Adenylate Cyclases of Trypanosoma brucei, Environmental Sensors and Controllers of Host Innate Immune Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
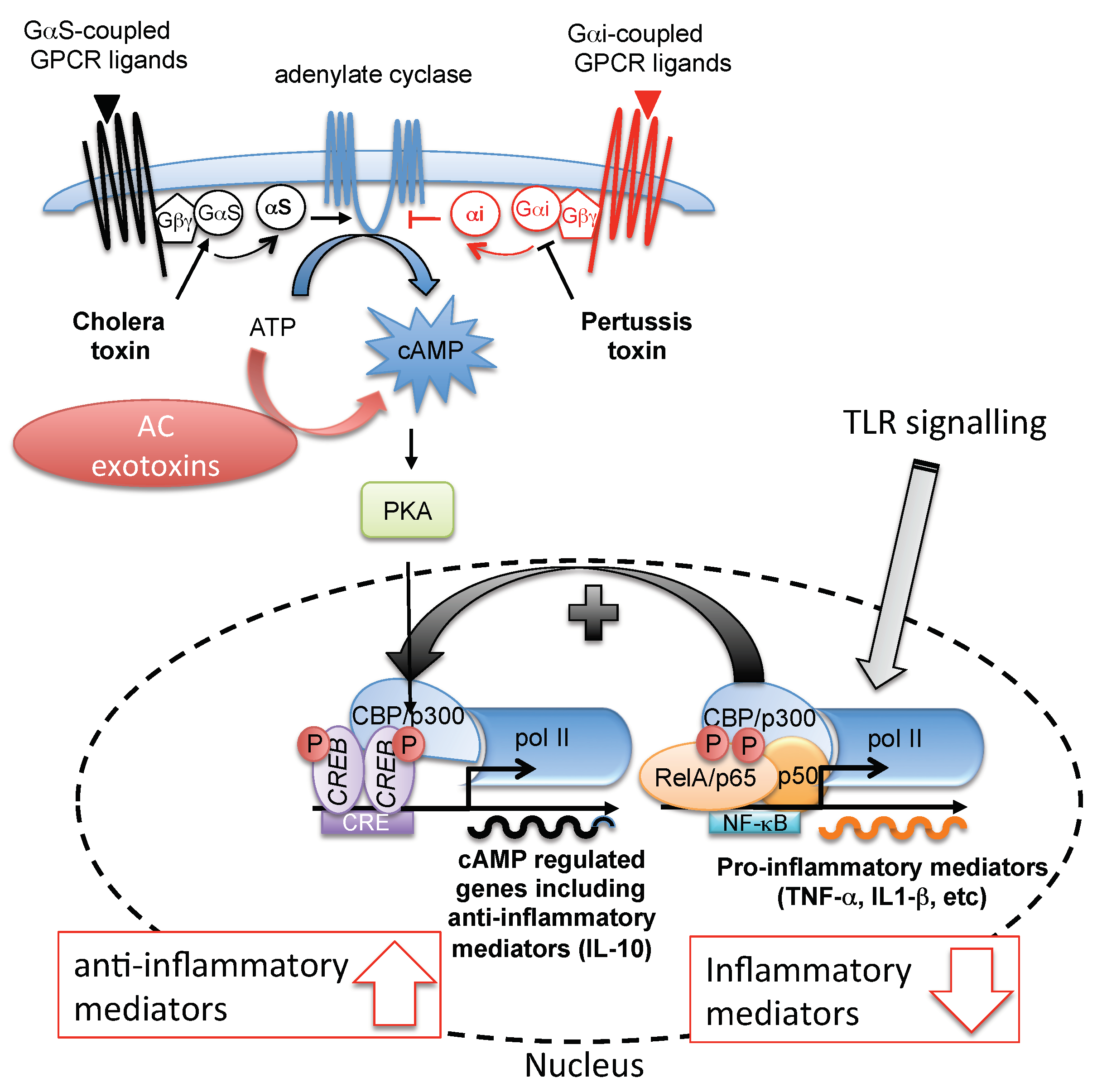
2. Role of cAMP in Innate and Adaptive Immunity and Pathogen Strategies to Counteract Immunity
3. T. brucei cAMP Signaling Pathway: From an Environmental Sensing Mechanism to an Innate Immune Evasion System
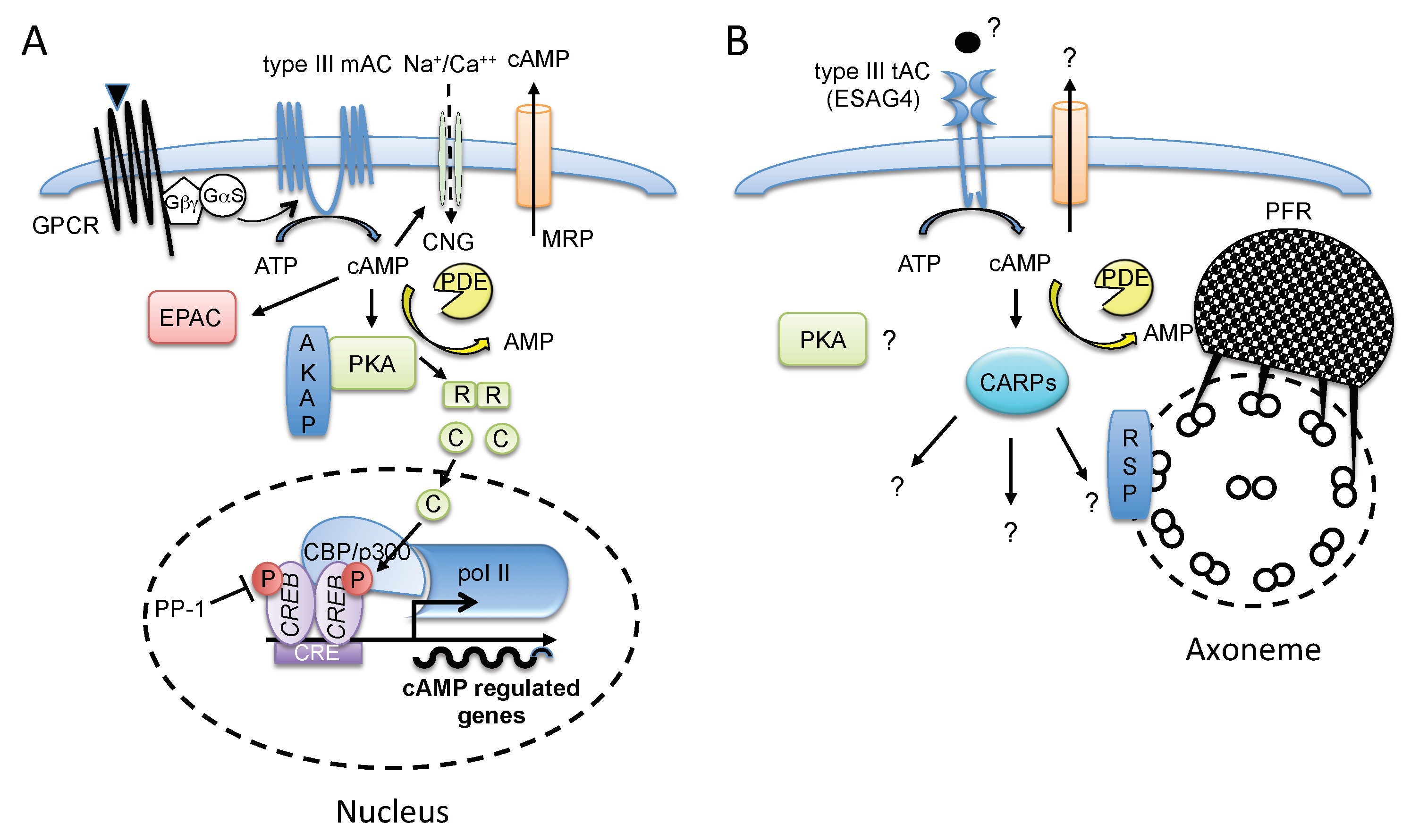
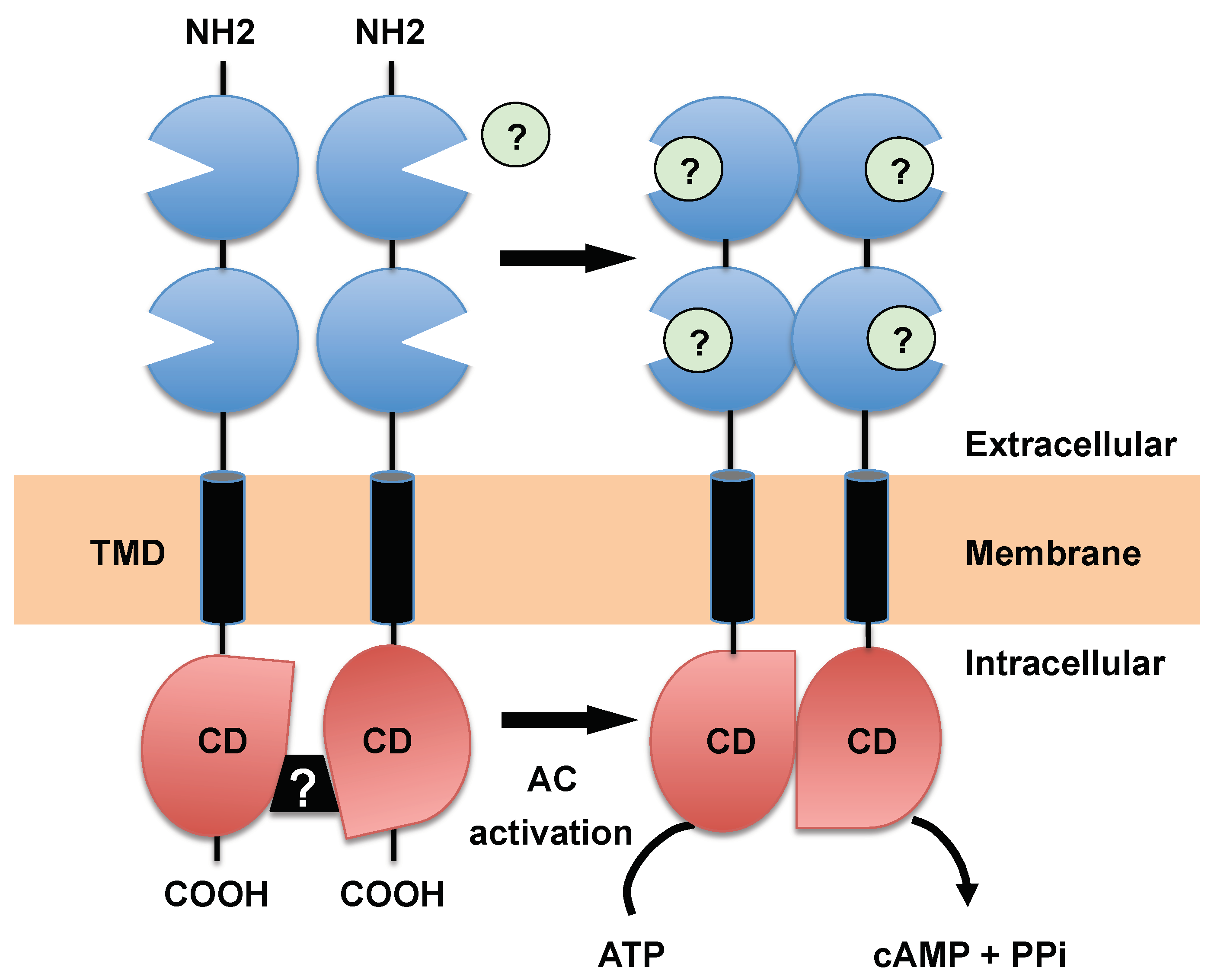
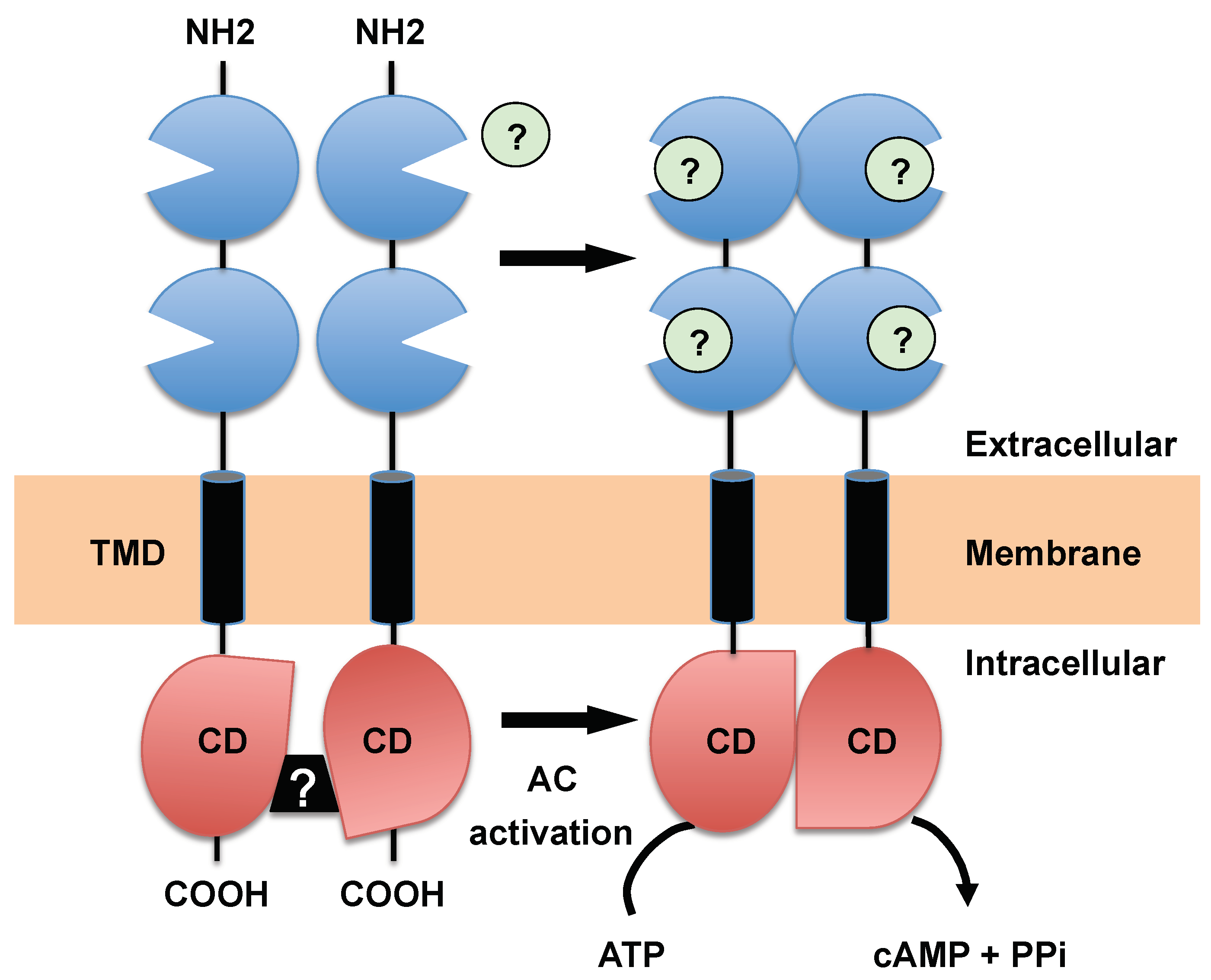
3.1. Receptor-Type ACs, a Hallmark of Trypanosomatids
3.2. cAMP Signal Integration in the T. brucei Flagellum, a Complex Organ for Sensing the Environment
3.3. Intracellular cAMP Function and a Novel cAMP Pathway in T. brucei
3.4. Role of ESAG4, a Receptor-Type AC Specific for the Bloodstream Form
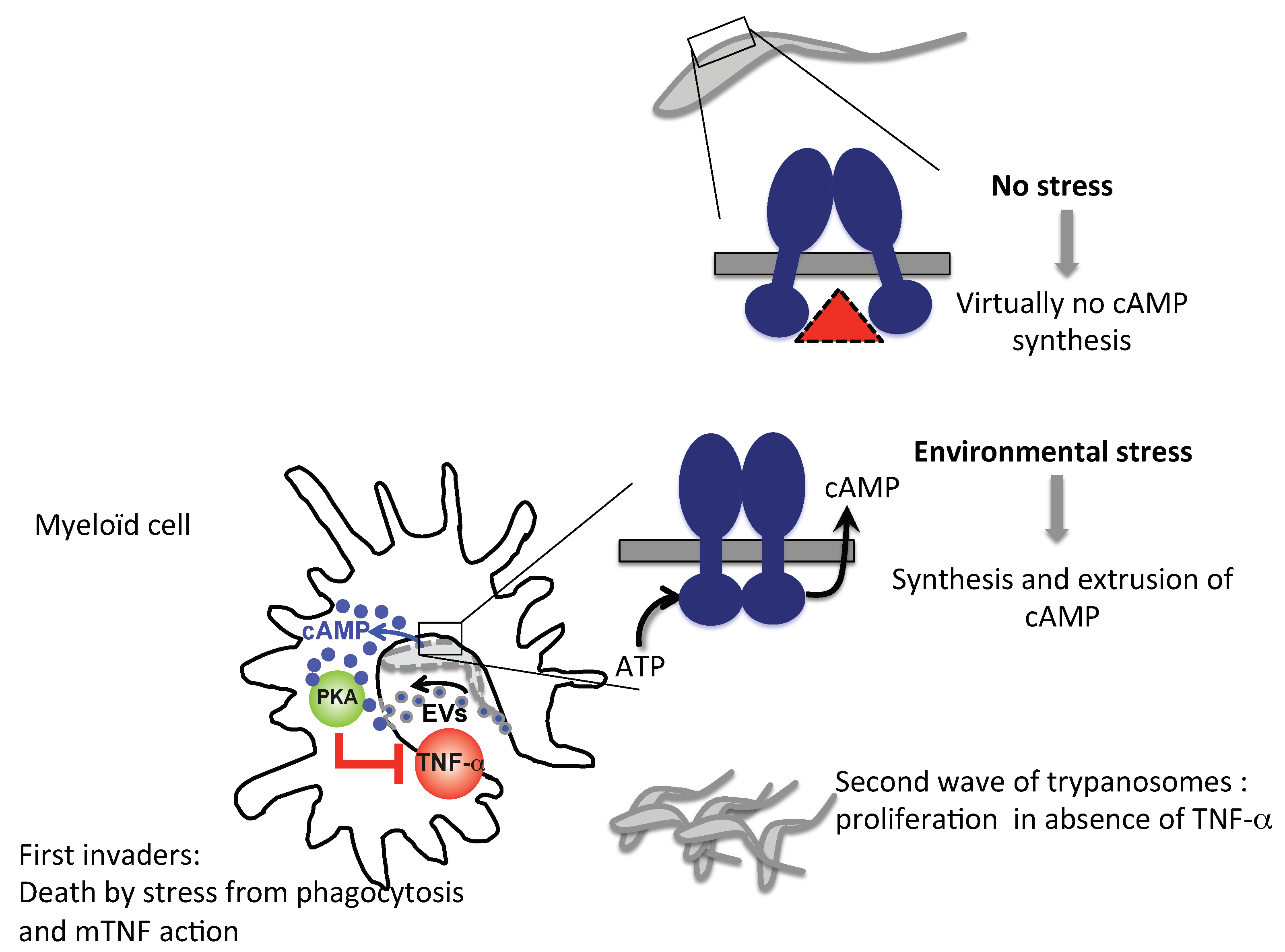
3.5. Trypanosomal ACs as a Tolerogenic Tool in Mammalian Host Innate Immunity
4. Concluding Remarks and Perspectives
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.; Agrebi, R.; Bellows, L.E.; Collet, J.F.; Kaever, V.; Grundling, A. Evolutionary adaptation of the essential trna methyltransferase trmd to the signaling molecule 3′,5′-camp in bacteria. J. Biol. Chem. 2017, 292, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Gorke, B.; Stulke, J. Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nat. Rev. Microbiol. 2008, 6, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Van Haastert, P.J. Transduction of the chemotactic camp signal across the plasma membrane of dictyostelium cells. Experientia 1995, 51, 1144–1154. [Google Scholar] [CrossRef] [PubMed]
- Gancedo, J.M. Biological roles of camp: Variations on a theme in the different kingdoms of life. Biol. Rev. Camb. Philos. Soc. 2013, 88, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, E.W., Jr.; Wosilait, W.D. Inactivation and activation of liver phosphorylase. Nature 1955, 175, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, S.A. Earl sutherland (1915–1974) [corrected] and the discovery of cyclic amp. Perspect. Biol. Med. 2012, 55, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Rodbell, M. The role of hormone receptors and gtp-regulatory proteins in membrane transduction. Nature 1980, 284, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.M.; Maguire, M.E.; Sturgill, T.W.; Biltonen, R.L.; Gilman, A.G. Relationship between the beta-adrenergic receptor and adenylate cyclase. J. Biol. Chem. 1977, 252, 5761–5775. [Google Scholar] [PubMed]
- Soderling, T.R.; Hickenbottom, J.P.; Reimann, E.M.; Hunkeler, F.L.; Walsh, D.A.; Krebs, E.G. Inactivation of glycogen synthetase and activation of phosphorylase kinase by muscle adenosine 3′,5′-monophosphate-dependent protein kinases. J. Biol. Chem. 1970, 245, 6317–6328. [Google Scholar] [PubMed]
- Walsh, D.A.; Perkins, J.P.; Krebs, E.G. An adenosine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. J. Biol. Chem. 1968, 243, 3763–3765. [Google Scholar] [PubMed]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a rap1 guanine-nucleotide-exchange factor directly activated by cyclic amp. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Toki, S.; Canales, J.J.; Harlan, P.; Blumenstiel, J.P.; Chen, E.J.; Bany, I.A.; Mochizuki, N.; Ashbacher, A.; et al. A rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. USA 1998, 95, 13278–13283. [Google Scholar] [CrossRef] [PubMed]
- Fesenko, E.E.; Kolesnikov, S.S.; Lyubarsky, A.L. Induction by cyclic gmp of cationic conductance in plasma membrane of retinal rod outer segment. Nature 1985, 313, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Rall, T.W.; Sutherland, E.W. Formation of a cyclic adenine ribonucleotide by tissue particles. J. Biol. Chem. 1958, 232, 1065–1076. [Google Scholar] [PubMed]
- Weiss, B. Differential activation and inhibition of the multiple forms of cyclic nucleotide phosphodiesterase. Adv. Cycl. Nucleotide Res. 1975, 5, 195–211. [Google Scholar]
- Godinho, R.O.; Duarte, T.; Pacini, E.S. New perspectives in signaling mediated by receptors coupled to stimulatory g protein: The emerging significance of camp e ffl ux and extracellular camp-adenosine pathway. Front. Pharmacol. 2015, 6, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danchin, A. Phylogeny of adenylyl cyclases. Adv. Second Messenger Phosphoprot. Res. 1993, 27, 109–162. [Google Scholar]
- Sunahara, R.K.; Taussig, R. Isoforms of mammalian adenylyl cyclase: Multiplicities of signaling. Mol. Interv. 2002, 2, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Coussen, F.; Bakalyar, H.A.; Tang, W.J.; Feinstein, P.G.; Orth, K.; Slaughter, C.; Reed, R.R.; Gilman, A.G. Adenylyl cyclase amino acid sequence: Possible channel- or transporter-like structure. Science 1989, 244, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Buck, J.; Sinclair, M.L.; Schapal, L.; Cann, M.J.; Levin, L.R. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. Proc. Natl. Acad. Sci. USA 1999, 96, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Omori, K.; Kotera, J. Overview of pdes and their regulation. Circ. Res. 2007, 100, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Scott, J.D. Akap signalling complexes: Focal points in space and time. Nat. Rev. Mol. Cell Biol. 2004, 5, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, V.O.; Bunemann, M.; Hein, L.; Hannawacker, A.; Lohse, M.J. Novel single chain camp sensors for receptor-induced signal propagation. J. Biol. Chem. 2004, 279, 37215–37218. [Google Scholar] [CrossRef] [PubMed]
- Zaccolo, M.; Pozzan, T. Discrete microdomains with high concentration of camp in stimulated rat neonatal cardiac myocytes. Science 2002, 295, 1711–1715. [Google Scholar] [CrossRef] [PubMed]
- Rich, T.C.; Fagan, K.A.; Nakata, H.; Schaack, J.; Cooper, D.M.; Karpen, J.W. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted camp diffusion. J. Gen. Physiol. 2000, 116, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Skalhegg, B.S.; Tasken, K. Specificity in the camp/pka signaling pathway. Differential expression, regulation, and subcellular localization of subunits of pka. Front. Biosci. 1997, 2, d331–d342. [Google Scholar] [CrossRef] [PubMed]
- Sassone-Corsi, P. Transcription factors responsive to camp. Ann. Review Cell Dev. Biol. 1995, 11, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor creb. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Alberts, A.; Brindle, P.; Meinkoth, J.; Feramisco, J.; Deng, T.; Karin, M.; Shenolikar, S.; Montminy, M. Transcriptional attenuation following camp induction requires pp-1-mediated dephosphorylation of creb. Cell 1992, 70, 105–113. [Google Scholar] [CrossRef]
- Wadzinski, B.E.; Wheat, W.H.; Jaspers, S.; Peruski, L.F., Jr.; Lickteig, R.L.; Johnson, G.L.; Klemm, D.J. Nuclear protein phosphatase 2a dephosphorylates protein kinase a-phosphorylated creb and regulates creb transcriptional stimulation. Mol. Cell. Biol. 1993, 13, 2822–2834. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.E. Life without transcriptional control? From fly to man and back again. EMBO J. 2002, 21, 1881–1888. [Google Scholar] [CrossRef] [PubMed]
- Clayton, C.E. Networks of gene expression regulation in trypanosoma brucei. Mol. Biochem. Parasitol. 2014, 195, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Mony, B.M.; MacGregor, P.; Ivens, A.; Rojas, F.; Cowton, A.; Young, J.; Horn, D.; Matthews, K. Genome-wide dissection of the quorum sensing signalling pathway in trypanosoma brucei. Nature 2014, 505, 681–685. [Google Scholar] [CrossRef] [PubMed]
- Sen Santara, S.; Roy, J.; Mukherjee, S.; Bose, M.; Saha, R.; Adak, S. Globin-coupled heme containing oxygen sensor soluble adenylate cyclase in leishmania prevents cell death during hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 16790–16795. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, T.; Liniger, M.; Seebeck, T. The regulatory subunit of a cgmp-regulated protein kinase a of trypanosoma brucei. Eur. J. Biochem. 2001, 268, 6197–6206. [Google Scholar] [CrossRef] [PubMed]
- Gould, M.K.; Bachmaier, S.; Ali, J.A.; Alsford, S.; Tagoe, D.N.; Munday, J.C.; Schnaufer, A.C.; Horn, D.; Boshart, M.; de Koning, H.P. Cyclic amp effectors in african trypanosomes revealed by genome-scale rna interference library screening for resistance to the phosphodiesterase inhibitor cpda. Antimicrob. Agents Chemother. 2013, 57, 4882–4893. [Google Scholar] [CrossRef] [PubMed]
- Jager, A.V.; De Gaudenzi, J.G.; Mild, J.G.; Mc Cormack, B.; Pantano, S.; Altschuler, D.L.; Edreira, M.M. Identification of novel cyclic nucleotide binding proteins in trypanosoma cruzi. Mol. Biochem. Parasitol. 2014, 198, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Serezani, C.H.; Ballinger, M.N.; Aronoff, D.M.; Peters-Golden, M. Cyclic amp: Master regulator of innate immune cell function. Am. J. Respir. Cell Mol. Biol. 2008, 39, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Aronoff, D.M.; Carstens, J.K.; Chen, G.H.; Toews, G.B.; Peters-Golden, M. Short communication: Differences between macrophages and dendritic cells in the cyclic amp-dependent regulation of lipopolysaccharide-induced cytokine and chemokine synthesis. J. Interf. Cytokine Res. 2006, 26, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Van der Pouw Kraan, T.C.; Boeije, L.C.; Smeenk, R.J.; Wijdenes, J.; Aarden, L.A. Prostaglandin-e2 is a potent inhibitor of human interleukin 12 production. J. Exp. Med. 1995, 181, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Jones, S.M.; Phare, S.M.; Coffey, M.J.; Peters-Golden, M.; Brock, T.G. Protein kinase a inhibits leukotriene synthesis by phosphorylation of 5-lipoxygenase on serine 523. J. Biol. Chem. 2004, 279, 41512–41520. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor creb in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed]
- Raker, V.K.; Becker, C.; Steinbrink, K. The camp pathway as therapeutic target in autoimmune and inflammatory diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, M.A.; Muscat, G.E. The nr4a subgroup: Immediate early response genes with pleiotropic physiological roles. Nuclear Recept. Signal. 2006, 4, e002. [Google Scholar] [CrossRef] [PubMed]
- Bystrom, J.; Evans, I.; Newson, J.; Stables, M.; Toor, I.; van Rooijen, N.; Crawford, M.; Colville-Nash, P.; Farrow, S.; Gilroy, D.W. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by camp. Blood 2008, 112, 4117–4127. [Google Scholar] [CrossRef] [PubMed]
- Baumer, W.; Hoppmann, J.; Rundfeldt, C.; Kietzmann, M. Highly selective phosphodiesterase 4 inhibitors for the treatment of allergic skin diseases and psoriasis. Inflamm. Allergy Drug Targets 2007, 6, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Oger, S.; Mehats, C.; Dallot, E.; Cabrol, D.; Leroy, M.J. Evidence for a role of phosphodiesterase 4 in lipopolysaccharide-stimulated prostaglandin e2 production and matrix metalloproteinase-9 activity in human amniochorionic membranes. J. Immunol. 2005, 174, 8082–8089. [Google Scholar] [CrossRef] [PubMed]
- Mary, D.; Aussel, C.; Ferrua, B.; Fehlmann, M. Regulation of interleukin 2 synthesis by camp in human t cells. J. Immunol. 1987, 139, 1179–1184. [Google Scholar] [PubMed]
- Munoz, E.; Zubiaga, A.M.; Merrow, M.; Sauter, N.P.; Huber, B.T. Cholera toxin discriminates between t helper 1 and 2 cells in t cell receptor-mediated activation: Role of camp in t cell proliferation. J. Exp. Med. 1990, 172, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Bopp, T. Cyclic amp represents a crucial component of treg cell-mediated immune regulation. Front. Immunol. 2016, 7, 315. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; SuYang, H.; Erdjument-Bromage, H.; Tempst, P.; Ghosh, S. The transcriptional activity of nf-kappab is regulated by the ikappab-associated pkac subunit through a cyclic amp-independent mechanism. Cell 1997, 89, 413–424. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The regulation of nf-kappab subunits by phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, V.; Parry, G.C.; Cobb, R.R.; de Prost, D.; Mackman, N. Elevated cyclic amp inhibits nf-kappab-mediated transcription in human monocytic cells and endothelial cells. J. Biol. Chem. 1996, 271, 20828–20835. [Google Scholar] [CrossRef] [PubMed]
- Parry, G.C.; Mackman, N. Role of cyclic amp response element-binding protein in cyclic amp inhibition of nf-kappab-mediated transcription. J. Immunol. 1997, 159, 5450–5456. [Google Scholar] [PubMed]
- Baker, D.A.; Kelly, J.M. Structure, function and evolution of microbial adenylyl and guanylyl cyclases. Mol. Microbiol. 2004, 52, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Pezard, C.; Weber, M.; Sirard, J.C.; Berche, P.; Mock, M. Protective immunity induced by bacillus anthracis toxin-deficient strains. Infect. Immun. 1995, 63, 1369–1372. [Google Scholar] [PubMed]
- Agarwal, N.; Lamichhane, G.; Gupta, R.; Nolan, S.; Bishai, W.R. Cyclic amp intoxication of macrophages by a mycobacterium tuberculosis adenylate cyclase. Nature 2009, 460, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Yahr, T.L.; Vallis, A.J.; Hancock, M.K.; Barbieri, J.T.; Frank, D.W. Exoy, an adenylate cyclase secreted by the pseudomonas aeruginosa type iii system. Proc. Natl. Acad. Sci. USA 1998, 95, 13899–13904. [Google Scholar] [CrossRef] [PubMed]
- Sory, M.P.; Cornelis, G.R. Translocation of a hybrid yope-adenylate cyclase from yersinia enterocolitica into hela cells. Mol. Microbiol. 1994, 14, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Coote, J.G. Structural and functional relationships among the rtx toxin determinants of gram-negative bacteria. FEMS Microbiol. Rev. 1992, 8, 137–161. [Google Scholar] [CrossRef] [PubMed]
- Krueger, K.M.; Barbieri, J.T. The family of bacterial adp-ribosylating exotoxins. Clin. Microbiol. Rev. 1995, 8, 34–47. [Google Scholar] [PubMed]
- Paindavoine, P.; Rolin, S.; Van Assel, S.; Geuskens, M.; Jauniaux, J.C.; Dinsart, C.; Huet, G.; Pays, E. A gene from the variant surface glycoprotein expression site encodes one of several transmembrane adenylate cyclases located on the flagellum of trypanosoma brucei. Mol. Cell. Biol. 1992, 12, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Salmon, D.; Bachmaier, S.; Krumbholz, C.; Kador, M.; Gossmann, J.A.; Uzureau, P.; Pays, E.; Boshart, M. Cytokinesis of trypanosoma brucei bloodstream forms depends on expression of adenylyl cyclases of the esag4 or esag4-like subfamily. Mol. Microbiol. 2012, 84, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Salmon, D.; Vanwalleghem, G.; Morias, Y.; Denoeud, J.; Krumbholz, C.; Lhomme, F.; Bachmaier, S.; Kador, M.; Gossmann, J.; Dias, F.B.; et al. Adenylate cyclases of trypanosoma brucei inhibit the innate immune response of the host. Science 2012, 337, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Magez, S.; Geuskens, M.; Beschin, A.; del Favero, H.; Verschueren, H.; Lucas, R.; Pays, E.; de Baetselier, P. Specific uptake of tumor necrosis factor-alpha is involved in growth control of trypanosoma brucei. J. Cell Biol. 1997, 137, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Magez, S.; De Leys, R.; Fransen, L.; Scheerlinck, J.P.; Rampelberg, M.; Sablon, E.; De Baetselier, P. Mapping the lectin-like activity of tumor necrosis factor. Science 1994, 263, 814–817. [Google Scholar] [CrossRef] [PubMed]
- McDonough, K.A.; Rodriguez, A. The myriad roles of cyclic amp in microbial pathogens: From signal to sword. Nat. Rev. Microbiol. 2011, 10, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Langousis, G.; Hill, K.L. Motility and more: The flagellum of trypanosoma brucei. Nat. Rev. Microbiol. 2014, 12, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y.; Wang, C.; Yuan, Y.A.; He, C.Y. An intracellular membrane junction consisting of flagellum adhesion glycoproteins links flagellum biogenesis to cell morphogenesis in trypanosoma brucei. J. Cell Sci. 2013, 126, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Ralston, K.S.; Lerner, A.G.; Diener, D.R.; Hill, K.L. Flagellar motility contributes to cytokinesis in trypanosoma brucei and is modulated by an evolutionarily conserved dynein regulatory system. Eukaryotic Cell 2006, 5, 696–711. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.; Worthey, E.A.; Ward, P.N.; Mottram, J.C. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, trypanosoma brucei and trypanosoma cruzi. BMC Genom. 2005, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Nett, I.R.; Martin, D.M.; Miranda-Saavedra, D.; Lamont, D.; Barber, J.D.; Mehlert, A.; Ferguson, M.A. The phosphoproteome of bloodstream form trypanosoma brucei, causative agent of african sleeping sickness. Mol. Cell. Proteom. 2009, 8, 1527–1538. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.; Ruben, L. Pathways involved in environmental sensing in trypanosomatids. Parasitol. Today 2000, 16, 56–62. [Google Scholar] [CrossRef]
- Garbers, D.L.; Koesling, D.; Schultz, G. Guanylyl cyclase receptors. Mol. Biol. Cell 1994, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Linder, J.U. Class iii adenylyl cyclases: Molecular mechanisms of catalysis and regulation. Cell. Mol. Life Sci. 2006, 63, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.C.; Sprang, S.R. Structures, mechanism, regulation and evolution of class iii nucleotidyl cyclases. Revi. Physiol. Biochem. Pharmacol. 2006, 157, 105–140. [Google Scholar]
- Zhang, G.; Liu, Y.; Ruoho, A.E.; Hurley, J.H. Structure of the adenylyl cyclase catalytic core. Nature 1997, 386, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, J.J.; Sprang, S.R. The structure, catalytic mechanism and regulation of adenylyl cyclase. Curr. Opin. Struct. Biol. 1998, 8, 713–719. [Google Scholar] [CrossRef]
- Tesmer, J.J.; Sunahara, R.K.; Johnson, R.A.; Gosselin, G.; Gilman, A.G.; Sprang, S.R. Two-metal-ion catalysis in adenylyl cyclase. Science 1999, 285, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Bieger, B.; Essen, L.O. Structural analysis of adenylate cyclases from trypanosoma brucei in their monomeric state. EMBO J. 2001, 20, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Rolin, S.; Hanocq-Quertier, J.; Paturiaux-Hanocq, F.; Nolan, D.; Salmon, D.; Webb, H.; Carrington, M.; Voorheis, P.; Pays, E. Simultaneous but independent activation of adenylate cyclase and glycosylphosphatidylinositol-phospholipase c under stress conditions in trypanosoma brucei. J. Biol. Chem. 1996, 271, 10844–10852. [Google Scholar] [CrossRef] [PubMed]
- Tesmer, J.J.; Dessauer, C.W.; Sunahara, R.K.; Murray, L.D.; Johnson, R.A.; Gilman, A.G.; Sprang, S.R. Molecular basis for p-site inhibition of adenylyl cyclase. Biochemistry 2000, 39, 14464–14471. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.A.; Shoshani, I. Inhibition of bordetella pertussis and bacillus anthracis adenylyl cyclases by polyadenylate and “p”-site agonists. J. Biol. Chem. 1990, 265, 19035–19039. [Google Scholar] [PubMed]
- Voorheis, H.P.; Martin, B.R. Characteristics of the calcium-mediated mechanism activating adenylate cyclase in trypanosoma brucei. Eur. J. Biochem. 1981, 116, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Rolin, S.; Halleux, S.; Van Sande, J.; Dumont, J.; Pays, E.; Steinert, M. Stage-specific adenylate cyclase activity in trypanosoma brucei. Exp. Parasitol. 1990, 71, 350–352. [Google Scholar] [CrossRef]
- Hamedi, A.; Botelho, L.; Britto, C.; Fragoso, S.P.; Umaki, A.C.; Goldenberg, S.; Bottu, G.; Salmon, D. In vitro metacyclogenesis of trypanosoma cruzi induced by starvation correlates with a transient adenylyl cyclase stimulation as well as with a constitutive upregulation of adenylyl cyclase expression. Mol. Biochem. Parasitol. 2015, 200, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.C.; Batista, M.; da Cunha, E.S.; Lucena, A.C.R.; Lima, C.V.P.; Sousa, K.; Krieger, M.A.; Marchini, F.K. Quantitative proteome and phosphoproteome analyses highlight the adherent population during trypanosoma cruzi metacyclogenesis. Sci. Rep. 2017, 7, 9899. [Google Scholar] [CrossRef] [PubMed]
- Potter, L.R.; Hunter, T. Phosphorylation of the kinase homology domain is essential for activation of the a-type natriuretic peptide receptor. Mol. Cell. Biol. 1998, 18, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Naula, C.; Schaub, R.; Leech, V.; Melville, S.; Seebeck, T. Spontaneous dimerization and leucine-zipper induced activation of the recombinant catalytic domain of a new adenylyl cyclase of trypanosoma brucei, gresag4.4b. Mol. Biochem. Parasitol. 2001, 112, 19–28. [Google Scholar] [CrossRef]
- Gould, M.K.; de Koning, H.P. Cyclic-nucleotide signalling in protozoa. FEMS Microbiol. Rev. 2011, 35, 515–541. [Google Scholar] [CrossRef] [PubMed]
- Nolan, D.P.; Rolin, S.; Rodriguez, J.R.; Van Den Abbeele, J.; Pays, E. Slender and stumpy bloodstream forms of trypanosoma brucei display a differential response to extracellular acidic and proteolytic stress. Eur. J. Biochem. 2000, 267, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Saada, E.A.; Kabututu, Z.P.; Lopez, M.; Shimogawa, M.M.; Langousis, G.; Oberholzer, M.; Riestra, A.; Jonsson, Z.O.; Wohlschlegel, J.A.; Hill, K.L. Insect stage-specific receptor adenylate cyclases are localized to distinct subdomains of the trypanosoma brucei flagellar membrane. Eukaryot Cell 2014, 13, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Garbers, D.L.; Chrisman, T.D.; Wiegn, P.; Katafuchi, T.; Albanesi, J.P.; Bielinski, V.; Barylko, B.; Redfield, M.M.; Burnett, J.C., Jr. Membrane guanylyl cyclase receptors: An update. Trends Endocrinol. Metab. 2006, 17, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Alexandre, S.; Paindavoine, P.; Tebabi, P.; Pays, A.; Halleux, S.; Steinert, M.; Pays, E. Differential expression of a family of putative adenylate/guanylate cyclase genes in trypanosoma brucei. Mol. Biochem. Parasitol. 1990, 43, 279–288. [Google Scholar] [CrossRef]
- Tam, R.; Saier, M.H., Jr. Structural, functional, and evolutionary relationships among extracellular solute-binding receptors of bacteria. Microbiol. Rev. 1993, 57, 320–346. [Google Scholar] [PubMed]
- Felder, C.B.; Graul, R.C.; Lee, A.Y.; Merkle, H.P.; Sadee, W. The venus flytrap of periplasmic binding proteins: An ancient protein module present in multiple drug receptors. AAPS PharmSci 1999, 1, E2. [Google Scholar] [CrossRef] [PubMed]
- Herrou, J.; Bompard, C.; Wintjens, R.; Dupre, E.; Willery, E.; Villeret, V.; Locht, C.; Antoine, R.; Jacob-Dubuisson, F. Periplasmic domain of the sensor-kinase bvgs reveals a new paradigm for the venus flytrap mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 17351–17355. [Google Scholar] [CrossRef] [PubMed]
- Pin, J.P.; Kniazeff, J.; Liu, J.; Binet, V.; Goudet, C.; Rondard, P.; Prezeau, L. Allosteric functioning of dimeric class c g-protein-coupled receptors. FEBS J. 2005, 272, 2947–2955. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Chow, D.; Martick, M.M.; Garcia, K.C. Allosteric activation of a spring-loaded natriuretic peptide receptor dimer by hormone. Science 2001, 293, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, F. Structural insights into the ligand binding domains of membrane bound guanylyl cyclases and natriuretic peptide receptors. J. Mol. Biol. 2001, 311, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Beschin, A.; Van Den Abbeele, J.; De Baetselier, P.; Pays, E. African trypanosome control in the insect vector and mammalian host. Trends Parasitol. 2014, 30, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Szurmant, H.; Ordal, G.W. Diversity in chemotaxis mechanisms among the bacteria and archaea. Microbiol. Mol. Biol. Rev. 2004, 68, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Swaney, K.F.; Huang, C.H.; Devreotes, P.N. Eukaryotic chemotaxis: A network of signaling pathways controls motility, directional sensing, and polarity. Ann. Rev. Biophys. 2010, 39, 265–289. [Google Scholar] [CrossRef] [PubMed]
- Ralston, K.S.; Kabututu, Z.P.; Melehani, J.H.; Oberholzer, M.; Hill, K.L. The trypanosoma brucei flagellum: Moving parasites in new directions. Ann. Rev. Microbiol. 2009, 63, 335–362. [Google Scholar] [CrossRef] [PubMed]
- Tyler, K.M.; Fridberg, A.; Toriello, K.M.; Olson, C.L.; Cieslak, J.A.; Hazlett, T.L.; Engman, D.M. Flagellar membrane localization via association with lipid rafts. J. Cell Sci. 2009, 122, 859–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.I.; Olson, C.L.; Engman, D.M. The lipid raft proteome of african trypanosomes contains many flagellar proteins. Pathogens 2017, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Hertz-Fowler, C.; Ersfeld, K.; Gull, K. Cap5.5, a life-cycle-regulated, cytoskeleton-associated protein is a member of a novel family of calpain-related proteins in trypanosoma brucei. Mol. Biochem. Parasitol. 2001, 116, 25–34. [Google Scholar] [CrossRef]
- Liu, W.; Apagyi, K.; McLeavy, L.; Ersfeld, K. Expression and cellular localisation of calpain-like proteins in trypanosoma brucei. Mol. Biochem. Parasitol. 2010, 169, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Clynes, M.A.; Masada, N.; Ciruela, A.; Ayling, L.J.; Wachten, S.; Cooper, D.M. Insights into the residence in lipid rafts of adenylyl cyclase ac8 and its regulation by capacitative calcium entry. Am. J. Physiol. Cell Physiol. 2009, 296, C607–C619. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, M.; Marti, G.; Baresic, M.; Kunz, S.; Hemphill, A.; Seebeck, T. The trypanosoma brucei camp phosphodiesterases tbrpdeb1 and tbrpdeb2: Flagellar enzymes that are essential for parasite virulence. FASEB J. 2007, 21, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Baron, D.M.; Ralston, K.S.; Kabututu, Z.P.; Hill, K.L. Functional genomics in trypanosoma brucei identifies evolutionarily conserved components of motile flagella. J. Cell Sci. 2007, 120, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, A.R.; Diener, D.R.; Rosenbaum, J.L.; Sale, W.S. Flagellar radial spoke protein 3 is an a-kinase anchoring protein (akap). J. Cell Biol. 2001, 153, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Kramer, S. Characterization of a Pka-Like Kinase from Trypanosoma Brucei. Ph.D. Thesis, LMU München, München, Germany, 2005. [Google Scholar]
- Oberholzer, M.; Langousis, G.; Nguyen, H.T.; Saada, E.A.; Shimogawa, M.M.; Jonsson, Z.O.; Nguyen, S.M.; Wohlschlegel, J.A.; Hill, K.L. Independent analysis of the flagellum surface and matrix proteomes provides insight into flagellum signaling in mammalian-infectious trypanosoma brucei. Mol. Cell. Proteom. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Engstler, M.; Boshart, M. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in trypanosoma brucei. Genes Dev. 2004, 18, 2798–2811. [Google Scholar] [CrossRef] [PubMed]
- Bridges, D.J.; Pitt, A.R.; Hanrahan, O.; Brennan, K.; Voorheis, H.P.; Herzyk, P.; de Koning, H.P.; Burchmore, R.J. Characterisation of the plasma membrane subproteome of bloodstream form trypanosoma brucei. Proteomics 2008, 8, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Broadhead, R.; Dawe, H.R.; Farr, H.; Griffiths, S.; Hart, S.R.; Portman, N.; Shaw, M.K.; Ginger, M.L.; Gaskell, S.J.; McKean, P.G.; et al. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 2006, 440, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Mancini, P.E.; Patton, C.L. Cyclic 3′,5′-adenosine monophosphate levels during the developmental cycle of trypanosoma brucei brucei in the rat. Mol. Biochem. Parasitol. 1981, 3, 19–31. [Google Scholar] [CrossRef]
- Rolin, S.; Paindavoine, P.; Hanocq-Quertier, J.; Hanocq, F.; Claes, Y.; Le Ray, D.; Overath, P.; Pays, E. Transient adenylate cyclase activation accompanies differentiation of trypanosoma brucei from bloodstream to procyclic forms. Mol. Biochem. Parasitol. 1993, 61, 115–125. [Google Scholar] [CrossRef]
- Gonzales-Perdomo, M.; Romero, P.; Goldenberg, S. Cyclic amp and adenylate cyclase activators stimulate trypanosoma cruzi differentiation. Exp. Parasitol. 1988, 66, 205–212. [Google Scholar] [CrossRef]
- Rangel-Aldao, R.; Triana, F.; Fernandez, V.; Comach, G.; Abate, T.; Montoreano, R. Cyclic amp as an inducer of the cell differentiation of trypanosoma cruzi. Biochem. Int. 1988, 17, 337–344. [Google Scholar] [PubMed]
- Vassella, E.; Reuner, B.; Yutzy, B.; Boshart, M. Differentiation of african trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the camp pathway. J. Cell Sci. 1997, 110 Pt 21, 2661–2671. [Google Scholar] [PubMed]
- Bhattacharya, A.; Biswas, A.; Das, P.K. Role of intracellular camp in differentiation-coupled induction of resistance against oxidative damage in leishmania donovani. Free Radic. Biol. Med. 2008, 44, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Laxman, S.; Riechers, A.; Sadilek, M.; Schwede, F.; Beavo, J.A. Hydrolysis products of camp analogs cause transformation of trypanosoma brucei from slender to stumpy-like forms. Proc. Natl. Acad. Sci. USA 2006, 103, 19194–19199. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, M.; Lopez, M.A.; McLelland, B.T.; Hill, K.L. Social motility in african trypanosomes. PLoS Pathog. 2010, 6, e1000739. [Google Scholar] [CrossRef] [PubMed]
- Imhof, S.; Knusel, S.; Gunasekera, K.; Vu, X.L.; Roditi, I. Social motility of african trypanosomes is a property of a distinct life-cycle stage that occurs early in tsetse fly transmission. PLoS Pathog. 2014, 10, e1004493. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.A.; Saada, E.A.; Hill, K.L. Insect stage-specific adenylate cyclases regulate social motility in african trypanosomes. Eukaryotic Cell 2015, 14, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, M.; Saada, E.A.; Hill, K.L. Cyclic amp regulates social behavior in african trypanosomes. mBio 2015, 6, e01954-14. [Google Scholar] [CrossRef] [PubMed]
- Saada, E.A.; DeMarco, S.F.; Shimogawa, M.M.; Hill, K.L. “With a little help from my friends”-social motility in trypanosoma brucei. PLoS Pathog. 2015, 11, e1005272. [Google Scholar] [CrossRef] [PubMed]
- Eliaz, D.; Kannan, S.; Shaked, H.; Arvatz, G.; Tkacz, I.D.; Binder, L.; Waldman Ben-Asher, H.; Okalang, U.; Chikne, V.; Cohen-Chalamish, S.; et al. Exosome secretion affects social motility in trypanosoma brucei. PLoS Pathog. 2017, 13, e1006245. [Google Scholar] [CrossRef] [PubMed]
- Imhof, S.; Vu, X.L.; Butikofer, P.; Roditi, I. A glycosylation mutant of trypanosoma brucei links social motility defects in vitro to impaired colonization of tsetse flies in vivo. Eukaryotic Cell 2015, 14, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Tebabi, P.; Pays, A.; Coquelet, H.; Revelard, P.; Salmon, D.; Steinert, M. The genes and transcripts of an antigen gene expression site from t. Brucei. Cell 1989, 57, 835–845. [Google Scholar] [CrossRef]
- Alexandre, S.; Paindavoine, P.; Hanocq-Quertier, J.; Paturiaux-Hanocq, F.; Tebabi, P.; Pays, E. Families of adenylate cyclase genes in trypanosoma brucei. Mol. Biochem. Parasitol. 1996, 77, 173–182. [Google Scholar] [CrossRef]
- Ross, D.T.; Raibaud, A.; Florent, I.C.; Sather, S.; Gross, M.K.; Storm, D.R.; Eisen, H. The trypanosome vsg expression site encodes adenylate cyclase and a leucine-rich putative regulatory gene. EMBO J. 1991, 10, 2047–2053. [Google Scholar] [PubMed]
- Palmer, G.H.; Bankhead, T.; Seifert, H.S. Antigenic variation in bacterial pathogens. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Dempsey, W.L.; Mansfield, J.M. Lymphocyte function in experimental african trypanosomiasis. V. Role of antibody and the mononuclear phagocyte system in variant-specific immunity. J. Immunol. 1983, 130, 405–411. [Google Scholar] [PubMed]
- Hertz, C.J.; Filutowicz, H.; Mansfield, J.M. Resistance to the african trypanosomes is ifn-gamma dependent. J. Immunol. 1998, 161, 6775–6783. [Google Scholar] [PubMed]
- De Muylder, G.; Daulouede, S.; Lecordier, L.; Uzureau, P.; Morias, Y.; Van Den Abbeele, J.; Caljon, G.; Herin, M.; Holzmuller, P.; Semballa, S.; et al. A trypanosoma brucei kinesin heavy chain promotes parasite growth by triggering host arginase activity. PLoS Pathog. 2013, 9, e1003731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stijlemans, B.; Caljon, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Magez, S.; De Trez, C. Immune evasion strategies of trypanosoma brucei within the mammalian host: Progression to pathogenicity. Front. Immunol. 2016, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Vanwalleghem, G.; Morias, Y.; Beschin, A.; Szymkowski, D.E.; Pays, E. Trypanosoma brucei growth control by tnf in mammalian host is independent of the soluble form of the cytokine. Sci. Rep. 2017, 7, 6165. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Bull, H.; Zhou, X.; Tabel, H. Intradermal infections of mice by low numbers of african trypanosomes are controlled by innate resistance but enhance susceptibility to reinfection. J. Infect. Dis. 2011, 203, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Szempruch, A.J.; Sykes, S.E.; Kieft, R.; Dennison, L.; Becker, A.C.; Gartrell, A.; Martin, W.J.; Nakayasu, E.S.; Almeida, I.C.; Hajduk, S.L.; et al. Extracellular vesicles from trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell 2016, 164, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Wall, E.A.; Zavzavadjian, J.R.; Chang, M.S.; Randhawa, B.; Zhu, X.; Hsueh, R.C.; Liu, J.; Driver, A.; Bao, X.R.; Sternweis, P.C.; et al. Suppression of lps-induced tnf-alpha production in macrophages by camp is mediated by pka-akap95-p105. Sci. Signal. 2009, 2, ra28. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Das, S.; Smith, T.F.; Samuelson, J. Trichomonas transmembrane cyclases result from massive gene duplication and concomitant development of pseudogenes. PLoS Negl. Trop. Dis. 2010, 4, e782. [Google Scholar] [CrossRef] [PubMed]
- Ratier, L.; Urrutia, M.; Paris, G.; Zarebski, L.; Frasch, A.C.; Goldbaum, F.A. Relevance of the diversity among members of the trypanosoma cruzi trans-sialidase family analyzed with camelids single-domain antibodies. PLoS ONE 2008, 3, e3524. [Google Scholar] [CrossRef] [PubMed]
- Mayer, D.C.; Mu, J.B.; Feng, X.; Su, X.Z.; Miller, L.H. Polymorphism in a plasmodium falciparum erythrocyte-binding ligand changes its receptor specificity. J. Exp. Med. 2002, 196, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Bitter, W.; Gerrits, H.; Kieft, R.; Borst, P. The role of transferrin-receptor variation in the host range of trypanosoma brucei. Nature 1998, 391, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Lips, S.; Nolan, D.; Vanhamme, L.; Perez-Morga, D. The vsg expression sites of trypanosoma brucei: Multipurpose tools for the adaptation of the parasite to mammalian hosts. Mol. Biochem. Parasitol. 2001, 114, 1–16. [Google Scholar] [CrossRef]
- Jansen, C.; Wang, H.; Kooistra, A.J.; de Graaf, C.; Orrling, K.M.; Tenor, H.; Seebeck, T.; Bailey, D.; de Esch, I.J.; Ke, H.; et al. Discovery of novel trypanosoma brucei phosphodiesterase b1 inhibitors by virtual screening against the unliganded tbrpdeb1 crystal structure. J. Med. Chem. 2013, 56, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Makin, L.; Gluenz, E. Camp signalling in trypanosomatids: Role in pathogenesis and as a drug target. Trends Parasitol. 2015, 31, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.; Cooper, D.M. Live-cell imaging of camp dynamics. Nat. Methods 2008, 5, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Karpen, J.W.; Rich, T.C. High-resolution measurements of cyclic adenosine monophosphate signals in 3d microdomains. Methods Mol. Biol. 2005, 307, 15–26. [Google Scholar] [PubMed]
- Voorheis, H.P.; Martin, B.R. ‘Swell dialysis‘ demonstrates that adenylate cyclase in trypanosoma brucei is regulated by calcium ions. Eur. J. Biochem. 1980, 113, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Jansen, V.; Alvarez, L.; Balbach, M.; Strunker, T.; Hegemann, P.; Kaupp, U.B.; Wachten, D. Controlling fertilization and camp signaling in sperm by optogenetics. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmon, D. Adenylate Cyclases of Trypanosoma brucei, Environmental Sensors and Controllers of Host Innate Immune Response. Pathogens 2018, 7, 48. https://doi.org/10.3390/pathogens7020048
Salmon D. Adenylate Cyclases of Trypanosoma brucei, Environmental Sensors and Controllers of Host Innate Immune Response. Pathogens. 2018; 7(2):48. https://doi.org/10.3390/pathogens7020048
Chicago/Turabian StyleSalmon, Didier. 2018. "Adenylate Cyclases of Trypanosoma brucei, Environmental Sensors and Controllers of Host Innate Immune Response" Pathogens 7, no. 2: 48. https://doi.org/10.3390/pathogens7020048
APA StyleSalmon, D. (2018). Adenylate Cyclases of Trypanosoma brucei, Environmental Sensors and Controllers of Host Innate Immune Response. Pathogens, 7(2), 48. https://doi.org/10.3390/pathogens7020048