How do PrPSc Prions Spread between Host Species, and within Hosts?


Abstract
:1. Introduction
2. Transmission of PrPSc Prions between Host Species
2.1. Horizontal Transmission
2.1.1. Oral/Ingestion
2.1.2. Prions Can Be Shed into the Environment and Can Remain Infectious
2.1.3. Nasal Cavity Is a Potential Portal for Prion Entry
2.1.4. Lesions to Skin and Mucous Membranes
2.1.5. Accidental Iatrogenic Transmission in Humans
2.2. Vertical Transmission
Milk and Colostrum
3. Transmission of PrPSc Prions within Host Species
3.1. Prions and the Prion Protein
Cellular Sites of PrPC to PrPSc Conversion
3.2. The Accumulation of PrPSc Prions in SLO is Essential for Their Efficient Spread to the CNS
3.3. The Cellular Dissemination of PrPSc Prions within the Host
3.3.1. Prions Cross the Gut Epithelium via M Cells
3.3.2. Conventional Dendritic Cells Aid the Delivery of Prions to SLO
3.3.3. Macrophages Can Phagocytose and Destroy Prions
3.3.4. Cell Free
3.3.5. B Cells Indirectly Support Prion Replication in SLO
3.3.6. Follicular Dendritic Cells Retain and Replicate Prions
3.3.7. FDC Acquire Prions as Complement-Opsonized Complexes
3.3.8. Conventional DC Can Shuttle Prions towards FDC
3.3.9. FDC-Independent Prion Accumulation and Neuroinvasion
3.4. Prion Infections Cause Limited Pathology in SLO
3.5. Orally-Acquired Prions Replicate First in the GALT of the upper Gastrointestinal Tract
3.6. B Cells Aid the Spread of Prions between SLO
3.7. Prion Infection of the CNS Occurs via the Sympathetic and Parasympathetic Nervous Systems
3.8. Effect of Inflammation and Pathogen Co-Infection on Prion Disease Pathogenesis
3.9. Effects of Host Age on Prion Disease Pathogenesis and Susceptibility
4. Opportunities for Prophylactic and Therapeutic Intervention
4.1. PrPSc as a Therapeutic Target
4.1.1. Quinacrine
4.1.2. Pentosan Polysulphate
4.1.3. Tetracyclic Antibiotics
4.2. Prions as Anti-Prions
4.3. Targeting Prion-Induced Neurodegeneration
4.4. Immunization
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bolton, D.C.; McKinley, M.P.; Prusiner, S.B. Identification of a protein that purifies with the scrapie prion. Science 1982, 218, 1309–1311. [Google Scholar] [CrossRef] [PubMed]
- Legname, G.; Baskakov, I.V.; Nguyen, H.-O.B.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Cassard, H.; Torres, J.M.; Lacroux, C.; Douet, J.Y.; Benenstad, S.L.; Lantier, F.; Lugan, S.; Lantier, I.; Costes, P.; Aron, N.; et al. Evidence for zoonotic potential of ovine scrapie prions. Nat. Commun. 2014, 5, 5821. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.E.; Mathews, L.; Coen, P.; Stringer, S.M.; Foster, J.D.; Hunter, N. Population dynamics of scrapie in a flock. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Williams, E.S. Prion disease: Horizontal prion transmission in mule deer. Nature 2003, 425, 35–36. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.J.; Kunkle, R.; Greenlee, M.H.; Nicholson, E.; Richt, J.; Hamir, A.; Waters, W.R.; Greenlee, J. Horizontal transmission of chronic wasting disease. Emerg. Infect. Dis. 2016, 22, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, N.M.; Donnelly, C.A.; Woolhouse, M.E.; Anderson, R.M. The epidemiology of BSE in cattle herds in Great Britain. II. Model construction and analysis of transmission dynamics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 352, 803–838. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.A.; Ferguson, N.M.; Ghani, A.C.; Woolhouse, M.E.; Watt, C.J.; Anderson, R.M. The epidemiology of BSE in cattle herds in Great Britain. I. Epidemiological processes, demography of cattle and approaches to control by culling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 352, 781–801. [Google Scholar] [CrossRef] [PubMed]
- Bruce, M.E.; Will, R.G.; Ironside, J.W.; McConnell, I.; Drummond, D.; Suttie, A.; McCardle, L.; Chree, A.; Hope, J.; Birkett, C.; et al. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997, 389, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.F.; Desbruslais, M.; Joiner, S.; Sidle, K.C.L.; Gowland, I.; Collinge, J. The same prion strain causes vCJD and BSE. Nature 1997, 389, 448–450. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Pearson, G.R.; McConnell, I.; Bruce, M.E.; Wyatt, M.E.; Gruffydd-Jones, T.J. Transmission of feline spongiform encephalopathy to mice. Vet. Rec. 1994, 134, 449. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, J.K.; Wells, G.A.; Wilesmith, J.W.; Cunningham, A.A.; Jackson, S.I. Spongiform encephalopathy in an arabian oryx (Oryx leucoryx) and a greater kudu (Tragelaphus strepsiceros). Vet. Rec. 1990, 127, 418–420. [Google Scholar] [PubMed]
- Jeffrey, M.; Scott, J.R.; Williams, A.; Fraser, H. Ultrastructural features of spongiform encephalopathy transmitted to mice from three species of bovidae. Acta Neuropathol. 1992, 84, 559–569. [Google Scholar] [CrossRef] [PubMed]
- John, T.R.; Schatzl, H.M.; Gilch, S. Early detection of chronic wasting disease prions in urine of pre-symptomatic deer by real-time quaking-induced coversion assay. Prion 2013, 7, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Haley, N.J.; Mathiason, C.K.; Carver, S.; Zabel, M.; Telling, G.C.; Hoover, E.A. Detection of chronic wasting disease prions in salivary, urinary, and intestinal tissues of deer: Potential mechanisms of prion shedding and transmission. J. Virol. 2011, 85, 6309–6318. [Google Scholar] [CrossRef] [PubMed]
- Gregori, L.; Kovacs, G.G.; Alexeeva, I.; Budka, H.; Rohwer, R.G. Excretion of transmissible spongiform encephalopathy infectivity in urine. Emerg. Infect. Dis. 2008, 14, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Murayama, Y.; Yoshioka, M.; Okada, H.; Takata, M.; Yokoyama, T.; Mohri, S. Urinary excretion and blood level of prions in scrapie-infected hamsters. J. Gen. Virol. 2007, 88, 2890–2898. [Google Scholar] [CrossRef] [PubMed]
- Ligios, C.; Cancedda, G.M.; Margalith, I.; Santucciu, C.; Madau, L.; Maestrale, C.; Basagni, M.; Saba, M.; Heikenwalder, M. Intraepithelial and interstitial deposition of pathological prion protein in kidneys of scrapie-affected sheep. PLoS ONE 2007, 2, e859. [Google Scholar] [CrossRef] [PubMed]
- Tamguney, G.; Miller, M.W.; Wolfe, L.L.; Sirochman, T.M.; Glidden, D.V.; Palmer, C.; Lemus, A.; DeArmond, S.J.; Prusiner, S.B. Asymptomatic deer excrete infectious prions in faeces. Nature 2009, 461, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G.; Lessar, P.; Tamguney, G.; Freyman, Y.; Deering, C.; Letessier, F.; DeArmond, S.J.; Prusiner, S.B. Transmission and detection of prions in feces. J. Infect. Dis. 2008, 198, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Tamguney, G.; Richt, J.A.; Hamir, A.N.; Greenlee, J.J.; Miller, M.W.; Wolfe, L.L.; Sirochman, T.M.; Young, A.J.; Gidden, D.V.; Johnson, N.L.; et al. Salivary prions in sheep and deer. Prion 2012, 6, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Gough, K.C.; Baker, C.A.; Rees, H.C.; Terry, L.A.; Spiropoulos, J.; Thorne, L.; Maddison, B.C. The oral secretion of infectious scrapie prions occurs in preclinical sheep with a range of PRNP genotypes. J. Virol. 2012, 86, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, M.; Nonno, R.; Mutinelli, F.; Bigolaro, M.; Di Bari, M.A.; Melchiotti, E.; Marcon, S.; D’Agostino, C.; Vaccari, G.; Conte, M.; et al. PrPSc in salivary glands of scrapie-affected sheep. J. Virol. 2007, 81, 4872–4876. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Shearin, H.; Martinka, S.; Boharski, R.; Lowe, D.; Wilham, J.M.; Caughey, B.; Wiley, J.A. Prion shedding from olfactory neurons into nasal secretions. PLoS Pathog. 2010, 6, e1000837. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Wilham, J.M.; Lowe, D.; Watschke, C.P.; Shearin, H.; Martinka, S.; Caughey, B.; Wiley, J.A. Accelerated shedding of prions following damage to the olfactory epithelium. J. Virol. 2012, 86, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Plummer, I.H.; Wright, S.D.; Johnson, C.J.; Pedersen, J.A.; Samuel, M.D. Temporal patterns of chronic wasting disease prion excretion in three cervid species. J. Gen. Virol. 2017, 98, 1932–1942. [Google Scholar] [CrossRef] [PubMed]
- Seeger, H.; Heikenwalder, M.; Zeller, N.; Kranich, J.; Schwarz, P.; Gaspert, A.; Seifert, B.; Miele, G.; Aguzzi, A. Coincident scrapie infection and nephritis lead to urinary prion excretion. Science 2005, 310, 324–326. [Google Scholar] [CrossRef] [PubMed]
- Maddison, B.C.; Baker, C.A.; Terry, L.A.; Bellworthy, S.J.; Thorne, L.; Rees, H.C.; Gough, K.C. Environmental sources of scrapie prions. J. Virol. 2010, 84, 11560–11562. [Google Scholar] [CrossRef] [PubMed]
- Maddison, B.C.; Owen, J.P.; Bishop, K.; Shaw, G.; Rees, H.C.; Gough, K.C. The interaction of ruminant PrP(Sc) with soils is influenced by prion source and soil type. Environ. Sci. Technol. 2010, 44, 8503–8508. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, A.C.; Kane, S.; Lockwood, K.; Seligman, J.; Michel, B.; Hill, D.; Ortega, A.; Mangalea, M.R.; Telling, G.C.; Miller, M.W.; et al. Clay components in soil dictate environmental stability and bioavailability of cervid prions in mice. Front. Microb. 2016, 7, 1885. [Google Scholar] [CrossRef] [PubMed]
- Pritzkow, S.; Morales, R.; Moda, F.; Khan, U.; Telling, G.C.; Hoover, E.; Soto, C. Grass plants bind, retain, uptake, and transport infectious prions. Cell Rep. 2015, 11, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.J.; Pedersen, J.A.; Chappell, R.J.; McKenzie, D.; Aiken, J.M. Oral transmissibility of prion disease is enhanced by binding soil particles. PLoS Pathog. 2007, 3, e93. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, A.E.; Bartz, J.C. The nasal cavity is a route for prion infection in hamsters. J. Virol. 2007, 81, 4482–4491. [Google Scholar] [CrossRef] [PubMed]
- Hamir, A.N.; Kunkle, R.A.; Richt, J.A.; MIller, J.M.; Greenlee, J.J. Experimental transmission of US scrapie agent by nasal, peritoneal, and conjuctival routes to genetically susceptible sheep. Vet. Pathol. 2008, 45, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Martinka, S.; Kelly, J.; Gonzales, D. Role of the lymphoreticular system in prion neuroinvasion from the oral and nasal mucosa. J. Virol. 2009, 83, 6435–6445. [Google Scholar] [CrossRef] [PubMed]
- Denkers, N.D.; Seelig, D.M.; Telling, G.C.; Hoover, E.A. Aerosol and nasal transmission of chronic wasting disease in cervidized mice. J. Gen. Virol. 2010, 91, 1651–1658. [Google Scholar] [CrossRef] [PubMed]
- Haybaeck, J.; Heikenwalder, M.; Klevenz, B.; Schwarz, P.; Margalith, I.; Bridel, C.; Mertz, K.; Zirdum, E.; Petsch, B.; Fuchs, T.J.; et al. Aerosols transmit prions to immunocompetent and immunodeficient mice. PLoS Pathog. 2011, 7, e1001257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, A.E.; Ayers, J.I.; Bartz, J.C. Specificity, size and frequency of spaces that characterize the mechanism of bulk transepithelial transport of prions in the nasal cavities of hamsters and mice. J. Virol. 2016, 90, 8293–8301. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.M.; McConnell, I.; Fraser, H. Scrapie infection can be established readily through skin scarification in immunocompetent but not immunodeficient mice. J. Gen. Virol. 1996, 77, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Gossner, A.; Hunter, N.; Hopkins, J. Role of lymph-borne cells in the early stages of scrapie agent dissemination from the skin. Vet. Immunol. Immunopathol. 2005, 109, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Bartz, J.C.; Kincaid, A.E.; Bessen, R.A. Rapid prion neuroinvasion following tongue infection. J. Virol. 2003, 77, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Carp, R. Transmission of scrapie by oral route: Effect of gingival scarification. Lancet 1982, 1, 170–171. [Google Scholar] [CrossRef]
- Denkers, N.D.; Telling, G.C.; Hoover, E.A. Minor oral lesions facilitate transmission of chronic wasting disease. J. Virol. 2011, 85, 1396–1399. [Google Scholar] [CrossRef] [PubMed]
- Crowell, J.; Wiley, J.A.; Bessen, R.A. Lesion of the alfactory epithelium accelerates prion neuroinvasion and disease onset when prion replication is restricted to neurons. PLoS ONE 2015, 10, e0119863. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Preece, M.A.; Will, R.G. “Friendly fire” in medicine: Hormones, homografts and Creutzfeldt-Jakob disease. Lancet 1992, 340, 24–27. [Google Scholar] [CrossRef]
- Frontzek, K.; Moos, R.; Schaper, E.; Jann, L.; Herfs, G.; ZImmermann, D.R.; Aguzzi, A.; Budka, H. Iatrogenic and sporadic Creutzfeldt-Jakob disease in 2 sisters without mutation in the prion protein gene. Prion 2015, 9, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Sakurai, M.; Yokoyama, T.; Mohri, S. Disease-associated prion protein in the dental tissue of mice infected with scrapie. J. Comp. Pathol. 2010, 143, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Everington, D.; Smith, A.J.; Ward, H.J.T.; Letters, S.; Will, R.G.; Bagg, J. Dental treatment and risk of variant CJD—A case control study. Br. Dent. J. 2007, 202, E19. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Rohwer, R.G.; Dunstan, B.C.; MacAuley, C.; Gajdusek, D.C.; Drohan, W.N. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion 1998, 38, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.; Cervenakova, L.; McShane, L.M.; Barber, P.; Rubenstein, R.; Drohan, W.N. Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion 1999, 39, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Brown, P. Creutzfeldt-Jakob disease: Blood infectivity and screeing tests. Semin. Hematol. 2001, 38 (Suppl. 9), 2–6. [Google Scholar] [CrossRef]
- Cervenakova, L.; Yakovleva, O.; McKenzie, C.; Kolchinsky, S.; McShane, L.M.; Drohan, W.N.; Brown, P. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 2003, 43, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Holada, K.; Vostal, J.G.; Theisen, P.W.; MacAuley, C.; Gregori, L.; Rohwer, R.G. Scrapie infectivity in hamster blood is not associated with platelets. J. Virol. 2002, 76, 4649–4650. [Google Scholar] [CrossRef] [PubMed]
- Houston, F.; Foster, J.D.; Chong, A.; Hunter, N.; Bostock, C.J. Transmission of BSE by blood transfusion in sheep. Lancet 2000, 356, 999. [Google Scholar] [CrossRef]
- Hunter, N.; Foster, J.; Chong, A.; McCutcheon, S.; Parnham, D.; Eaton, S.; MacKenzie, C.; Houston, F. Transmission of prion diseases by blood transfusion. J. Gen. Virol. 2002, 83, 2897–2905. [Google Scholar] [CrossRef] [PubMed]
- Brown, P. The risk of blood-borne Creutzfeldt-Jakob Disease. In Transmissible Subacute Spongiform Encephalopathies: Prion Diseases; Court, L., Dodet, B., Eds.; Elsevier: Paris, France, 1996; pp. 447–450. [Google Scholar]
- Wietjens, D.P.W.M.; Davanipour, Z.; Hofman, A.; Kondo, K.; Martthews, W.B.; Will, R.G.; van Duijn, C.M. Risk factors for Creutzfeldt-Jakob disease: A reanalysis of case-control studies. Neurology 1996, 47, 1287–1291. [Google Scholar] [CrossRef]
- Van Duijn, C.M.; Delasnerie-Lauprêtre, N.; Masullo, C.; Zerr, I.; De Silva, R.; Wietjens, D.P.W.M.; Brandel, J.-P.; Weber, T.; Bonavita, V.; Zeilder, M.; et al. Case-control study of risk factors of Creutzfeldt-Jakob disease in Europe during 1993–1995. Lancet 1998, 351, 1081–1085. [Google Scholar] [CrossRef]
- Collins, S.; Law, M.G.; Fletcher, A.; Boyd, A.; Kaldor, J.; Masters, C.L. Surgical treatment and risk of sporadic Creutzfeldt-Jakob disease: A case-control study. Lancet 1999, 353, 693–697. [Google Scholar] [CrossRef]
- Unwin, P.J.; Mackenzie, J.M.; Llewelyn, C.A.; Will, R.G.; Hewitt, P.E. Creutzfeldt-Jakob disease and blood transfusion: Updated results of the UK transfusion medicine epidemiology review study. Vox Sang. 2016, 110, 310–316. [Google Scholar]
- Douet, J.Y.; Zafar, S.; Perret-Liaudet, A.; Lacroux, C.; Lugan, S.; Aron, N.; Cassard, H.; Ponto, C.; Corbiere, F.; Torres, J.M.; et al. Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt-Jakob disease. Emerg. Infect. Dis. 2014, 20, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Llewelyn, C.A.; Hewitt, P.E.; Knight, R.S.G.; Amar, K.; Cousens, S.; Mackenzie, J.; Will, R.G. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004, 363, 417–421. [Google Scholar] [CrossRef]
- Peden, A.H.; Head, M.W.; Ritchie, D.L.; Bell, J.E.; Ironside, J.W. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004, 354, 527–529. [Google Scholar] [CrossRef]
- Wroe, S.J.; Pal, S.; Siddique, D.; Hyare, H.; Macfarlane, R.; Joiner, S.; Lineham, J.M.; Brandner, S.; Wadsworth, J.D.F.; Hewitt, P.; et al. Clinical presentation and pre-mortem diagnosis of variant Creutzfeldt-Jakob disease associated with blood transfusion: A case report. Lancet 2006, 368, 2061–2067. [Google Scholar] [CrossRef]
- Health Protection Agency. vCJD Abnormal Protein Found in a Patient with Haemophilia at Post Mortem. Available online: http://webarchive.nationalarchives.gov.uk/20140714094822tf_/http://www.hpa.org.uk/NewsCentre/NationalPressReleases/2009PressReleases/090217vCJDABNORMALPRIONPROTEINFOUNDINAPATIENTWITH/ (accessed on 23 November 2017).
- Hill, A.F.; Zeidler, M.; Ironside, J.; Collinge, J. Diagnosis of new variant Creutzfeldt-Jakob disease by tonsil biopsy. Lancet 1997, 349, 99–100. [Google Scholar] [CrossRef]
- Hill, A.F.; Butterworth, R.J.; Joiner, S.; Jackson, G.; Rossor, M.N.; Thomas, D.J.; Frosh, A.; Tolley, N.; Bell, J.E.; Spencer, M.; et al. Investigation of variant Creutzfeldt-Jakob disease and other prion diseases with tonsil biopsy samples. Lancet 1999, 353, 183–189. [Google Scholar] [CrossRef]
- Gregori, L.; McCombie, N.; Palmer, D.; Birch, P.; Sowemimo-Coker, S.O.; Giulivi, A.; Rohwer, R.G. Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathy. Lancet 2004, 364, 529–531. [Google Scholar] [CrossRef]
- Hoinville, L.J.; Tongue, S.C.; Wilesmith, J.W. Evidence for maternal transmission of scrapie in naturally affected flocks. Prev. Vet. Med. 2010, 93, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Sarradin, P.; Melo, S.; Barc, C.; Lecomte, C.; Andréoletti, O.; Lantier, F.; Dacheux, J.L.; Gatti, J.L. Semen from scrapie-infected rams does not transmit prion infection to transgenic mice. Reproduction 2008, 135, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Nalls, A.V.; McNulty, E.; Powers, J.; Seelig, D.M.; Hoover, C.; Haley, N.J.; Hayes-Klug, J.; Anderson, K.; Stewart, P.; Goldmann, W.; et al. Mother to offspring transmission of chronic wasting disease in reeves’ muntjac deer. PLoS ONE 2013, 8, e71844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selariu, A.; Powers, J.G.; Nalls, A.; Brandhuber, M.; Mayfield, A.; Fullaway, S.; Wyckoff, C.A.; Goldmann, W.; Zabel, M.M.; Wild, M.A.; et al. In utero transmission and tissue distribution of chronic wasting disease-associated prions in free-ranging Rocky Mountain elk. J. Gen. Virol. 2015, 96, 3444–3455. [Google Scholar] [CrossRef] [PubMed]
- Wrathall, A.E.; Brown, K.F.; Sayers, A.R.; WElls, G.A.; Simmons, M.M.; Farrelly, S.S.; Bellerby, P.; Squirrell, J.; Spencer, Y.I.; Wells, M.; et al. Studies of embryo transfer from cattle clinically affected by bovine spongiform encephalopathy (BSE). Vet. Rec. 2002, 150, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Brun, A.; Díaz-San Segundo, F.; Gutiérrez-Adán, A.; Pintado, B.; Ramírez, M.A.; del Riego, L.; Torres, J.M. Vertical transmission of bovine spongiform encephalopathy prions evaluated in a transgenic mouse model. J. Virol. 2005, 79, 8665–8668. [Google Scholar] [CrossRef] [PubMed]
- Wilesmith, J.W.; Wells, G.A.; Ryan, J.B.; Gavier-Widen, D.; Simmons, M.M. A cohort study to examine maternally-associated risk factors for bovine spongiform encephalopathy. Vet. Rec. 1997, 141, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.A.; Ferguson, N.M.; Ghani, A.C.; Wilesmith, J.W.; Anderson, R.M. Analysis of dam-calf pairs of BSE cases: Confirmation of maternal risk enhancement. Proc. R. Soc. Lond. B 1997, 264, 1647–1656. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.; McKelvey, W.; Fraser, H.; Chong, A.; Ross, A.; Parnham, D.; Goldmann, W.; Hunter, N. Experimentally induced bovine spongiform encephalopathy did not transmit via goat embryos. J. Gen. Virol. 1999, 80, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; Goldmann, W.; McKenzie, C.; Smith, A.; Parnham, D.; Hunter, N. Maternal transmission studies of BSE in sheep. J. Gen. Virol. 2004, 85, 3159–3163. [Google Scholar] [CrossRef] [PubMed]
- Foote, W.C.; Clarke, W.; Maciulis, A.; Call, J.W.; Hourrigan, J.; Evans, R.C.; Marshall, M.R.; de Camp, M. Prevention of scrapie transmission in sheep, using embryo transfer. Am. J. Vet. Res. 1993, 54, 1863–1868. [Google Scholar] [PubMed]
- Wang, S.; Foote, W.C.; Sutton, D.L.; Maciulis, A.; Miller, J.M.; Evans, R.C.; Holyoak, G.R.; Call, J.W.; Bunch, T.D.; Taylor, W.D.; et al. Preventing experimental vertical transmission of scrapie by embryo transfer. Theriogenology 2001, 56, 315–327. [Google Scholar] [CrossRef]
- Low, J.C.; Chambers, J.; McKelvey, W.A.; McKendrick, I.J.; Jeffrey, M. Failure to transmit scrapie infection by transferring preimplantation embryos from naturally infected donor sheep. Theriogenology 2009, 72, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cockett, N.E.; MIller, J.M.; Shay, T.L.; Maciulis, A.; Sutton, D.L.; Foote, W.C.; Holyoak, G.R.; Evans, R.C.; Bunc, T.D.; et al. Polymorphic distribution of the ovine prion protein (PrP) gene in scrapie-infected sheep flocks in which embryo transfer was used to circumvent the transmission of scrapie. Theriogenology 2002, 57, 1865–1875. [Google Scholar] [CrossRef]
- Andréoletti, O.; Lacroux, C.; Chabert, A.; Monnereau, L.; Tabouret, G.; Lantier, F.; Berthon, P.; Eychenne, P.; Lafond-Benestad, S.; Elsen, J.M.; et al. PrP(Sc) accumulation in placentas of ewes exposed to natural scrapie: Influence of foetal PrP genotype and effect on ewe-to-lamb transmission. J. Gen. Virol. 2002, 83, 2607–2616. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.A.; Madsen-Bouterse, S.A.; Zhuang, D.; Truscott, T.C.; Dassayake, R.P.; O’Rourke, K.I. The placenta shed from goats with classical scrapie is infectious to goat kids and lambs. J. Gen. Virol. 2015, 96, 2464–2469. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, L.; Dalgleish, M.P.; Martin, S.; Finlayson, J.; Siso, S.; Eaton, S.L.; Goldmann, W.; Witz, J.; Hamilton, S.; Stewart, P.; et al. Factors influencing temporal variation of scrapie incidence within a clsoed Suffolk sheep flock. J. Gen. Virol. 2012, 93, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; Hunter, N.; Williams, A.; Mylne, M.J.; McKelvey, W.A.; Hope, J.; Fraser, H.; Bostock, C. Observations on the transmission of scrapie in experiments using embryo transfer. Vet. Rec. 1996, 138, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; McKelvey, W.A.; Mylne, M.J.; Williams, A.; Hunter, N.; Hope, J.; Fraser, H. Studies on maternal transmission of scarpie in sheep by embryo transfer. Vet. Rec. 1992, 130, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; Goldmann, W.; Hunter, N. Evidence in sheep for pre-natal transmission of scrapie to lambs from infected mothers. PLoS ONE 2013, 8, e79433. [Google Scholar] [CrossRef] [PubMed]
- Garza, M.C.; Fernandez-Borges, N.; Bolea, R.; Badiola, J.J.; Castilla, J.; Monleon, E. Detection of PrPres in genetically susceptible fetuses from sheep with natural scrapie. PLoS ONE 2011, 6, e27525. [Google Scholar] [CrossRef] [PubMed]
- Spiropoulos, J.; Hawkins, S.A.; Simmons, M.M.; Bellworthy, S.J. Evidence of in utero transmission of classical scrapie in sheep. J. Virol. 2014, 88, 4591–4594. [Google Scholar] [CrossRef] [PubMed]
- Alverson, J.; O’Rourke, K.I.; Baszler, T.V. PrPSc accumulation in fetal cotyledons of scrapie-resistant lambs is influenced by fetus location in the uterus. J. Gen. Virol. 2006, 87, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Peters, J.; Stellitano, L.; Winstone, A.M.; Verity, C.; Will, R.G. Is there evidence of vertical transmission of variant Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 2011, 82, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Amyx, H.L.; Gibbs, C.J., Jr.; Gadjusek, D.C.; Greer, W.E. Absence of vertical transmission of subacute spongiform viral encephalopathies in experimental primates. Proc. Soc. Exp. Biol. Med. 1981, 166, 469–471. [Google Scholar] [CrossRef]
- Xiao, X.; Miravalle, L.; Yuan, J.; McGheehan, J.; Dong, Z.; Wyza, R.; MacLennan, G.T.; Golichowski, A.M.; Kneale, G.; King, N.; et al. Failure to detect the presence of prions in the uterine and gestational tissues from a Gravida with Creutzfeldt-Jakob disease. Am. J. Pathol. 2009, 174, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Garza, M.C.; Monzon, M.; Marin, B.; Badiola, J.J.; Monleon, E. Distribution of peripheral PrP(Sc) in sheep with naturally acquired scrapie. PLoS ONE 2014, 9, e97768. [Google Scholar] [CrossRef] [PubMed]
- Maestrale, C.; Di Guardo, G.; Cancedda, M.G.; Marruchella, G.; Masia, M.; Sechi, S.; Macciocu, S.; Santucciu, C.; Petruzzi, M.; Ligios, C. A lympho-follicular microenvironment is required for pathological prion protein deposition in chronically inflamed tissues from scrapie-affected sheep. PLoS ONE 2013, 8, e62830. [Google Scholar] [CrossRef] [PubMed]
- Ligios, C.; Cancedda, M.G.; Carta, A.; Santucciu, C.; Maestrale, C.; Demontis, F.; Saba, M.; Patta, C.; DeMartini, J.C.; Aguzzi, A.; et al. Sheep with scrapie and mastitis transmit infectious prions through the milk. J. Virol. 2011, 85, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Salazar, E.; Monleon, E.; Bolea, R.; Acin, C.; Perez, M.; Alvarez, N.; Leginagoikoa, I.; Juste, R.; Minguijon, E.; Reina, R.; et al. Detection of PrPSc in lung and mammary gland is favoured by the presence of Visna/maedi virus lesions in naturally coinfected sheep. Vet. Res. 2010, 41, 58. [Google Scholar] [CrossRef] [PubMed]
- Konold, T.; Moore, S.J.; Bellworthy, S.J.; Terry, L.A.; Thorne, L.; Ramsay, A.; Salguerro, F.J.; Simmons, M.M.; Simmons, H.A. Evidence of effective scrapie transmission via colostrum and milk in sheep. BMC Vet. Res. 2013, 9, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacroux, C.; Simon, S.; Benenstad, S.L.; Maillet, S.; Mathey, J.; Lugan, S.; Corbiere, F.; Cassard, H.; Costes, P.; Bergonier, D.; et al. Prions in milk from ewes incubating natural scrapie. PLoS Pathog. 2008, 4, e1000238. [Google Scholar] [CrossRef] [PubMed]
- Everest, S.J.; Thorne, L.T.; Hawthorne, J.A.; Jenkins, R.; Hammersley, C.; Ramsay, A.M.; Hawkins, S.A.; Venables, L.; Flynn, L.; Sayers, R.; et al. No abnormal prion protein detected in the milk of cattle infected with the bovine spongiform encephalopathy agent. J. Gen. Virol. 2006, 87, 2433–2441. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Clarke, A.R.; McBride, P.A.; McConnell, I.; Hope, J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994, 3, 331–340. [Google Scholar] [PubMed]
- Prusiner, S.B.; Groth, D.; Serban, A.; Koehler, R.; Foster, D.; Torchia, M.; Burton, D.; Yang, S.-L.; DeArmond, S.J. Ablation of the Prion Protein (PrP) Gene in Mice Prevents Scrapie and Facilitates Production of Anti-PrP Antibodies. Proc. Natl. Acad. Sci. USA 1993, 90, 10608–10612. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, X.; Yuan, C.G.; Ma, J. Generating a prion with bacterially expressed recombinant prion protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol gylcolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Pan, K.-M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion protein. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wuthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP. FEBS Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein PrP 27–30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef] [PubMed]
- Deleault, N.R.; Lucassen, R.W.; Supattapone, S. RNA molecules stimulate prion protein conversion. Nature 2003, 425, 717–720. [Google Scholar] [CrossRef] [PubMed]
- Ma, J. The role of cofactors in prion propagation and infectivity. PLoS Pathog. 2012, 8, e1002589. [Google Scholar] [CrossRef] [PubMed]
- Küffer, A.; Lakkraju, A.K.; Mogha, A.; Petersen, S.C.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rulicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered circadian activity rythyms and sleep in mice devoid of prion protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Whittington, M.A.; Sidle, K.C.; Smith, C.J.; Palmer, M.S.; Clarke, A.R.; Jefferys, J.G.R. Prion protein is necessary for normal synaptic function. Nature 1994, 370, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Coitinho, A.S.; Roesler, R.; Martins, V.R.; Brentani, R.R.; Izquierdo, I. Cellular prion protein ablation impairs behaviour as a function of age. Neuroreport 2003, 14, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Walz, R.; Amaral, O.B.; Rockenbach, I.C.; Roesler, R.; Izquierdo, I.; Cavalheiro, E.A.; Martins, V.R.; Brentani, R.R. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilesia 1999, 40, 1679–1682. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Spielhaupter, C.; Schatzl, H.M. PrPC directly interacts with proteins involved in signalling pathways. J. Biol. Chem. 2001, 276, 44604–44612. [Google Scholar] [CrossRef] [PubMed]
- Bounar, Y.; Zhang, Y.; Goodyer, C.G.; LeBlanc, A. Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 2001, 276, 39145–39149. [Google Scholar] [CrossRef] [PubMed]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Nolting, S.; Hamann, G.F.; Kretzschmar, H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Young, J.; McConnell, I.; Bruce, M.E. Follicular dendritic cell dedifferentiation by treatment with an inhibitor of the lymphotoxin pathway dramatically reduces scrapie susceptibility. J. Virol. 2003, 77, 6845–6854. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Huber, G.; Macpherson, A.J.S.; Heppner, F.L.; Glatzel, M.; Eugster, H.-P.; Wagner, N.; Aguzzi, A. Oral prion infection requires normal numbers of Peyer’s patches but not of enteric lymphocytes. Am. J. Pathol. 2003, 162, 1103–1111. [Google Scholar] [CrossRef]
- Glaysher, B.R.; Mabbott, N.A. Role of the GALT in scrapie agent neuroinvasion from the intestine. J. Immunol. 2007, 178, 3757–3766. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Williams, E.S.; Miller, M.W.; Spraker, T.R.; O’Rourke, K.I.; Hoover, E.A. Oral transmission and early lymphoid tropism of chronic wasting disease PrPres in mule deer fawns (Odocoileus hemionus). J. Gen. Virol. 1999, 80, 2757–2764. [Google Scholar] [CrossRef] [PubMed]
- Andreoletti, O.; Berthon, P.; Marc, D.; Sarradin, P.; Grosclaude, J.; van Keulen, L.; Schelcher, F.; Elsen, J.-M.; Lantier, F. Early accumulation of PrPSc in gut-associated lymphoid and nervous tissues of susceptible sheep from a Romanov flock with natural scrapie. J. Gen. Virol. 2000, 81, 3115–3126. [Google Scholar] [CrossRef] [PubMed]
- Heggebø, R.; Press, C.M.; Gunnes, G.; Lie, K.I.; Tranulis, M.A.; Ulvund, M.; Groschup, M.H.; Landsverk, T. Distribution of prion protein in the ileal Peyer’s patch of scrapie-free lambs and lambs naturally and experimentally exposed to the scrapie agent. J. Gen. Virol. 2000, 81, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- van Keulen, L.J.M.; Schreuder, B.E.G.; Vromans, M.E.W.; Langeveld, J.P.M.; Smits, M.A. Scrapie-associated prion protein in the gastro-intestinal tract of sheep with scrapie. J. Comp. Pathol. 1999, 121, 55–63. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.; Brown, K.L.; Mabbott, N.A. Ablation of the cellular prion protein, PrPC, specifcally on follicular dendritic cells has no effect on their maturation or function. Immunology 2013, 138, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Cashman, N.R.; Loertscher, R.; Nalbantoglu, J.; Shaw, I.; Kascsak, R.J.; Bolton, D.C.; Bendheim, P.E. Cellular isoform of the scrapie agent protein participates in lymphocyte activation. Cell 1990, 61, 185–192. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Brown, K.L.; Manson, J.; Bruce, M.E. T lymphocyte activation and the cellular form of the prion protein. Immunology 1997, 92, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Martínez del Hoyo, G.; López-Bravo, M.; Metharom, P.; Ardavín, C.; Aucouturier, P. Prion protein expression by mouse dendritic cells is restricted to the nonplasmacytoid subsets and correlates with the maturation state. J. Immunol. 2006, 177, 6137–6142. [Google Scholar] [CrossRef] [PubMed]
- Jouvin-Marche, E.; Attuli-Audenis, V.; Aude-Garcia, C.; Rachidi, W.; Zabel, M.; Podevin-Dimster, V.; Siret, C.; Huber, C.; Martinic, M.; Riondel, J.; et al. Overexpression of cellular prion protein induces and antioxidant environment altering T cell development in the thymus. J. Immunol. 2006, 176, 3490–3497. [Google Scholar] [CrossRef] [PubMed]
- Ballerini, C.; Gourdain, P.; Bachy, V.; Blanchard, N.; Levavasseur, E.; Gregoire, S.; Fontes, P.; Aucouturier, P.; Hivroz, C.; Carnaud, C. Funcitonal implication of cellular prion protein in antigen-driven interactions between T cells and dendritic cells. J. Immunol. 2006, 176, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Nakato, G.; Hase, K.; Suzuki, M.; Kimura, M.; Ato, M.; Hanazato, M.; Tobiume, M.; Horiuchi, M.; Atarashi, R.; Nishida, N.; et al. Cutting edge: Brucella abortus exploits a cellular prion protein on intestinal M cells as an invasive receptor. J. Immunol. 2012, 189, 1540–1544. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, C.J.G.; Chiarini, L.B.; da Silva, J.P.; e Silva, P.M.R.; Martins, M.A.; Linden, R. The cellular prion protein modulates phagocytosis and inflammatory response. J. Leukoc. Biol. 2005, 77, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T.; Diringer, H.; Ludwig, H. Absence of autoantibodies against neurofilament proteins in the sera of scarpie infected mice. Tohoku J. Exp. Med. 1985, 4, 483–484. [Google Scholar] [CrossRef]
- Clarke, M.C.; Haig, D.A. Attempts to demonstrate neutralising antibodies in the sera of scrapie-infected animals. Vet. Rec. 1966, 19, 647–649. [Google Scholar] [CrossRef]
- Sassa, Y.; Kataoka, N.; Inoshima, Y.; Ishiguro, N. Anti-PrP antibodies detected at terminal stage of prion-affected mouse. Cell. Immunol. 2010, 263, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A. Trafficking, turnover and membrane topology of PrP. Br. Med. Bull. 2003, 66, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 188–199. [Google Scholar]
- Arnold, J.E.; Tipler, C.; Laszlo, L.; Hope, J.; Landon, M.; Mayer, R.J. The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J. Pathol. 1995, 176, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar] [CrossRef] [PubMed]
- Godsave, S.F.; Wille, H.; Kujala, P.; Latawiec, D.; DeArmond, S.J.; Serban, A.; Prusiner, S.B.; Peters, P.J. Cryo-immunogold EM for prions: Towards identification of a conversion site. J. Neurosci. 2008, 28, 12489–12499. [Google Scholar] [CrossRef] [PubMed]
- Beranger, F.; Mange, A.; Goud, B.; Lehmann, S. Stimulation of PrPC retrograde transport toward the endoplasmic reticulum increases accumulation of PrPSc in prion -infected cells. J. Biol. Chem. 2002, 277, 38972–38977. [Google Scholar] [CrossRef] [PubMed]
- Kujala, P.; Raymond, C.R.; Romeijn, M.; Godsave, S.F.; van Kasteren, S.I.; Wille, H.; Prusiner, S.B.; Mabbott, N.A.; Peters, P.J. Prion uptake in the gut: Identification of the first uptake and replication sites. PLoS Pathog. 2011, 7, e1002449. [Google Scholar] [CrossRef] [PubMed]
- McGovern, G.; Mabbott, N.A.; Jeffrey, M. Scrapie affects the maturation cycle and immune complex trapping by follicular dendritic cells in mice. PLoS ONE 2009, 4, e8186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goold, R.; Rabbanian, S.; Sutton, L.; Andre, R.; Arora, P.; Moonga, J.; Clarke, A.R.; Schiavo, G.; Jat, P.; Collinge, J.; et al. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat. Commun 2011, 2, 281. [Google Scholar] [CrossRef] [PubMed]
- Goold, R.; McKinnon, C.; Rabbanian, S.; Collinge, J.; Schiavo, G.; Tabrizi, S.J. Alternative sites of newly formed PrPSc upon prion conversion on the plasma membrane. J. Cell Sci. 2013, 126, 3552–3562. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Dickinson, A.G. Pathogenesis of scrapie in the mouse: The role of the spleen. Nature 1970, 226, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Dickinson, A.G. Studies on the lymphoreticular system in the pathogenesis of scrapie: The role of spleen and thymus. J. Comp. Pathol. 1978, 88, 563–573. [Google Scholar] [CrossRef]
- Horiuchi, M.; Furuoka, H.; Kitamura, N.; Shinagawa, M. Alymphoplasia mice are resistant to prion infection via oral route. Jpn. J. Vet. Res. 2006, 53, 149–157. [Google Scholar] [PubMed]
- Donaldson, D.S.; Else, K.J.; Mabbott, N.A. The gut-associated lymphoid tissues in the small intestine, not the large intestine, play a major role in oral prion disease pathogenesis. J. Virol. 2015, 15, 9532–9547. [Google Scholar] [CrossRef] [PubMed]
- Glaysher, B.R.; Mabbott, N.A. Role of the draining lymph node in scrapie agent transmission from the skin. Immunol. Lett. 2007, 109, 64–71. [Google Scholar] [CrossRef] [PubMed]
- González, L.; Dagleish, M.P.; Bellworthy, S.J.; Sisó, S.; Stack, M.J.; Chaplin, M.J.; Davis, L.A.; Hawkins, S.A.C.; Hughes, J.; Jeffrey, M. Postmortem diagnosis of preclinical and clinical scrapie in sheep by the detection of disease-associated PrP in their rectal mucosa. Vet. Rec. 2006, 158, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Espenes, A.; Press, C.M.; Landsverk, T.; Tranulis, M.A.; Aleksandersen, M.; Gunnes, G.; Benestad, S.L.; Fuglestveit, R.; Ulvund, M.J. Detection of PrPSc in rectal biopsy and necroscopy samples from sheep with experimental scrapie. J. Comp. Pathol. 2006, 134, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; Gidlewski, T.L.; Balachandran, A.; VerCauteren, K.C.; Creekmore, L.; Munger, R.D. Detection of PrPCWD in postmortem rectal lymphoid tissues in Rocky Mountain elk (Cervus elaphus nelsoni) infected with chronic wasting disease. J. Vet. Diagn. Investig. 2006, 18, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, L.L.; Spraker, T.R.; González, L.; Dagleish, M.P.; Sirochman, T.M.; Brown, J.C.; Jeffrey, M.; Miller, M.W. PrPCWD in rectal lymphoid tissue of deer (Odocoileus spp.). J. Gen. Virol. 2007, 88, 2078–2082. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.M.; Thomsen, B.V.; Marshall, K.L.; Hall, S.M.; Wagner, B.A.; Salman, M.D.; Norden, D.K.; Gaiser, C.; Sutton, D.L. Evaluation of immunohistochemical detection of prion protein in rectoanal mucosa-associated lymphoid tissue for diagnosis of scrapie in sheep. Am. J. Vet. Res. 2009, 70, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.; Fathers, E.; Edwards, P.; Ironside, J.; Zajicek, J. Prion immunoreactivity in appendix before clinical onset of variant Creutzfeldt-Jakob disease. Lancet 1998, 352, 703–704. [Google Scholar] [CrossRef]
- Hilton, D.A.; Ghani, A.C.; Conyers, L.; Edwards, P.; McCardle, L.; Ritchie, D.; Penney, M.; Hegazy, D.; Ironside, J.W. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J. Pathol. 2004, 203, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.A.; Ghani, A.C.; Conyers, L.; Edwards, P.; McCardle, L.; Penney, M.; Ritchie, D.; Ironside, J.W. Accumulation of prion protein in tonsil and appendix: Review of tissue samples. Br. Med. J. 2002, 325, 633–634. [Google Scholar] [CrossRef] [Green Version]
- Gill, O.N.; Spencer, Y.; Richard-Loendt, A.; Kelly, C.; Dabaghian, R.; Boyes, L.; Lineham, J.; Simmons, M.; Webb, P.; Bellerby, P.; et al. Prevelent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: Large scale survey. Br. Med. J. 2013, 347, f5675. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Donaldson, D.S.; Ohno, H.; Williams, I.R.; Mahajan, A. Microfold (M) cells: Important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013, 6, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hase, K.; Kawano, K.; Nochi, T.; Pontes, G.S.; Fukuda, S.; Ebisawa, M.; Kadokura, K.; Tobe, T.; Fujimura, Y.; Kawano, S.; et al. Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune responses. Nature 2009, 462, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Rios, D.; Wood, M.B.; Li, J.; Chassaing, B.; Gewirtz, A.T.; Williams, I.R. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol. 2016, 9, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Tahoun, A.; Mahajan, S.; Paxton, E.; Malterer, G.; Donaldson, D.S.; Wang, D.; Tan, A.; Gillespie, T.L.; O’Shea, M.; Rose, A.; et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe 2012, 12, 645–666. [Google Scholar] [CrossRef] [PubMed]
- Westphal, S.; Lugering, A.; von Wedel, J.; von Eiff, C.; Maaser, C.; Spahn, T.; Heusipp, G.; Schmidt, M.A.; Herbst, H.; Williams, I.R.; et al. Resistance of chemokine receptor 6-deficient mice to Yersinia enterocolitica infection: Evidence on defective M-cell formation in vivo. Am. J. Pathol. 2008, 172, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, A.O.; Gonzalez-Hernandez, M.B.; Turula, H.; Yu, C.; Elftman, M.D.; Wobus, C.E. Oral norovirus infection is blocked in mice lacking Peyer’s patches and mature M cells. J. Virol. 2015, 90, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hernandez, M.B.; Liu, T.; Payne, H.C.; Stencel-Baerenwald, J.E.; Ikizler, M.; Yagita, H.; Dermody, T.S.; Williams, I.R.; Wobus, C.E. Efficient norovirus and reovirus replication in the mouse intestine requires microfold (M) cells. J. Virol. 2014, 88, 6934–6943. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Christ, A.D.; Klein, M.A.; Prinz, M.; Fried, M.; Kraehenbuhl, J.-P.; Aguzzi, A. Transepithelial prion transport by M cells. Nat. Med. 2001, 7, 976–977. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Kanaya, T.; Takakura, I.; Tanaka, S.; Hondo, T.; Watanabe, H.; Rose, M.T.; Kitazawa, H.; Yamaguchi, T.; Katamine, S.; et al. Transcytosis of murine-adapted bovine spongiform encephalopathy agents in an in vitro bovine M cell model. J. Virol. 2010, 84, 12285–12291. [Google Scholar] [CrossRef] [PubMed]
- Takakura, I.; Miyazawa, K.; Kanaya, T.; Itani, W.; Watanabe, K.; Ohwada, S.; Watanabe, H.; Hondo, T.; Rose, M.T.; Mori, T.; et al. Orally administered prion protein is incorporated by M cells and spreads to lymphoid tissues with macrophages in prion protein knockout mice. Am. J. Pathol. 2011, 179, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Kobayashi, A.; Ohno, H.; Yagita, H.; Williams, I.R.; Mabbott, N.A. M cell depletion blocks oral prion disease pathogenesis. Mucosal Immunol. 2012, 5, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, D.S.; Sehgal, A.; Rios, D.; Williams, I.R.; Mabbott, N.A. Increased abundance of M cells in the gut epithelium dramatically enhances oral prion disease susceptibility. PLoS Pathog. 2016, 12, e1006075. [Google Scholar] [CrossRef] [PubMed]
- Beekes, M.; McBride, P.A. Early accumulation of pathological PrP in the enteric nervous system and gut-associated lymphoid tissue of hamsters orally infected with scrapie. Neurosci. Lett. 2000, 278, 181–184. [Google Scholar] [CrossRef]
- Bennet, K.M.; Parnell, E.A.; Sanscartier, C.; Parks, S.; Chen, G.; Nair, M.G.; Lo, D.D. Induction of colonic M cells during intestinal inflammation. Am. J. Pathol. 2016, 186, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Terahara, K.; Yoshida, M.; Igarashi, O.; Nochi, T.; Soares Pontes, G.; Hase, K.; Ohno, H.; Kurokawa, S.; Mejima, M.; Takayama, N.; et al. Comprehensive gene expression profiling of Peyer’s patch M cells, villous M-like cells, and intestinal epithelial cells. J. Immunol. 2008, 180, 7840–7846. [Google Scholar] [CrossRef] [PubMed]
- Knoop, K.A.; Kumar, N.; Butler, B.R.; Sakthivel, S.K.; Taylor, R.T.; Nochi, T.; Akiba, H.; Yagita, H.; Kiyono, H.; Williams, I.R. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 2009, 183, 5738–5747. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.; Sawa, S.; Nitta, T.; Tsutsumi, M.; Okamura, T.; Penninger, J.M.; Nakashima, T.; Takayanagi, H. Identification of subepithelial mesenchymal cells that induce IgA and diversify gut microbiota. Nat. Immunol. 2017, 18, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Foster, N.; Macpherson, G.G. Murine cecal patch M cells transport infectious prions in vivo. J. Infect. Dis. 2010, 202, 1916–1919. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; González, L.; Espenes, A.; Press, C.M.; Martin, S.; Chaplin, M.; Davis, L.; Landsverk, T.; MacAldowie, C.; Eaton, S.; et al. Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J. Pathol. 2006, 209, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.S.; Basu, S.; Gu, Y.; Luo, X.; Zou, W.-Q.; Mishra, R.; Li, R.; Chen, S.G.; Gambetti, P.; Fujioka, H.; et al. Protease-resistant human prion protein and ferritin are cotransported across Caco-2 epithelial cells: Implications for species barrier in prion uptake from the intestine. J. Neurosci. 2004, 24, 11280–11290. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, M.; Kimura, S.; Takashi-Iwanaga, H.; Hisamoto, M.; Iwanaga, T.; Iida, J. RANKL regulates differentiation of microfold cells in mouse nasopharynx-associated lymphoid tissue (NALT). Cell Tissue Res. 2016, 364, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.R.; Franco, L.H.; Zacharia, V.M.; Khan, H.S.; Stamm, C.E.; You, W.; Marciano, D.K.; Yagita, H.; Levine, B.; Shiloh, M.U. Microfold cells actively translocate Mycobacterium tuberculosis to initiate infection. Cell Rep. 2016, 16, 1253–1258. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, A.E.; Hudson, K.F.; Richey, M.W.; Bartz, J.C. Rapid transepithelial transport of prions following inhalation. J. Virol. 2012, 86, 12731–12740. [Google Scholar] [CrossRef] [PubMed]
- Elder, A.M.; Henderson, D.M.; Nalls, A.V.; Hoover, E.A.; Kincaid, A.E.; Bartz, J.C.; Mathiason, C.K. Immediate and ongoing detection of prions in the blood of hamsters and deer following oral, nasal and blood inoculations. J. Virol. 2015, 89, 7421–7424. [Google Scholar] [CrossRef] [PubMed]
- Urayama, A.; Concha-Marambio, L.; Khan, U.; Bravo-Alegria, J.; Kharat, V.; Soto, C. Prions efficiently cross the intestinal barrier after oral administration: Study of the bioavailability, and cellular tissue distribution in vivo. Sci. Rep. 2016, 6, 32338. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.R.; Aucouturier, P.; Mabbott, N.A. In vivo depletion of CD11c+ cells impairs scrapie agent neuroinvasion from the intestine. J. Immunol. 2007, 179, 7758–7766. [Google Scholar] [CrossRef] [PubMed]
- Nakato, G.; Fukuda, S.; Hase, K.; Goitsuka, R.; Cooper, M.D.; Ohno, H. New approach for M-cell-specific molecules by screening comprehensive transcriptome analysis. DNA Res. 2009, 16, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Sakhon, O.S.; Ross, B.; Gusti, V.; Pham, A.J.; Vu, K.; Lo, D.D. M cell-derived vesicles suggest a unique pathway for trans-epithelial antigen delivery. Tissue Barriers 2015, 3, e1004975. [Google Scholar] [CrossRef] [PubMed]
- Delamarre, L.; Pack, M.; Chang, H.; Mellman, I.; Trombetta, E.S. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 2005, 307, 1630–1634. [Google Scholar] [CrossRef] [PubMed]
- Bergtold, A.; Desai, D.D.; Gavhane, A.; Clynes, R. Cell surface recycling of internalized antigen permits dendritic cell priming to B cells. Immunity 2005, 23, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; MacPherson, G.G. Antigen acquisition by dendritic cells: Intestinal dendritic cells acquire antigen administered orally and can prime naive T cells in vivo. J. Exp. Med. 1993, 177, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Cerovic, V.; Houston, S.A.; Westlund, J.; Utriainen, L.; Davison, E.S.; Scott, C.L.; Bain, C.; Joeris, T.; Agace, W.W.; Kroczek, R.A.; et al. Lymph-borne CD8a+ dendritic cells are uniquely able to cross-prime CD8+ T cells with antigen acquired from intestinal epithelial cells. Mucosal Immunol. 2014, 8, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-J.L.; Grouard-Vogel, G.; Sun, W.; Mascola, J.R.; Brachtel, E.; Putvatana, R.; Louder, M.K.; Filgueira, L.; Marovich, M.A.; Wong, H.K.; et al. Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 2000, 6, 816–820. [Google Scholar] [PubMed]
- Ho, L.-J.; Wang, J.-J.; Shaio, M.-F.; Kao, C.-L.; Chang, D.-M.; Han, S.-W.; Lai, J.-H. Infection of human dendritic cells by dengue virus causes cell maturation and cytokine production. J. Immunol. 2001, 166, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Granelli-Piperno, A.; Pope, M.; Trumpfheller, C.; Ignatius, R.; Arrode, G.; Racz, P.; Tenner-Racz, K. The interaction of immunodeficiency viruses with dendritic cells. Curr. Top. Microbiol. Immunol. 2003, 276, 1–30. [Google Scholar] [PubMed]
- Ho, A.W.; Prabhu, N.; Betts, R.J.; Ge, M.Q.; Dai, X.; Hutchinson, P.E.; Lew, F.C.; Wong, K.L.; Hanson, B.J.; Macary, P.A.; et al. Lung CD103+ dendritic cells efficiently transport infuenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J. Immunol. 2011, 187, 6011–6021. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.; Pombo, A.; Jenkins, C.; MacPherson, G.G. Dendritic cells interact directly with Naive B lymphocytes to transfer antigen and initiate class switching in a primary T-dependent response. J. Immunol. 1998, 161, 1313–1319. [Google Scholar] [PubMed]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science 2004, 303, 1662–1665. [Google Scholar] [CrossRef] [PubMed]
- Saeki, H.; Wu, M.; Olasz, E.; Hwang, S.T. A migratory population of skin-derived dendritic cells expresses CXCR5, responds to B lymphocyte chemoattractant in vitro, and co-localizes to B cell zones in lymph nodes in vivo. Eur. J. Immunol. 2000, 30, 2808–2814. [Google Scholar] [CrossRef]
- Berney, C.; Herren, S.; Power, C.A.; Gordon, S.; Martinez-Pomares, L.; Kosco-Vilbois, M.H. A member of the dendritic cell family that enters B cell follicles and stimulates primary antibody responses identified by a mannose receptor fusion protein. J. Exp. Med. 1999, 190, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Leon, B.; Ballesteros-Tato, A.; Browning, J.L.; Dunn, R.; Randall, T.D.; Lund, F.E. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat. Immunol. 2012, 13, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.-P.; Farquhar, C.F.; Mabbott, N.A.; Bruce, M.E.; MacPherson, G.G. Migrating intestinal dendritic cells transport PrPSc from the gut. J. Gen. Virol. 2002, 83, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Cordier-Dirikoc, S.; Chabry, J. Temporary depletion of CD11c+ dendritic cells delays lymphoinvasion after intraperitoneal scrapie infection. J. Virol. 2008, 82, 8933–8936. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Kerksiek, K.M.; Brocker, T.; Kretzschmar, H. Role of the CD8+ dendritic cell subset in transmission of prions. J. Virol. 2007, 81, 4877–4880. [Google Scholar] [CrossRef] [PubMed]
- Wathne, G.J.; Kissenpfennig, A.; Malissen, B.; Zurzolo, C.; Mabbott, N.A. Determining the role of mononuclear phagocytes in prion neuroinvasion from the skin. J. Leukoc. Biol. 2012, 91, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Kelsalla, B.A. Localization of distinct Peyer’s patch dendritic cell subsets and their recruitment by chemokines macrophage inflammatory protein (MIP)-3, MIP-3ß, and secondary lymphoid organ chemokine. J. Exp. Med. 2000, 191, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Castro-Seoane, R.; Hummerich, H.; Sweeting, T.; Tattum, M.H.; Lineham, J.M.; Fernandez de Marco, M.; Brandner, S.; Collinge, J.; Klöhn, P.C. Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog. 2012, 8, e1002538. [Google Scholar] [CrossRef] [PubMed]
- Yrlid, U.; Cerovic, V.; Milling, S.; Jenkins, C.D.; Zhang, J.; Crocker, P.R.; Klavinskis, L.S.; MacPherson, G.G. Plasmacytoid dendritic cells do not migrate in intestinal or hepatic lymph. J. Immunol. 2006, 177, 6115–6121. [Google Scholar] [CrossRef] [PubMed]
- Burthem, J.; Urban, B.; Pain, A.; Roberts, D.J. The normal cellular prion protein is strongly expressed by myeloid dendritic cells. Blood 2001, 98, 3733–3738. [Google Scholar] [CrossRef] [PubMed]
- Cordier-Dirikoc, S.; Zsürger, N.; Cazareth, J.; Ménard, B.; Chabry, J. Expression profiles of prion and doppel proteins and of their receptors in mo`use splenocytes. Eur. J. Immunol. 2008, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Kanaya, T.; Tanaka, S.; Takakura, I.; Watanabe, K.; Ohwada, S.; Kitazawa, H.; Rose, M.T.; Sakaguchi, S.; Katamine, S.; et al. Immunohistochemical characterization of cell types expressing the cellular prion protein in the small intestine of cattle and mice. Histochem. Cell Biol. 2007, 127, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Stewart, K.; Ritchie, D.; Mabbott, N.A.; Williams, A.; Fraser, H.; Morrison, W.I.; Bruce, M.E. Scrapie replication in lymphoid tissues depends on PrP-expressing follicular dendritic cells. Nat. Med. 1999, 5, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.A.; Frigg, R.; Raeber, A.J.; Flechsig, E.; Hegyi, I.; Zinkernagel, R.M.; Weissmann, C.; Aguzzi, A. PrP expression in B lymphocytes is not required for prion neuroinvasion. Nat. Med. 1998, 4, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Mohan, J.; Brown, K.L.; Farquhar, C.F.; Bruce, M.E.; Mabbott, N.A. Scrapie transmission following exposure through the skin is dependent on follicular dendritic cells in lymphoid tissues. J. Dermatol. Sci. 2004, 35, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Lemaire-Vielle, C.; Naquet, P.; Cesbron-Delauw, M.-F.; Gagnon, J.; Cesbron, J.-Y. Prion replication in the hematopoietic compartment is not required for neuroinvasion in scrapie mouse model. PLoS ONE 2010, 5, e13166. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, L.; Brown, K.L.; Bradford, B.M.; Hopkins, J.; Bailey, M.; Rajewsky, K.; Manson, J.C.; Mabbott, N.A. Follicular dendritic cell-specific prion protein (PrPC) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathog. 2011, 7, e1002402. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Meyerett-Reid, C.; Johnson, T.; Ferguson, A.; Wycoff, C.; Pulford, B.; Bender, H.; Avery, A.; Telling, G.; Dow, S.; et al. Incunabular immunological events in prion trafficking. Sci. Rep. 2012, 2, 440. [Google Scholar] [CrossRef] [PubMed]
- Flores-Lagnarica, A.; Sebti, Y.; Mitchell, D.A.; Sim, R.B.; MacPherson, G.G. Scrapie pathogenesis: The role of complement C1q in scrapie agent uptake by conventional dendritic cells. J. Immunol. 2009, 182, 1305–1313. [Google Scholar] [CrossRef]
- Bradford, B.M.; Brown, K.L.; Mabbott, N.A. Prion pathogenesis is unaltered following down-regulation of SIGN-R1. Virology 2016, 497, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Kohler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C.; et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Santini, P.A.; Sullivan, J.S.; He, B.; Shan, M.; Ball, S.C.; Dyer, W.B.; Ketas, T.J.; Chadburn, A.; Cohen-Gould, L.; et al. HIV-1 evades virus-specific IgG2 and IgA responses by targeting systemic conduits and intestinal B cells via long-range intercellular conduits. Nat. Immunol. 2009, 10, 1008. [Google Scholar] [CrossRef] [PubMed]
- Hase, K.; Kimura, S.; Takatsu, H.; Ohmae, M.; Kawano, S.; Kitamura, H.; Ito, M.; Watarai, H.; Hazelett, C.C.; Yeaman, C.; et al. M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 2009, 11, 1427–1432. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Victoria, G.S.; Marzo, L.; Ghosh, R.; Zurzolo, C. Prion aggregates transfer through tunneling nanotubes in endocytic vesicles. Prion 2015, 9, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chanouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Langevin, C.; Gousset, K.; Costanzo, M.; Richard-Le Goff, O.; Zurzolo, C. Characterization of the role of dendritic cells in prion transfer to primary neurons. Biochem. J. 2010, 431, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Sadaike, T.; Inoshima, Y.; Ishiguro, N. Characterisation of PrPSc transmission from immune cells to neuronal cells. Cell. Immunol. 2012, 279, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed]
- Arellano-Anaya, Z.E.; Huor, A.; Leblanc, P.; Lehmann, S.; Provansal, M.; Raposo, G.; Andréoletti, O.; Viette, D. Prion strains are differentially released through the exosomal pathway. Cell. Mol. Life Sci. 2015, 72, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Carp, R.I.; Callahan, S.M. In vitro interaction of scrapie agent and mouse peritoneal macrophages. Intervirology 1981, 16, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Carp, R.I.; Callahan, S.M. Effect of mouse peritoneal macrophages on scrapie infectivity during extended in vitro incubation. Intervirology 1982, 17, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Maignien, T.; Shakweh, M.; Calvo, P.; Marce, D.; Sales, N.; Fattal, E.; Deslys, J.-P.; Couvreur, P.; Lasmezas, C.I. Role of gut macrophages in mice orally contaminated with scrapie or BSE. Int. J. Pharm. 2005, 298, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Beringue, V.; Demoy, M.; Lasmezas, C.I.; Gouritin, B.; Weingarten, C.; Deslys, J.-P.; Andreux, J.P.; Couvreur, P.; Dormont, D. Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. J. Pathol. 2000, 190, 495–502. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Williams, A.; Farquhar, C.F.; Pasparakis, M.; Kollias, G.; Bruce, M.E. Tumor necrosis factor-alpha-deficient, but not interleukin-6-deficient, mice resist peripheral infection with scrapie. J. Virol. 2000, 74, 3338–3344. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, T.; Muramoto, T.; Mohri, S.; Doh-Ura, K.; Tateishi, J. Abnormal Isoform of Prion Protein Accumulates in Follicular Dendritic Cells in Mice with Creutzfeldt-Jakob Disease. J. Virol. 1991, 65, 6292–6295. [Google Scholar] [PubMed]
- Fraser, H.; Brown, K. Peripheral Pathogenesis of Scrapie in Normal and Immunocompromised Mice. Anim. Technol. 1994, 45, 21–22. [Google Scholar]
- Klein, M.A.; Frigg, R.; Flechsig, E.; Raeber, A.J.; Kalinke, U.; Bluethman, H.; Bootz, F.; Suter, M.; Zinkernagel, R.M.; Aguzzi, A. A crucial role for B cells in neuroinvasive scrapie. Nature 1997, 390, 687–691. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.; Brown, K.L.; Stewart, K.; McConnell, I.; McBride, P.; Williams, A. Replication of scrapie in spleens of SCID mice follows reconstitution with wild-type mouse bone marrow. J. Gen. Virol. 1996, 77, 1935–1940. [Google Scholar] [CrossRef] [PubMed]
- McFarlin, D.E.; Raff, M.C.; Simpson, E.; Nehlsen, S.H. Scrapie in immunologically deficient mice. Nature 1971, 233, 336. [Google Scholar] [CrossRef] [PubMed]
- Raeber, A.J.; Sailer, A.; Hegyi, I.; Klein, M.A.; Rulicke, T.; Fischer, M.; Brandner, S.; Aguzzi, A.; Weissmann, C. Ectopic expression of prion protein (PrP) in T lymphocytes or hepatocytes of PrP knockout mice is insufficient to sustain prion replication. Proc. Natl. Acad. Sci. USA 1999, 96, 3987–3992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, P.; Eikelenboom, P.; Kraal, G.; Fraser, H.; Bruce, M.E. PrP protein is associated with follicular dendritic cells of spleens and lymph nodes in uninfected and scrapie-infected mice. J. Pathol. 1992, 168, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Montrasio, F.; Cozzio, A.; Flechsig, E.; Rossi, D.; Klein, M.A.; Rulicke, T.; Raeber, A.J.; Vosshenrich, C.A.J.; Proft, J.; Aguzzi, A.; Weissmann, C. B-lymphocyte-restricted expression of the prion protein does not enable prion replication in PrP knockout mice. Proc. Natl. Acad. Sci. USA 2001, 98, 4034–4037. [Google Scholar] [CrossRef] [PubMed]
- Kapasi, Z.F.; Burton, G.F.; Schultz, L.D.; Tew, J.G.; Szakal, A.K. Induction of functional follicular dendritic cell development in severe combined immunodeficiency mice. J. Immunol. 1993, 150, 2648–2658. [Google Scholar] [PubMed]
- Chaplin, D.D.; Fu, Y.-X. Cytokine regulation of secondary lymphoid organ development. Curr. Opin. Immunol. 1998, 10, 289–297. [Google Scholar] [CrossRef]
- Mackay, F.; Browning, J.L. Turning off follicular dendritic cells. Nature 1998, 395, 26–27. [Google Scholar] [CrossRef] [PubMed]
- Tumanov, A.V.; Kuprash, D.V.; Lagarkova, M.A.; Grivennikov, S.I.; Abe, K.; Shakhov, A.; Drutskaya, L.N.; Stewart, C.L.; Chervonsky, A.V.; Nedospasov, S.A. Distinct role of surface lymphotoxin epxressed by B cells in the organization of secondary lymphoid tissues. Immunity 2002, 239, 239–250. [Google Scholar] [CrossRef]
- Krautler, N.J.; Kana, V.; Kranich, J.; Tian, Y.; Perera, D.; Lemm, D.; Schwarz, P.; Armulik, A.; Browning, J.L.; Tallquist, M.; et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell 2012, 150, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Shortman, K.; Liu, Y.-J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Bailie, J.K.; Kobayashi, A.; Donaldson, D.S.; Ohmori, H.; Yoon, S.-O.; Freedman, A.S.; Freeman, T.C.; Summers, K.M. Expression of mesenchyme-specific gene signatures by follicular dendritic cells: Insights from the meta-analysis of microarray data from multiple mouse cell populations. Immunology 2011, 133, 482–498. [Google Scholar] [CrossRef] [PubMed]
- Fütterer, A.; Mink, K.; Luz, A.; Kosco-Vilbois, M.H.; Pfeffer, K. The lymphotoxin b receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 1998, 9, 59–70. [Google Scholar] [CrossRef]
- Matsumoto, M.; Lo, S.F.; Carruthers, C.J.L.; Min, J.; Mariathasan, S.; Huang, G.; Plas, D.R.; Martin, S.M.; Geha, R.S.; Nahm, M.H.; et al. Affinity maturation without germinal centres in lymphotoxin-a-deficient mice. Nature 1996, 382, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Koni, P.A.; Sacca, R.; Lawton, P.; Browning, J.L.; Ruddle, N.H.; Flavell, R.A. Distinct roles in lymphoid organogenesis for lymphotoxins a and b revealed in lymphotoxin b-deficient mice. Immunity 1997, 6, 491–500. [Google Scholar] [CrossRef]
- Pasparakis, M.; Alexopoulo, L.; Episkopou, V.; Kollias, G. Immune and inflammatory responses in TNFa-deficient mice: A critical requirement for TNFa in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centres, and in the maturation of the humoral immune response. J. Exp. Med. 1996, 184, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Tkachuk, M.; Bolliger, S.; Ryffel, B.; Pluschke, G.; Banks, T.A.; Herren, S.; Gisler, R.H.; Kosco-Vilbois, M.H. Crucial role of tumour necrosis factor receptor 1 expression on nonhematopoietic cells for B cell localization within the splenic white pulp. J. Exp. Med. 1998, 187, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Montrasio, F.; Klein, M.A.; Schwarz, P.; Priller, J.; Odermatt, B.; Pfeffer, K.; Aguzzi, A. Lymph nodal prion replication and neuroinvasion in mice devoid of follicular dendritic cells. Proc. Natl. Acad. Sci. USA 2002, 99, 919–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabbott, N.A.; Mackay, F.; Minns, F.; Bruce, M.E. Temporary inactivation of follicular dendritic cells delays neuroinvasion of scrapie. Nat. Med. 2000, 6, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Montrasio, F.; Frigg, R.; Glatzel, M.; Klein, M.A.; Mackay, F.; Aguzzi, A.; Weissmann, C. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science 2000, 288, 1257–1259. [Google Scholar] [CrossRef] [PubMed]
- Mohan, J.; Bruce, M.E.; Mabbott, N.A. Follicular dendritic cell dedifferentiation reduces scrapie susceptibility following inoculation via the skin. Immunology 2005, 114, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Heikenwalder, M.; Junt, T.; Schwarz, P.; Glatzel, M.; Heppner, F.L.; Fu, Y.-X.; Lipp, M.; Aguzzi, A. Positioning of follicular dendritic cells within the spleen controls prion neuroinvasion. Nature 2003, 425, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; McGovern, G.; Jeffrey, M.; Bruce, M.E. Temporary blockade of the tumour necrosis factor signaling pathway impedes the spread of scrapie to the brain. J. Virol. 2002, 76, 5131–5139. [Google Scholar] [CrossRef] [PubMed]
- Heikenwalder, M.; Federau, C.; von Boehmer, L.; Schwarz, P.; Wagner, M.; Zeller, N.; Haybaeck, J.; Prinz, M.; Becher, B.; Aguzzi, A. Germinal centre B cells are dispensible for prion transport and neuroinvasion. J. Neuroimmunol. 2007, in press. [Google Scholar] [CrossRef] [PubMed]
- Helm, S.L.T.; Burton, G.F.; Szakal, A.K.; Tew, J.G. Follicular Dendritic Cells and the Maintenance of IgE Responses. Eur. J. Immunol. 1995, 25, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-X.; Molina, H.; Matsumoto, M.; Huang, G.; Min, J.; Chaplin, D.D. Lymphotoxin-a (LTa) supports development of splenic follicular structure that is required for IgG response. J. Exp. Med. 1997, 185, 2111–2120. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-X.; Huang, G.; Wang, Y.; Chaplin, D.D. B lymphocytes induce the formation of follicular dendritic cell clusters in a lymphotoxin a-dependent fashion. J. Exp. Med. 1998, 187, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Endres, R.; Alimzhanov, M.B.; Plitz, T.; Futterer, A.; Kosco-Vilbois, M.H.; Nedospasov, S.A.; Rajewsky, K.; Pfeffer, K. Mature follicular dendritic cell networks depend on expression of lymphotoxin b receptor by radioresistant stromal cells and of lymphotoxin b and tumour necrosis factor by B cells. J. Exp. Med. 1999, 189, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-X.; Huang, G.; Wang, Y.; Chaplin, D.D. Lymphotoxin-a-dependent spleen microenvironment supports the generation of memory B cells and is required for their subsequent antigen-induced activation. J. Immunol. 2000, 164, 2508–2514. [Google Scholar] [CrossRef] [PubMed]
- Aydar, Y.; Sukumar, A.; Szakal, A.K.; Tew, J.G. The influence of immune complex-bearing follicular dendritic cells on the IgM response, Ig class switching, and production of high affinity IgG. J. Immunol. 2005, 174, 5358–5366. [Google Scholar] [CrossRef] [PubMed]
- Heesters, B.A.; Myers, R.C.; Carroll, M.C. Follicular dendritic cells: Dynamic antigen libraries. Nat. Rev. Immunol. 2014, 14, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Kranich, J.; Krautler, N.J.; Heinen, E.; Polymenidou, M.; Bridel, C.; Schildknecht, A.; Huber, C.; Kosco-Vilbois, M.H.; Zinkernagel, R.; Miele, G.; et al. Follicular dendritic cells control engulfment of apoptotic bodies by secreting Mfge8. J. Exp. Med. 2008, 205, 1293–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victoratos, P.; Lagnel, J.; Tzima, S.; Alimzhanov, M.B.; Rajewsky, K.; Pasparakis, M.; Kollias, G. FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity 2006, 24, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.D.; Heikenwalder, M.; Prinz, M.; Arright, I.; Schwarz, P.; Kranich, J.; Von Teichman, A.; Haas, K.M.; Zeller, N.; Tedder, T.F.; et al. Stromal complement receptor CD21/35 facilitates lymphoid prion colonization and pathogenesis. J. Immunol. 2007, 179, 6144–6152. [Google Scholar] [CrossRef] [PubMed]
- McBride, P.A.; Schulz-Shaeffer, W.J.; Donaldson, M.; Bruce, M.; Diringer, H.; Kretzschmar, H.A.; Beekes, M. Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J. Virol. 2001, 75, 9320–9327. [Google Scholar] [CrossRef] [PubMed]
- Glatzel, M.; Heppner, F.L.; Albers, K.M.; Aguzzi, A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron 2001, 31, 25–34. [Google Scholar] [CrossRef]
- Brown, K.L.; Gossner, A.; Mok, S.; Mabbott, N.A. The effects of host age on the transport of complement-bound complexes to the spleen and the pathogenesis of intravenous scrapie infection. J. Virol. 2012, 86, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Sim, R.B.; Kishore, U.; Villiers, C.L.; Marche, P.N.; Mitchell, D.A. C1q binding and complement activation by prions and amyloid. Immunobiology 2007, 212, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Kirby, L.; Paulin, S.M.; Villiers, C.L.; Sim, R.B. Prion protein activates and fixes complement directly via the classical pathway: Implications for the mechanism of scrapie agent propagation in lymphoid tissue. Mol. Immunol. 2007, 44, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.A.; Kaeser, P.S.; Schwarz, P.; Weyd, H.; Xenarios, I.; Zinkernagel, R.M.; Carroll, M.C.; Verbeek, J.S.; Botto, M.; Walport, M.J.; et al. Complement facilitates early prion pathogenesis. Nat. Med. 2001, 7, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Bruce, M.E.; Botto, M.; Walport, M.J.; Pepys, M.B. Temporary depletion of complement component C3 or genetic deficiency of C1q significantly delays onset of scrapie. Nat. Med. 2001, 7, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Ferguson, A.; Johnson, T.; Bender, H.; Meyerett-Reid, C.; Pulford, B.; von Teichman, A.; Seelig, D.; Weiss, J.H.; Telling, G.C.; et al. Genetic depletion of complement receptors CD21/35 prevents terminal prion disease in a mouse model of chronic wasting disease. J. Immunol. 2012, 189, 4520–4527. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Ferguson, A.; Johnson, T.; Bender, H.; Meyerett-Reid, C.; Wycoff, A.C.; Pulford, B.; Telling, G.C.; Zabel, M.D. Complement protein C3 exacerbates prion disease in a mouse model of chronic wasting disease. Int. Immunol. 2013, 25, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Kane, S.J.; Farley, T.K.; Gordon, E.O.; Estep, J.; Bender, H.R.; Moreno, J.A.; Bartz, J.; Telling, G.C.; Pickering, M.C.; Zabel, M.D. Complement regulatory protein factor H is a soluble prion receptor that potentiates peripheral prion pathogenesis. J. Immunol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Kane, S.J.; Swanson, E.; Gordon, E.O.; Rocha, S.; Bender, H.R.; Donius, L.R.; Hannan, J.P.; Zabel, M.D. Relative impact of complement receptors CD21/35 (Cr2/1) on scrapie pathogenesis in mice. mSphere 2017, in press. [Google Scholar] [CrossRef]
- Mabbott, N.A.; Bruce, M.E. Complement component C5 is not involved in scrapie pathogenesis. Immunobiology 2004, 209, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Heesters, B.A.; Chatterjee, P.; Kim, Y.A.; Kuligowski, M.P.; Kirchhausen, T.; Carroll, M.C. Endocytosis and recycling of immune complexes by follicular dendritic cells enhances B cell antigen binding and activation. Immunity 2013, 38, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, M.; McGovern, G.; Goodsir, C.M.; Brown, K.L.; Bruce, M.E. Sites of prion protein accumulation in scrapie-infected mouse spleen revealed by immuno-electron microscopy. J. Pathol. 2000, 191, 323–332. [Google Scholar] [CrossRef]
- McGovern, G.; Brown, K.L.; Bruce, M.E.; Jeffrey, M. Murine scrapie infection causes an abnormal germinal centre reaction in the spleen. J. Comp. Pathol. 2004, 130, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Barillas-Mury, C.; Miller, M.W.; Oesch, B.; van Keulen, L.J.M.; Langeveld, J.P.M.; Hoover, E.A. PrPCWD lymphoid cell targets in early and advanced chronic wasting disease of mule deer. J. Gen. Virol. 2002, 83, 2617–2628. [Google Scholar] [CrossRef] [PubMed]
- Gunn, M.D.; Ngo, V.N.; Ansel, K.M.; Ekland, E.H.; Cyster, J.G.; Williams, L.T. A B-cell-homing chemokine made in lymphoid follicles activates Burkitt’s lymphoma receptor-1. Nature 1998, 391, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Ansel, K.M.; Ngo, V.N.; Hyman, P.L.; Luther, S.A.; Forster, R.; Sedgwick, J.D.; Browning, J.L.; Lipp, M.; Cyster, J. A chemokine-driven feedback loop organizes lymphoid follicles. Nature 2000, 406, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wang, Y.; Chin, R.K.; Martinez-Pomares, L.; Gordon, S.; Kosco-Vilbois, M.H.; Cyster, J.; Fu, Y.-X. B cells control the migration of a subset of dendritic cells into B cell follicles via CXC chemokine ligand 13 in a lymphotoxin-dependent fashion. J. Immunol. 2002, 168, 5117–5123. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Reizis, B.; Mabbott, N.A. Oral prion disease pathogenesis is impeded in the specific absence of CXCR5-expressing dendritic cells. J. Virol. 2017, 91, e00124-17. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.H.; Sougawa, N.; Tanaka, T.; Hirata, T.; Hiroi, T.; Tohya, K.; Guo, Z.; Umemoto, E.; Ebisuno, Y.; Yang, B.-G.; et al. CCR7 is critically important for migration of dendritic cells in intestinal lamina propria to mesenteric lymph nodes. J. Immunol. 2006, 176, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Levavasseur, E.; Matharom, P.; Dorban, G.; Nakano, H.; Kakiuchi, T.; Carnaud, C.; Sarradin, P.; Aucouturier, P. Experimental scrapie in ‘plt’ mice: An assessment of the role of dendritic-cell migration in the pathogenesis of prion diseases. J. Gen. Virol. 2007, 88, 2353–2360. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Grigorova, I.; Okada, T.; Cyster, J.G. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat. Immunol. 2007, 8, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, Y.R.; Batista, F.D. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 2007, 27, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Green, J.A.; Gray, E.E.; Xu, Y.; Cyster, J.G. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat. Immunol. 2009, 10, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Cinamon, G.; Zachariah, M.A.; Lam, O.M.; Foss Jr, F.W.; Cyster, J.G. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008, 9, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Heggebø, R.; Press, C.M.; Gunnes, G.; González, L.; Jeffrey, M. Distribution and accumulation of PrP in gut-associated and peripheral lymphoid tissue of scrapie-affected Suffolk sheep. J. Gen. Virol. 2002, 83, 479–489. [Google Scholar]
- Glatzel, M.; Abela, E.; Maissen, M.; Aguzzi, A. Extraneural pathological prion protein in sporadic Creutzfeldt-Jakob disease. N. Engl. J. Med. 2003, 349, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Schlomchik, M.J.; Radebold, K.; Duclos, N.; Manuelidis, L. Neuroinvasion by a Creutzfeldt-Jakob disease agent in the absence of B cells and follicular dendritic cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9289–9294. [Google Scholar] [CrossRef] [PubMed]
- Somerville, R.A.; Birkett, C.R.; Farquhar, C.F.; Hunter, N.; Goldmann, W.; Dornan, J.; Grover, D.; Hennion, R.M.; Percy, C.; Foster, J.; et al. Immunodetection of PrPSc in spleens of some scrapie-infected sheep but not BSE-infected cows. J. Gen. Virol. 1997, 78, 2389–2396. [Google Scholar] [CrossRef] [PubMed]
- Terry, L.A.; Marsh, S.; Ryder, S.J.; Hawkins, S.A.C.; Wells, G.A.H.; Spencer, Y.I. Detection of disease-specific PrP in the distal ileum of cattle exposed orally to the agent of bovine spongiform encephalopathy. Vet. Rec. 2003, 152, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.A.H.; Dawson, M.; Hawkins, S.A.C.; Green, R.B.; Dexter, I.; Francis, M.E.; Simmons, M.M.; Austin, A.R.; Horigan, M.W. Infectivity in the ileum of cattle challenged orally with bovine spongiform encephalopathy. Vet. Rec. 1994, 135, 40–41. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Eiden, M.; Kaatz, M.; Keller, M.; Ziegler, U.; Rogers, R.; Hills, B.; Balkema-Buschmann, A.; Van Keulen, L.; Jacobs, J.G.; et al. BSE infectivity in jejunum, ileum and ileocaecal junction of incubating cattle. Vet. Res. 2011, 42, 21. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.D.; Parnham, D.W.; Hunter, N.; Bruce, M. Distribution of the prion protein in sheep terminally affected with BSE following experimental oral transmission. J. Gen. Virol. 2001, 82, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Mabbott, N.A. Evidence of subclinical prion disease in aged mice following exposure to bovine spongiform encephalopathy. J. Gen. Virol. 2014, 95, 231–243. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Frei, N.; Sponarova, J.; Schwarz, P.; Heikenwalder, M.; Agguzi, A. Lymphotxin, but not TNF, is required for prion invasion of lymph nodes. PLoS Pathog. 2012, 8, e1002867. [Google Scholar]
- Heikenwalder, M.; Kurrer, M.O.; Margalith, I.; Kranich, J.; Zeller, N.; Haybaeck, J.; Polymenidou, M.; Matter, M.; Bremer, J.; Jackson, W.S.; et al. Lymphotoxin-dependent prion replication in inflammatory stromal cells of granulomas. Immunity 2008, 29, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.C. The antibody response of scrapie-affected mice to immunisation with sheep red blood cells. Res.Vet. Sci. 1968, 9, 595–597. [Google Scholar] [PubMed]
- Garfin, D.E.; Stites, D.P.; Perlman, J.D.; Cochran, S.P.; Prusiner, S.B. Mitogen stimulation of splenocytes from mice infected with scrapie agent. J. Infect. Dis. 1978, 138, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Kingsbury, D.T.; Smeltzer, D.A.; Gibbs, C.J.; Gadjusek, D.C. Evidence for Normal Cell-Mediated Immunity in Scrapie-Infected Mice. Infect. Immun. 1981, 32, 1176–1180. [Google Scholar]
- Elleman, C.J. ConA induced suppressor cells in scrapie-infected mice. Vet. Immunol. Immunopathol. 1985, 8, 79–82. [Google Scholar] [CrossRef]
- Gonzalez, L.; Martin, S.; Siso, S.; Konold, T.; Ortiz-Pelaez, A.; Phelan, L.; Goldmann, W.; Stewart, P.; Saunders, G.; Windl, O.; et al. High prevalence of scrapie in a dairy goat herd: Tissue distribution of disease-associated PrP and effect of PRNP genotype and age. Vet. Res. 2009, 40, 65. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, B.V.; Schneider, D.A.; O’Rourke, K.I.; Gidlewski, T.; McLane, J.; Allen, R.W.; mcIsaac, A.A.; Mitchell, G.B.; Keane, D.P.; Spraker, T.R.; et al. Diagnostic accuracy of rectal mucosa biopsy testing for chronic wasting disease within white-tailed deer (Odocoileus virginianus) herds in North America: Effects of age, sex, polymorphism at PRNP codon 96, and disease progression. J. Vet. Diagn. Intestig. 2012, 24, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Monello, R.J.; Powers, J.G.; Hobbs, N.T.; Spraker, T.R.; O’Rourke, K.I.; Wild, M.A. Efficacy of antemortem rectal biopsies to diagnose and estimate prevalence of chronic wasting disease in free-ranging cow elk (Cervus elaphus nelsoni). J. Wildl. Dis. 2013, 49, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, L.J.; Schreuder, B.E.; Vromans, M.E.; Langeveld, J.P.; Smits, M.A. Pathogenesis of natural scrapie in sheep. Arch. Virol. Suppl. 2000, 16, 57–71. [Google Scholar]
- Van Keulen, L.J.M.; Vromans, M.E.W.; van Zijderveld, F.G. Ealry and late pathogenesis of natural scrapie infection in sheep. APMIS 2002, 110, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, L.J.M.; Bossers, A.; Van Zijderveld, F.G. TSE pathogenesis in cattle and sheep. Vet. Res. 2008, 39, 24. [Google Scholar] [CrossRef] [PubMed]
- Tabouret, G.; Lacroux, C.; Lugan, S.; Costes, P.; Corbiere, F.; Weisbecker, J.L.; Schelcher, F.; Andréoletti, O. Relevance of oral experimental challenge with classical scrapie in sheep. J. Gen. Virol. 2010, 91, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Van Keulen, L.J.M.; Vromans, M.E.W.; Dolstra, C.H.; Bossers, A.; van Zijderveld, F.G. Pathogenesis of bovine spongiform encephalopathy in sheep. Arch. Virol. 2008, 153, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Keane, D.; Barr, D.; Osborn, R.; Langenberg, J.; O’Rourke, K.; Schneider, D.; Bochsler, P. Validation of use of rectoanal mucosa-associated lymphoid tissue for immunohistochemical diagnosis of chronic wasting disease in white-tailed deer (Odocoileus virginianus). J. Clin. Microbiol. 2009, 47, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; VerCauteren, K.C.; Gidlewski, T.; Schneider, D.A.; Munger, R.; Balachandran, A.; O’Rourke, K.I. Antermortem detection of PrPCWD in preclinical, ranch-raised Rocky Mountain elk (Cervus elaphus nelsoni) by biopsy of the rectal mucosa. J. Vet. Diagn. Intestig. 2009, 21, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hoover, C.E.; Davenport, K.A.; Henderson, D.M.; Denkers, N.D.; Mathiason, C.K.; Soto, C.; Zabel, M.D.; Hoover, E.A. Pathways of prion spread during early chronic wasting disease in deer. J. Virol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.-P.; Platt, N.; Wykes, M.; Major, J.R.; Powell, T.J.; Jenkins, C.D.; MacPherson, G.G. A discrete subpopulation of dendritic cells transports apoptotic intestinal epithelial cells to T cell areas of mesenteric lymph nodes. J. Exp. Med. 2000, 191, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Houston, S.A.; Cerovic, V.; Thomson, C.; Brewer, J.; Mowat, A.M.; Milling, S. The lymph nodes draining the small intestine and colon are anatomically separate and immunologically distinct. Mucosal Immunol. 2016, 9, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, R.H.; Walker, C.A. Pathogenesis of scrapie in mice after intragastric infection. Virus Res. 1989, 12, 213–220. [Google Scholar] [CrossRef]
- Schmidt, T.H.; Bannard, O.; Gray, E.E.; Cyster, J.G. CXCR4 promotes B cell egress from Peyer’s patches. J. Exp. Med. 2013, 210, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Gulbranson-Judge, A.; Quinn, M.E.; Walters, A.E.; MacLennan, I.C.; Tybulewicz, V.L.J. Syk tyrosine kinase is required for the positive selection of immature B cells into the recirculating B cell pool. J. Exp. Med. 1997, 186, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Grigorova, I.; Phan, T.G.; Kelly, L.M.; Cyster, J.G. Visualizing B cell capture of cognate antigen from follicular dendritic cells. J. Exp. Med. 2009, 206, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.W.; Proia, R.L.; Brinkmann, V.; Mabbott, N.A. B cell-specific S1PR1 deficiency blocks prion dissemination between secondary lymphoid organs. J. Immunol. 2012, 188, 5032–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreoletti, O.; Litaise, C.; Simmons, H.; Corbiere, F.; Lugan, S.; Costes, P.; Schelcher, F.; Viette, D.; Grassi, J.; Lacroux, C. Highly efficient prion transmission by blood transfusion. PLoS Pathog. 2012, 8, e1002782. [Google Scholar] [CrossRef] [PubMed]
- Douet, J.Y.; Lacroux, C.; Litaise, C.; Lugan, S.; Corbiere, F.; Arnold, M.; Simmons, H.; Aron, N.; Costes, P.; Tillier, C.; et al. Mononucleated blood cell populations display different abilities to transmit prion disease by the transfusion route. J. Virol. 2016, 90, 3439–3445. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.C.; Moore, S.J.; Hawthorne, J.A.; Neale, M.H.; Terry, L.A. PrP(Sc) is associated with B cells in the blood of scrapie-infected sheep. Virology 2010, 405, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Mathiason, C.K.; Hayes-Klug, J.; Hays, S.A.; Powers, J.; Osborn, D.A.; Dahmes, S.J.; Miller, K.V.; Warren, R.J.; Mason, G.L.; Telling, G.C.; et al. B cells and platelets harbour prion infectivity in the blood of deer infected with chronic wasting disease. J. Virol. 2010, 84, 5097–5107. [Google Scholar] [CrossRef] [PubMed]
- Sisó, S.; González, L.; Jeffrey, M.; Martin, S.; Chianini, F.; Steele, P. Prion protein in kidneys of scrapie-infected sheep. Vet. Rec. 2006, 159, 327–328. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Nicola, D.; Schetters, S.T.T.; Perry, V.H. Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia 2014, 62, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A.; Cairns, N.J.; Ironside, J.W.; Lantos, P.L. Does the neuropathology of human patients with variant Creutzfeldt-Jakob disease reflect haematogenous spread of the disease. Neurosci. Lett. 2003, 348, 37–40. [Google Scholar] [CrossRef]
- Felten, S.Y.; Felten, D.L. Innervation of Lymphoid Tissue. In Psychoneuroimmunology, 2nd ed.; Academic Press Inc.: Cambridge, MA, USA, 1991; pp. 27–69. [Google Scholar]
- Beekes, M.; Baldauf, E.; Diringer, H. Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie. J. Gen. Virol. 1996, 77, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, E.; Beekes, M.; Diringer, H. Evidence for an alternative direct route of access for the scrapie agent to the brain bypassing the spinal cord. J. Gen. Virol. 1997, 78, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Beekes, M.; McBride, P.A.; Baldauf, E. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J. Gen. Virol. 1998, 79, 601–607. [Google Scholar] [CrossRef] [PubMed]
- McBride, P.A.; Beekes, M. Pathological PrP is abundant in sympathetic and sensory ganglia of hamsters fed with scrapie. Neurosci. Lett. 1999, 265, 135–138. [Google Scholar] [CrossRef]
- Flores-Lagnarica, A.; Meza-Perez, S.; Calderon-Amador, J.; Estrada-Garcia, T.; Macpherson, G.; Saeland, S.; Steinman, R.M.; Flores-Romo, L. Network of dendritic cells within the muscular layer of the mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 19039–19044. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.; Koscso, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between muscularis macrophages and enteric neurones regulates gastrointestinal motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Aucouturier, P.; Geissmann, F.; Damotte, D.; Saborio, G.P.; Meeker, H.C.; Kascsak, R.; Kascsak, R.; Carp, R.I.; Wisniewski, T. Infected splenic dendritic cells are sufficient for prion transmission to the CNS in mouse scrapie. J. Clin. Investig. 2001, 108, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Sigurdson, C.J.; Heikenwalder, M.; Manco, G.; Barthel, M.; Schwarz, P.; Stecher, B.; Krautler, N.J.; Hardt, W.-D.; Seifert, B.; MacPherson, A.J.S.; et al. Bacterial colitis increases susceptibility to oral prion pathogenesis. J. Infect. Dis. 2009, 199, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, A.G.; Fraser, H.; McConnell, I.; Outram, G.W. Mitogenic Stimulation of the Host Enhances Susceptibility to Scrapie. Nature 1978, 272, 54–55. [Google Scholar] [CrossRef] [PubMed]
- Bremer, J.; Heikenwalder, M.; Haybaeck, J.; Tiberi, C.; Krautler, N.J.; Kurrer, M.O.; Aguzzi, A. Repetitive immunization enhances the susceptibility of mice to peripherally administered prions. PLoS ONE 2009, 4, e7160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heikenwalder, M.; Zeller, N.; Seeger, H.; Prinz, M.; Klöhn, P.-C.; Schwarz, P.; Ruddle, N.H.; Weissmann, C.; Aguzzi, A. Chronic lymphocytic inflammation specifies the organ tropism of prions. Science 2005, 307, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Ligios, C.; Sigurdson, C.; Santucciu, C.; Carcassola, G.; Manco, G.; Basagni, M.; Maestrale, C.; Cancedda, M.G.; Madau, L.; Aguzzi, A. PrPSc in mammary glands of sheep affected by scrapie and mastitis. Nat. Med. 2005, 11, 1137–1138. [Google Scholar] [CrossRef] [PubMed]
- Valleron, A.-J.; Boelle, P.-Y.; Will, R.; Cesbron, J.-Y. Estimation of epidemic size and incubation time based on age characteristics of vCJD in the United Kingdom. Science 2001, 294, 1726–1728. [Google Scholar] [CrossRef] [PubMed]
- Diack, A.B.; Head, M.W.; McCutcheon, S.; Boyle, A.; Knight, R.; Ironside, J.W.; Manson, J.C.; Will, R.G. Variant CJD. 18 years of research and surveillance. Prion 2014, 2014, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.T.; Hart, P.; Aitchison, L.; Baybutt, H.N.; Plinston, C.; Thomson, V.; Tuzi, N.L.; Head, M.W.; Ironside, J.W.; Will, R.G.; et al. Predicting susceptibility and incubation time of human-to-human transmission of vCJD. Lancet Neurol. 2006, 5, 393–398. [Google Scholar] [CrossRef]
- Brown, K.L.; Stewart, K.; Bruce, M.E.; Fraser, H. Severly combined immunodeficient (SCID) mice resist infection with bovine spongiform encephalopathy. J. Gen. Virol. 1997, 78, 2707–2710. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Makarava, N.; Katorcha, E.; Savtchenko, R.; Brossmer, R.; Baskakov, I.V. Post-conversion sialylation of prions in lymphoid tissues. Proc. Natl. Acad. Sci. USA 2015, 112, E6654–E6662. [Google Scholar] [CrossRef] [PubMed]
- Boelle, P.-Y.; Cesbron, J.-Y.; Valleron, A.-J. Epidemiological evidence of higher susceptibility to vCJD in the young. BMC Infect. Dis. 2004, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, K.L.; Wu, Y.-C.; Barnett, Y.; Duggan, O.; Vaughan, R.; Kondeatis, E.; Nilsson, B.-O.; Wikby, A.; Kipling, D.; Dunn-Walters, D.K. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 2009, 8, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Akbar, A.N. Memory T-cell homeostasis and senescence during aging. Adv. Exp. Med. Biol. 2010, 684, 189–197. [Google Scholar] [PubMed]
- Bradford, B.M.; Crocker, P.R.; Mabbott, N.A. Peripheral prion disease pathogenesis is unaltered in the absence of sialoadhesin (Siglec-1/CD169). Immunology 2014, 143, 120–129. [Google Scholar] [CrossRef] [PubMed]
- St. Rose, S.; Hunter, N.; Matthews, D.; Foster, J.; Chase-Topping, M.E.; Kruuk, L.E.B.; Shaw, D.J.; Rhind, S.M.; Will, R.G.; Woolhouse, M.E.J. Comparative evidence for a link between Peyer’s patch development and susceptibility to transmissible spongiform encephalopathies. BMC Infect. Dis. 2006, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- St. Rose, S.G.; Hunter, N.; Foster, J.D.; Drummond, D.; McKenzie, C.; Parnham, D.; Will, R.G.; Woolhouse, M.E.J.; Rhind, S.M. Quantification of Peyer’s patches in Cheviot sheep for future scrapie pathogenesis studies. Vet. Immunol. Immunopathol. 2007, 116, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.L.; Wathne, G.J.; Sales, J.; Bruce, M.E.; Mabbott, N.A. The effects of host age on follicular dendritic cell status dramatically impair scrapie agent neuroinvasion in aged mice. J. Immunol. 2009, 183, 5199–5207. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Donaldson, D.S.; Erridge, C.; Kanaya, T.; Williams, I.R.; Ohno, H.; Mahajan, A.; Mabbott, N.A. The functional maturation of M cells is dramatically reduced in the Peyer’s patches of aged mice. Mucosal Immunol. 2013, 6, 1027–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebius, R.E.; Kraal, G. Structure and function of the spleen. Nat. Rev. Immunol. 2005, 5, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Turner, V.M.; Mabbott, N.A. Ageing adversely affects the migration and function of marginal zone B cells. Immunology 2017, 151, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Spraker, T.R.; VerCauteren, K.C.; Gidlewski, T.L.; Munger, R.D.; Walter, W.D.; Balachandran, A. Impact of age and sex of Rocky Mountain elk (Cervus elaphus nelsoni) on follicle counts from rectal mucosal biopsies for preclinical detection of chronic wasting disease. J. Vet. Diagn. Intestig. 2009, 21, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Geremia, C.; Hoeting, J.A.; Wolfe, L.L.; Galloway, N.L.; Antolin, M.F.; Spraker, T.R.; Miller, M.W.; Hobbs, N.T. Age and repeated biopsy influence antemortem PrPCWD testing in mule deer (Odocoileus hemionus) in Colorado, USA. J. Wildl. Dis. 2015, 51, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Outram, G.W.; Dickinson, A.G.; Fraser, H. Developmental maturation of susceptibility to scrapie in mice. Nature 1973, 241, 536–537. [Google Scholar] [CrossRef] [PubMed]
- Ierna, M.I.; Farquhar, C.F.; Outram, G.W.; Bruce, M.E. Resistance of neonatal mice to scrapie is associated with inefficient infection of the immature spleen. J. Virol. 2006, 80, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Hunter, N.; Houston, F.; Foster, J.; Goldmann, W.; Drummond, D.; Parnham, D.; Kennedy, I.; Green, A.; Stewart, P.; Chong, A. Susceptibility of young sheep to oral infection with bovine spongiform encephalopathy decreases significantly after weaning. J. Virol. 2012, 86, 11856–11862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ano, Y.; Sakudo, A.; Uraki, R.; Sato, Y.; Kono, J.; Sugiura, K.; Yokoyama, T.; Itohara, S.; Nakayama, H.; Yukawa, M.; et al. Enhanced enteric invasion of scrapie agents into the villous columnar epithelium via maternal immunoglobulin. Int. J. Mol. Med. 2010, 26, 845–851. [Google Scholar] [PubMed]
- Korth, C.; May, B.C.; Cohen, F.E.; Prusiner, S.B. Acridine and phenothiazine derivatives as pharmocotherapeutics for prion disease. Proc. Natl. Acad. Sci. USA 2001, 98, 9836–9841. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Lewis, V.; Brazier, M.; Hill, A.F.; Fletcher, A.; Masters, C.L. Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann. Neurol. 2002, 52, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Gorham, M.; Hudson, F.; Kennedy, A.; Keogh, G.; Pal, S.; Rossor, M.; Rudge, P.; Siddique, D.; Spyer, M.; et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): A patient-preference trial. Lancet Neurol. 2009, 8, 334–344. [Google Scholar] [CrossRef]
- Geschwind, M.D.; Kuo, A.L.; Wong, K.S.; Haman, A.; Devereux, G.; Raudabaugh, B.J.; Johnson, D.Y.; Torres-Chae, C.C.; Finley, R.; Garcia, P.; et al. Quinacrine treatment for sporadic Creutzfeldt-Jakob disease. Neurology 2013, 81, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Farquhar, C.; Dickinson, A.; Bruce, M. Prophylactic potential of pentosan polysulphate in transmissible spongiform encephalopathies. Lancet 1999, 353, 117. [Google Scholar] [CrossRef]
- Doh-ura, K.; Ishiwaki, K.; Murakami-Kubo, I.; Sasaki, K.; Mohri, S.; Race, R.; Iwaki, T. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J. Virol. 2004, 78, 4999–5006. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Doh-Ura, K.; Yamada, T. Continuous intraventricular infusion of pentosan polysulfate: Clinical trial against prion diseases. Neuropathology 2009, 29, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Tagliavini, F.; Forloni, G.; Colombo, L.; Rossi, G.; Girola, L.; Canciani, B.; Angretti, N.; Giampaolo, L.; Pressini, E.; Awan, T.; et al. Tetracycline affects abnormal properties of synthetic PrP peptides and PrPSc in vitro. J. Mol. Biol. 2000, 300, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Forloni, G.; Iussich, S.; Awan, T.; Colombo, L.; Angeretti, N.; Girola, L.; Bertani, I.; Poli, G.; Caramelli, M.; Grazia Bruzzone, M.; et al. Tetracyclines affect prion infectivity. Proc. Natl. Acad. Sci. USA 2002, 99, 10849–10854. [Google Scholar] [CrossRef] [PubMed]
- Haik, S.; Marcon, G.; Tettamanti, M.; Welaratne, A.; Giaccone, G.; Azimi, S.; Pietrini, V.; Fabrequettes, J.R.; Imperiale, D.; Cesaro, P.; et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014, 13, 150–158. [Google Scholar] [CrossRef]
- Dickinson, A.G.; Fraser, H.; McConnell, I.; Outram, G.W.; Sales, D.I.; Taylor, D.M. Extraneural competition between different scrapie agents leading to loss of infectivity. Nature 1975, 253, 556. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, A.G.; Fraser, H.; Meikle, V.M.H.; Outram, G.W. Competition between different scrapie agents in mice. Nat. New Biol. 1972, 237, 244–245. [Google Scholar] [CrossRef] [PubMed]
- Manuelidis, L. Vaccination with an attenuated Creutzfeldt-Jakob disease strain prevents expression of a virulent agent. Proc. Natl. Acad. Sci. USA 1998, 95, 2520–2525. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Espinoza, R.; Morales, R.; Concha-Marambio, L.; Moreno-Gonzalez, I.; Moda, F.; Soto, C. Treatment with a non-toxic, self-replicating anti-prion delays or prevents prion disease in vivo. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2aP mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [PubMed]
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A. Prospects for safe and effective vaccines against prion diseases. Exp. Rev. Vaccines 2015, 14, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Roettger, Y.; Du, Y.; Bacher, M.; Zerr, I.; Dodel, R.; Back, J.-P. Immunotherapy in prion disease. Nat. Rev. Neurol. 2013, 9, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Musahl, C.; Arrighi, I.; Klein, M.A.; Rulicke, T.; Oesch, B.; Zinkernagel, R.M.; Kalinke, U.; Aguzzi, A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 2001, 294, 178–182. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Enever, P.; Tayebi, M.; Mushens, R.; Lineham, J.; Brandner, S.; Anstee, D.; Collinge, J.; Hawke, S. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 2003, 422, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Goñi, F.; Knudsen, E.; Schreiber, F.; Scholtzova, H.; Pankiewicz, J.; Carp, R.; Meeker, H.C.; Rubenstein, R.; Brown, D.R.; Sy, M.S.; et al. Mucosal vaccination delays or prevents prion infection via the oral route. Neuroscience 2005, 133, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Goñi, F.; Chablagoity, J.A.; Prelli, F.; Schreiber, F.; Scholtzova, H.; Chung, E.; Kascsak, R.; Brown, D.R.; Sigurdsson, E.M.; Wisniewski, T. High titres of mucosal and systemic anti-PrP antibodies abrogates oral prion infection in mucosal vaccinated mice. Neuroscience 2008, 153, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Sonati, T.; Reimann, R.R.; Falsig, J.; Baral, P.K.; O’Connor, T.; Hornemann, S.; Yaganoglu, S.; Li, B.; Herrmann, U.S.; Wieland, B.; et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature 2013, 501, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, R.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Lineham, J.M.; et al. A naturally occuring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478–481. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
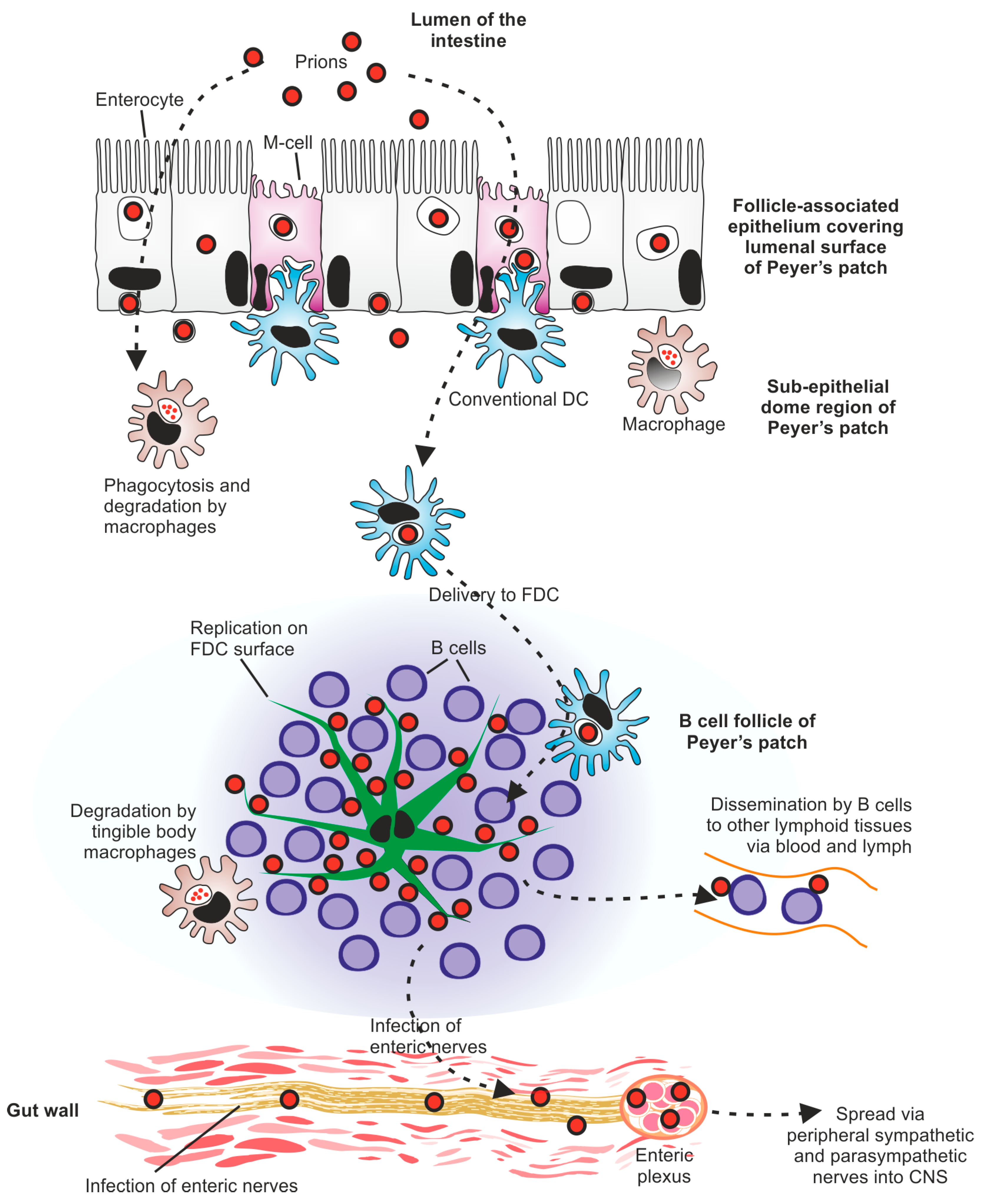{kind=link}
| Prion Disease | Affected Species | Transmission Route |
|---|---|---|
| Iatrogenic Creutzfeldt-Jakob disease (CJD) | Human | Accidental medical exposure to CJD-contaminated tissues or tissue products |
| Sporadic Creutzfeldt-Jakob disease | Human | Unknown. Theories include somatic mutation or spontaneous conversion of PrPc to PrPSc |
| Variant Creutzfeldt-Jakob disease | Human | Ingestion of BSE-contaminated food or transfusion of blood or blood products from variant CJD-infected blood donor |
| Familial Creutzfeldt-Jakob disease | Human | Germ-line mutations of the PRNP gene |
| Gerstmann-Straussler-Scheinker syndrome | Human | Germ-line mutations of the PRNP gene |
| Kuru | Human | Ritualistic cannibalism |
| Fatal familial insomnia | Human | Germ-line mutations of the PRNP gene |
| Bovine spongiform encephalopathy | Cattle | Ingestion of contaminated food |
| Scrapie | Sheep, goats, mouflon | Acquired. Ingestion, horizontal transmission, vertical transmission unclear |
| Chronic wasting disease | Elk, deer, moose | Acquired, ingestion, horizontal transmission, vertical transmission unclear |
| Transmissible mink encephalopathy | Mink | Acquired (ingestion) source unknown |
| Feline spongiform encephalopathy | Domestic and zoological cats | Ingestion of BSE-contaminated food |
| Exotic ungulate encephalopathy | Nyala, kudu | Ingestion of BSE-contaminated food |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabbott, N.A. How do PrPSc Prions Spread between Host Species, and within Hosts? Pathogens 2017, 6, 60. https://doi.org/10.3390/pathogens6040060
Mabbott NA. How do PrPSc Prions Spread between Host Species, and within Hosts? Pathogens. 2017; 6(4):60. https://doi.org/10.3390/pathogens6040060
Chicago/Turabian StyleMabbott, Neil A. 2017. "How do PrPSc Prions Spread between Host Species, and within Hosts?" Pathogens 6, no. 4: 60. https://doi.org/10.3390/pathogens6040060
APA StyleMabbott, N. A. (2017). How do PrPSc Prions Spread between Host Species, and within Hosts? Pathogens, 6(4), 60. https://doi.org/10.3390/pathogens6040060