease Progression" Pathogens 6, no. 3: 34. https://doi.org/10.3390/pathogens6030034
APA StyleDeCotiis, J. L., & Lukac, D. M. (2017). KSHV and the Role of Notch Receptor Dysregulation in Disease Progression. Pathogens, 6(3), 34. https://doi.org/10.3390/pathogens6030034