Molecular Characterisation of Equine Herpesvirus 1 Isolates from Cases of Abortion, Respiratory and Neurological Disease in Ireland between 1990 and 2017

 , ,
, ,
Abstract
:1. Introduction
2. Results
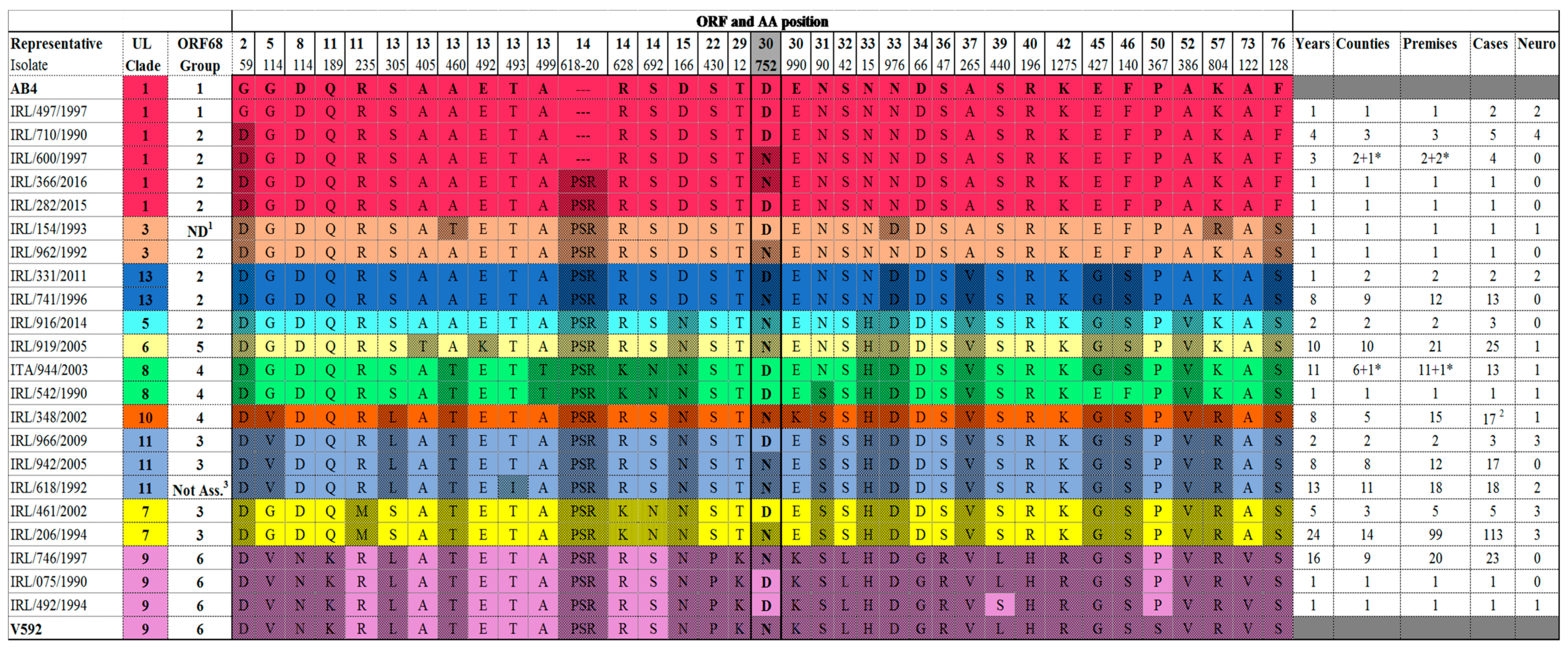
2.1. Multi-Locus Analysis
2.2. ORF68
3. Discussion
4. Materials and Methods
4.1. Viruses
4.2. Extraction of DNA
4.3. PCR of multiple loci of EHV-1
4.4. Multi-Locus Sequence Analysis
4.5. PCR of EHV-1 ORF68
4.6. ORF68 Sequence Analysis
4.7. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lunn, D.P.; Davis-Poynter, N.; Flaminio, M.J.; Horohov, D.W.; Osterrieder, K.; Pusterla, N.; Townsend, H.G. Equine herpesvirus-1 consensus statement. J. Vet. Intern. Med. 2009, 23, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Crowhurst, F.A.; Dickinson, G.; Burrows, R. An outbreak of paresis in mares and geldings associated with equid herpesvirus 1. Vet. Rec. 1981, 109, 527–528. [Google Scholar] [PubMed]
- McCartan, C.G.; Russell, M.M.; Wood, J.L.; Mumford, J.A. Clinical, serological and virological characteristics of an outbreak of paresis and neonatal foal disease due to equine herpesvirus-1 on a stud farm. Vet. Rec. 1995, 136, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mumford, J.A.; Edington, N. EHV1 and equine paresis. Vet. Rec. 1980, 106, 277. [Google Scholar] [CrossRef] [PubMed]
- Whitwell, K.E.; Blunden, A.S. Pathological findings in horses dying during an outbreak of the paralytic form of Equid herpesvirus type 1 (EHV-1) infection. Equine Vet. J. 1992, 24, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, A.; Franz, M.; Schattschneider, A.; Damiani, A.M.; Bischofberger, S.; Osterrieder, N.; Greenwood, A.D. Long term stability and infectivity of herpesviruses in water. Sci. Rep. 2017, 7, 46559. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.R.; Heldens, J. Equine herpesviruses 1 (EHV-1) and 4 (EHV-4)—Epidemiology, disease and immunoprophylaxis: A brief review. Vet. J. 2005, 170, 14–23. [Google Scholar] [CrossRef]
- Kydd, J.H.; Smith, K.C.; Hannant, D.; Livesay, G.J.; Mumford, J.A. Distribution of equid herpesvirus-1 (EHV-1) in respiratory tract associated lymphoid tissue: Implications for cellular immunity. Equine Vet. J. 1994, 26, 470–473. [Google Scholar] [CrossRef]
- Smith, D.J.; Hamblin, A.S.; Edington, N. Infection of endothelial cells with equine herpesvirus-1 (EHV-1) occurs where there is activation of putative adhesion molecules: A mechanism for transfer of virus. Equine Vet. J. 2001, 33, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Edington, N.; Bridges, C.G.; Patel, J.R. Endothelial cell infection and thrombosis in paralysis caused by equid herpesvirus-1: Equine stroke. Arch. Virol. 1986, 90, 111–124. [Google Scholar] [CrossRef] [PubMed]
- USDA-APHIS 2007. Equine Herpesvirus Myeloencephalopathy: A Potentially Emerging Disease. Available online: http://www.aphis.usda.gov/vs/ceah/cei/taf/emergingdiseasenoticefiles/EHV-1final.pdf (accessed on 29 August 2018).
- OIE. OIE-Listed Diseases, Infections and Infestations in Force in 2018. Available online: http://www.oie.int/animal-health-in-the-world/oie-listed-diseases-2018/ (accessed on 1 October 2018).
- Telford, E.A.; Watson, M.S.; McBride, K.; Davison, A.J. The DNA sequence of equine herpesvirus-1. Virology 1992, 189, 304–316. [Google Scholar] [CrossRef]
- Shakya, A.K.; O’Callaghan, D.J.; Kim, S.K. Comparative Genomic Sequencing and Pathogenic Properties of Equine Herpesvirus 1 KyA and RacL11. Front. Vet. Sci. 2017, 4, 211. [Google Scholar] [CrossRef] [PubMed]
- Mumford, J.A.; Hannant, D.; Jessett, D.M.; O’Neill, T.; Smith, K.C.; Ostlund, E.N. Abortigenic and neurological disease caused by experimental infection with equid herpesvirus-1. In Proceedings of the Seventh International Conference on Equine Infectious Diseases, Newmarket, UK, 8–11 June 1994; pp. 261–275. [Google Scholar]
- Gardiner, D.W.; Lunn, D.P.; Goehring, L.S.; Chiang, Y.W.; Cook, C.; Osterrieder, N.; McCue, P.; Del Piero, F.; Hussey, S.B.; Hussey, G.S. Strain impact on equine herpesvirus type 1 (EHV-1) abortion models: Viral loads in fetal and placental tissues and foals. Vaccine 2012, 30, 6564–6572. [Google Scholar] [CrossRef]
- Smith, K.C.; Whitwell, K.E.; Binns, M.M.; Dolby, C.A.; Hannant, D.; Mumford, J.A. Abortion of virologically negative foetuses following experimental challenge of pregnant pony mares with equid herpesvirus 1. Equine Vet. J. 1992, 24, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.C.; Whitwell, K.E.; Mumford, J.A.; Hannant, D.; Blunden, A.S.; Tearle, J.P. Virulence of the V592 isolate of equid herpesvirus-1 in ponies. J. Comp. Pathol. 2000, 122, 288–297. [Google Scholar] [CrossRef]
- Mumford, J.A.; Rossdale, P.D.; Jessett, D.M.; Gann, S.J.; Ousey, J.; Cook, R.F. Serological and virological investigations of an equid herpesvirus 1 (EHV-1) abortion storm on a stud farm in 1985. J. Reprod. Fertil. Suppl. 1987, 35, 509–518. [Google Scholar]
- Nugent, J.; Birch-Machin, I.; Smith, K.C.; Mumford, J.A.; Swann, Z.; Newton, J.R.; Bowden, R.J.; Allen, G.P.; Davis-Poynter, N. Analysis of equid herpesvirus 1 strain variation reveals a point mutation of the DNA polymerase strongly associated with neuropathogenic versus nonneuropathogenic disease outbreaks. J. Virol. 2006, 80, 4047–4060. [Google Scholar] [CrossRef] [PubMed]
- Goodman, L.B.; Loregian, A.; Perkins, G.A.; Nugent, J.; Buckles, E.L.; Mercorelli, B.; Kydd, J.H.; Palu, G.; Smith, K.C.; Osterrieder, N.; et al. A point mutation in a herpesvirus polymerase determines neuropathogenicity. PLoS Pathog. 2007, 3, e160. [Google Scholar] [CrossRef]
- Van de Walle, G.R.; Goupil, R.; Wishon, C.; Damiani, A.; Perkins, G.A.; Osterrieder, N. A single-nucleotide polymorphism in a herpesvirus DNA polymerase is sufficient to cause lethal neurological disease. J. Infect. Dis. 2009, 200, 20–25. [Google Scholar] [CrossRef]
- Cuxson, J.L.; Hartley, C.A.; Ficorilli, N.P.; Symes, S.J.; Devlin, J.M.; Gilkerson, J.R. Comparing the genetic diversity of ORF30 of Australian isolates of 3 equid alphaherpesviruses. Vet. Microbiol. 2014, 169, 50–57. [Google Scholar] [CrossRef]
- Fritsche, A.K.; Borchers, K. Detection of neuropathogenic strains of Equid Herpesvirus 1 (EHV-1) associated with abortions in Germany. Vet. Microbiol. 2011, 147, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.A.; Goodman, L.B.; Tsujimura, K.; Van de Walle, G.R.; Kim, S.G.; Dubovi, E.J.; Osterrieder, N. Investigation of the prevalence of neurologic equine herpes virus type 1 (EHV-1) in a 23-year retrospective analysis (1984–2007). Vet. Microbiol. 2009, 139, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Pronost, S.; Cook, R.F.; Fortier, G.; Timoney, P.J.; Balasuriya, U.B.R. Relationship between equine herpesvirus-1 myeloencephalopathy and viral genotype. Equine Vet. J. 2010, 42, 672–674. [Google Scholar] [CrossRef]
- Meindl, A.; Osterrieder, N. The equine herpesvirus 1 Us2 homolog encodes a nonessential membrane-associated virion component. J. Virol. 1999, 73, 3430–3437. [Google Scholar]
- Anagha, G.; Gulati, B.R.; Riyesh, T.; Virmani, N. Genetic characterization of equine herpesvirus 1 isolates from abortion outbreaks in India. Arch. Virol. 2017, 162, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Balint, A.; Dan, A.; Palfi, V. Molecular characterisation of the ORF68 region of equine herpesvirus-1 strains isolated from aborted fetuses in Hungary between 1977 and 2008. Acta Vet. Hung. 2012, 60, 175–187. [Google Scholar] [CrossRef]
- Negussie, H.; Gizaw, D.; Tessema, T.S.; Nauwynck, H.J. Equine Herpesvirus-1 Myeloencephalopathy, an Emerging Threat of Working Equids in Ethiopia. Transbound. Emerg. Dis. 2017, 64, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, K.; Dunowska, M.; Hills, S.F.; Rola, J. Genetic characterization of equid herpesvirus type 1 from cases of abortion in Poland. Arch. Virol. 2017, 162, 2329–2335. [Google Scholar] [CrossRef]
- Tsujimura, K.; Oyama, T.; Katayama, Y.; Muranaka, M.; Bannai, H.; Nemoto, M.; Yamanaka, T.; Kondo, T.; Kato, M.; Matsumura, T. Prevalence of Equine Herpesvirus Type 1 Strains of Neuropathogenic Genotype in a Major Breeding Area of Japan. J. Vet. Med. Sci. 2011, 73, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Vaz, P.K.; Horsington, J.; Hartley, C.A.; Browning, G.F.; Ficorilli, N.P.; Studdert, M.J.; Gilkerson, J.R.; Devlin, J.M. Evidence of widespread natural recombination among field isolates of equine herpesvirus 4 but not among field isolates of equine herpesvirus 1. J. Gen. Virol. 2016, 97, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Bryant, N.A.; Wilkie, G.S.; Russell, C.A.; Compston, L.; Grafham, D.; Clissold, L.; McLay, K.; Medcalf, L.; Newton, R.; Davison, A.J.; et al. Genetic diversity of equine herpesvirus 1 isolated from neurological, abortigenic and respiratory disease outbreaks. Transbound. Emerg. Dis. 2018, 65, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Loncoman, C.A.; Vaz, P.K.; Coppo, M.J.C.; Hartley, C.A.; Morera, F.J.; Browning, G.F.; Devlin, J.M. Natural recombination in alphaherpesviruses: Insights into viral evolution through full genome sequencing and sequence analysis. Infect. Genet. Evol. 2017, 49, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Pagamjav, O.; Sakata, T.; Matsumura, T.; Yamaguchi, T.; Fukushi, H. Natural recombinant between equine herpesviruses 1 and 4 in the ICP4 gene. Microbiol. Immunol. 2005, 49, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.D.; Tsangaras, K.; Ho, S.Y.; Szentiks, C.A.; Nikolin, V.M.; Ma, G.; Damiani, A.; East, M.L.; Lawrenz, A.; Hofer, H.; et al. A potentially fatal mix of herpes in zoos. Curr. Biol. CB 2012, 22, 1727–1731. [Google Scholar] [CrossRef] [PubMed]
- Jensen, N.J.; Rivailler, P.; Tseng, H.F.; Quinlivan, M.L.; Radford, K.; Folster, J.; Harpaz, R.; LaRussa, P.; Jacobsen, S.; Scott Schmid, D. Revisiting the genotyping scheme for varicella-zoster viruses based on whole-genome comparisons. J. Gen. Virol. 2017, 98, 1434–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norberg, P. Divergence and genotyping of human alpha-herpesviruses: An overview. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2010, 10, 14–25. [Google Scholar] [CrossRef]
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 2017, 15, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Allen, G.P. Development of a real-time polymerase chain reaction assay for rapid diagnosis of neuropathogenic strains of equine herpesvirus-1. J. Vet. Diagn. Investig. 2007, 19, 69–72. [Google Scholar] [CrossRef]
- Smith, K.L.; Li, Y.; Breheny, P.; Cook, R.F.; Henney, P.J.; Sells, S.; Pronost, S.; Lu, Z.; Crossley, B.M.; Timoney, P.J.; et al. New Real-Time PCR Assay Using Allelic Discrimination for Detection and Differentiation of Equine Herpesvirus-1 Strains with A(2254) and G(2254) Polymorphisms. J. Clin. Microbiol. 2012, 50, 1981–1988. [Google Scholar] [CrossRef]
- Burgess, B.A.; Tokateloff, N.; Manning, S.; Lohmann, K.; Lunn, D.P.; Hussey, S.B.; Morley, P.S. Nasal shedding of equine herpesvirus-1 from horses in an outbreak of equine herpes myeloencephalopathy in Western Canada. J. Vet. Intern. Med. 2012, 26, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Franz, M.; Goodman, L.B.; Van de Walle, G.R.; Osterrieder, N.; Greenwood, A.D. A Point Mutation in a Herpesvirus Co-Determines Neuropathogenicity and Viral Shedding. Viruses 2017, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Holz, C.L.; Nelli, R.K.; Wilson, M.E.; Zarski, L.M.; Azab, W.; Baumgardner, R.; Osterrieder, N.; Pease, A.; Zhang, L.; Hession, S.; et al. Viral genes and cellular markers associated with neurological complications during herpesvirus infections. J. Gen. Virol. 2017, 98, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.P. Risk factors for development of neurologic disease after experimental exposure to equine herpesvirus-1 in horses. Am. J. Vet. Res. 2008, 69, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.L.; Allen, G.P.; Branscum, A.J.; Frank Cook, R.; Vickers, M.L.; Timoney, P.J.; Balasuriya, U.B. The increased prevalence of neuropathogenic strains of EHV-1 in equine abortions. Vet. Microbiol. 2010, 141, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronost, S.; Legrand, L.; Pitel, P.H.; Wegge, B.; Lissens, J.; Freymuth, F.; Richard, E.; Fortier, G. Outbreak of equine herpesvirus myeloencephalopathy in France: A clinical and molecular investigation. Transbound. Emerg. Dis. 2012, 59, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Kydd, J.H.; Slater, J.; Osterrieder, N.; Lunn, D.P.; Antczak, D.F.; Azab, W.; Balasuriya, U.B.; Barnett, C.; Brosnahan, M.; Cook, C.; et al. Third International Havemeyer Workshop on Equine Herpesvirus type 1. Equine Vet. J. 2012, 44, 513–517. [Google Scholar] [CrossRef]
- Stasiak, K.; Rola, J.; Ploszay, G.; Socha, W.; Zmudzinski, J.F. Detection of the neuropathogenic variant of equine herpesvirus 1 associated with abortions in mares in Poland. BMC Vet. Res. 2015, 11, 102. [Google Scholar] [CrossRef]
- Vissani, M.A.; Becerra, M.L.; Olguin Perglione, C.; Tordoya, M.S.; Mino, S.; Barrandeguy, M. Neuropathogenic and non-neuropathogenic genotypes of Equid Herpesvirus type 1 in Argentina. Vet. Microbiol. 2009, 139, 361–364. [Google Scholar] [CrossRef]
- Edington, N.; Smyth, B.; Griffiths, L. The role of endothelial cell infection in the endometrium, placenta and foetus of equid herpesvirus 1 (EHV-1) abortions. J. Comp. Pathol. 1991, 104, 379–387. [Google Scholar] [CrossRef]
- Allen, G.P.; Breathnach, C.C. Quantification by real-time PCR of the magnitude and duration of leucocyte-associated viraemia in horses infected with neuropathogenic vs. non-neuropathogenic strains of EHV-1. Equine Vet. J. 2006, 38, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, A.P.; Glorieux, S.; Gryspeerdt, A.C.; Steukers, L.; Duchateau, L.; Osterrieder, N.; Van de Walle, G.R.; Nauwynck, H.J. Replication kinetics of neurovirulent versus non-neurovirulent equine herpesvirus type 1 strains in equine nasal mucosal explants. J. Gen. Virol. 2010, 91, 2019–2028. [Google Scholar] [CrossRef]
- Barbic, L.; Lojkic, I.; Stevanovic, V.; Bedekovic, T.; Staresina, V.; Lemo, N.; Lojkic, M.; Madic, J. Two outbreaks of neuropathogenic equine herpesvirus type 1 with breed-dependent clinical signs. Vet. Rec. 2012, 170, 227. [Google Scholar] [CrossRef] [PubMed]
- Damiani, A.M.; de Vries, M.; Reimers, G.; Winkler, S.; Osterrieder, N. A severe equine herpesvirus type 1 (EHV-1) abortion outbreak caused by a neuropathogenic strain at a breeding farm in northern Germany. Vet. Microbiol. 2014, 172, 555–562. [Google Scholar] [CrossRef]
- Walter, J.; Seeh, C.; Fey, K.; Bleul, U.; Osterrieder, N. Clinical observations and management of a severe equine herpesvirus type 1 outbreak with abortion and encephalomyelitis. Acta Vet. Scand. 2013, 55, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadden, A.M.; Hanlon, D.; McKenzie, R.K.; Gibson, I.; Bueno, I.M.; Pulford, D.J.; Orr, D.; Dunowska, M.; Stanislawek, W.L.; Spence, R.P.; et al. The first reported outbreak of equine herpesvirus myeloencephalopathy in New Zealand. N. Z. Vet. J. 2016, 64, 125–134. [Google Scholar] [CrossRef]
- Pronost, S.; Léon, A.; Legrand, L.; Fortier, C.; Miszczak, F.; Freymuth, F.; Fortier, G. Neuropathogenic and non-neuropathogenic variants of equine herpesvirus 1 in France. Vet. Microbiol. 2010, 145, 329–333. [Google Scholar] [CrossRef]
- Goehring, L.S.; van Winden, S.C.; van Maanen, C.; Sloet van Oldruitenborgh-Oosterbaan, M.M. Equine herpesvirus type 1-associated myeloencephalopathy in The Netherlands: A four-year retrospective study (1999–2003). J. Vet. Intern. Med. 2006, 20, 601–607. [Google Scholar]
- Brosnahan, M.M.; Al Abri, M.A.; Brooks, S.A.; Antczak, D.F.; Osterrieder, N. Genome-wide association study of equine herpesvirus type 1-induced myeloencephalopathy identifies a significant single nucleotide polymorphism in a platelet-related gene. Vet. J. 2018. [Google Scholar] [CrossRef]
- Gryspeerdt, A.; Vanderkerckhove, A.; Van Doorsselaere, J.; Van de Walle, G.; Nauwynck, H. Description of an unusually large outbreak of nervous system disorders caused by equine herpesvirus 1 (EHV1) in 2009 in Belgium. Vlaams Diergeneeskundig Tijdschrif 2011, 80, 147–153. [Google Scholar]
- Irish Thoroughbred Marketing. Available online: http://www.itm.ie (accessed on 1 September 2018).
- Allen, G.P.; Kydd, J.H.; Slater, J.D.; Smith, K.C. Equid Herpesvirus 1 and Equid Herpesvirus 4 Infections. In Infectious Diseaases of Livestock, 2nd ed.; Oxford University Press: Newmarket, UK, 2004; Chapter 76; pp. 829–859. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Rozen, S.; Skaletsky, H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000, 132, 365–386. [Google Scholar] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. TIG 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Leigh, J.; Bryant, D. PopART: Full-Feature Software for Haplotype Network Construction. Methods Ecol Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abortion | Neurological | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| UL Clade 1 | Isolates | Outbreaks | Premises | ORF30 D752 3 | Respiratory | Single 4 | Multiple 4 | Single | Multiple |
| 1 | 13 | 10 | 9 | 8 | 0 | 4 | 2 | 1 | 3 |
| 3 | 2 | 2 | 2 | 1 | 0 | 1 | 0 | 1 | 0 |
| 5 | 3 | 2 | 2 | 0 | 0 | 1 | 1 | 0 | 0 |
| 6 | 25 | 21 | 21 | 0 | 1 | 19 * | 1 | 1 * | 0 |
| 7 | 118 | 106 | 104 | 5 | 1 | 80 | 19 | 5 | 1 |
| 8 | 14 | 13 | 13 | 14 | 0 | 7 | 4 | 0 | 2 |
| 9 | 25 | 22 | 22 | 2 | 0 | 16 | 5 | 0 | 1 |
| 10 | 19 | 16 | 15 | 0 | 0 | 12 | 3 | 1 | 0 |
| 11 | 38 | 32 | 32 | 3 | 1 | 23 | 4 | 2 | 2 |
| 13 | 15 | 14 | 14 | 2 | 0 | 8 | 4 | 0 | 2 |
| Total | 272 | 238 | 220 2 | 35 | 3 | 170 * | 43 | 11 * | 11 |
| Year | Clade 1 | Clade 3 | Clade 5 | Clade 6 | Clade 7 | Clade 8 | Clade 9 | Clade 10 | Clade 11 | Clade 13 | Yearly Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1990 | 1 | 1 | 1 | 3 | |||||||
| 1991 | 2 | 2 | |||||||||
| 1992 | 1 | 1 | 1 | 1 | 4 | ||||||
| 1993 | 1 | 1 | 1 | 3 | |||||||
| 1994 | 1 | 3 | 1 | 2 | 7 | ||||||
| 1995 | 1 | 2 | 1 | 4 | |||||||
| 1996 | 1 | 1 | 1 | 3 | |||||||
| 1997 | 3 | 2 | 5 | ||||||||
| 1998 | 4 | 4 | |||||||||
| 1999 | 3 | 1 | 1 | 5 | |||||||
| 2000 | 3 | 1 | 4 | ||||||||
| 2001 | 1 | 2 | 2 | 5 | |||||||
| 2002 | 2 | 1 | 1 | 4 | |||||||
| 2003 | 2 | 1 | 3 | ||||||||
| 2004 | 3 | 1 | 1 | 5 | |||||||
| 2005 | 2 | 2 | 17 | 3 | 3 | 3 | 30 | ||||
| 2006 | 1 | 13 | 2 | 1 | 3 | 3 | 23 | ||||
| 2007 | 1 | 7 | 3 | 1 | 1 | 13 | |||||
| 2008 | 5 | 4 | 1 | 5 | 8 | 1 | 24 | ||||
| 2009 | 1 | 1 | 7 | 2 | 11 | ||||||
| 2010 | 1 | 9 | 1 | 1 | 2 | 1 | 15 | ||||
| 2011 | 2 | 5 | 1 | 1 | 3 | 12 | |||||
| 2012 | 5 | 3 | 1 | 1 | 10 | ||||||
| 2013 | 5 | 1 | 2 | 2 | 1 | 11 | |||||
| 2014 | 2 | 2 | 6 | 1 | 1 | 12 | |||||
| 2015 | 1 | 1 | 2 | 8 | 1 | 1 | 4 | 18 | |||
| 2016 | 1 | 2 | 7 | 1 | 1 | 1 | 2 | 2 | 17 | ||
| 2017 | 3 | 10 | 2 | 15 | |||||||
| Total | 13 | 2 | 3 | 25 | 118 | 14 | 25 | 19 | 38 | 15 | 272 |
| Disease Expression | Isolates | Horses | Outbreaks | Premises 2 | Multi-Locus Typing | ORF68 Typing |
|---|---|---|---|---|---|---|
| Respiratory | 3 | 3 | 3 | 3 | 3 | 3 |
| Single abortion/ neonatal foal death | 171 | 170 1 | 170 | 163 | 171 | 164 |
| Multiple abortion/ neonatal foal death | 73 | 71 | 43 | 42 | 73 | 38 |
| Single neurological disease | 11 | 11 1 | 11 | 11 | 11 | 7 |
| Multiple neurological disease | 14 | 14 | 11 | 11 | 14 | 10 |
| Total | 272 | 269 | 238 | 220 2 | 272 | 222 |
| AA Variation | ||||
|---|---|---|---|---|
| ORF | AA Position 1 | Ab4 | V592 | Other 2 |
| 2 | 59 | G | D | /3 |
| 5 | 114 | G | V | / |
| 8 | 114 | D | N | / |
| 11 | 189 | Q | K | / |
| 11 | 235 | R | R | M |
| 13 | 305 | S | L | / |
| 13 | 405 | A | A | T |
| 13 | 460 | A | T | / |
| 13 | 492 | E | E | K |
| 13 | 493 | T | T | I |
| 13 | 499 | A | A | T |
| 14 | 618–620 | -4 | PSR | / |
| 14 | 628 | R | R | K |
| 14 | 692 | S | S | N |
| 15 | 166 | D | N | / |
| 22 | 430 | S | P | / |
| 29 | 12 | T | K | / |
| 30 | 752 | D | N | / |
| 30 | 990 | E | K | / |
| 31 | 90 | N | S | / |
| 32 | 42 | S | L | / |
| 33 | 15 | N | H | / |
| 33 | 976 | N | D | / |
| 34 | 66 | D | G | / |
| 36 | 47 | S | R | / |
| 37 | 265 | A | V | / |
| 39 | 440 | S | L | / |
| 40 | 196 | R | H | / |
| 42 | 1275 | K | R | / |
| 45 | 427 | E | G | / |
| 46 | 140 | F | S | / |
| 50 | 367 | P | S | / |
| 52 | 386 | A | V | / |
| 57 | 804 | K | R | / |
| 73 | 122 | A | V | / |
| 76 | 128 | F | S | / |
| Primer | Description | Primer Sequence 5′ to 3′ | Nucleotide Position on Strain Ab4 |
|---|---|---|---|
| Forward | PCR | ATGGGTGTGGTCTTAATTAC | 126275–126256 |
| Reverse | PCR | GACACCGCCTGAAGTAGGAG | 124963–124982 |
| 68R2 | Sequencing | ACCGTTGAGCATAATCATCC [21] | 125730–125710 |
| 68S1 | Sequencing | GAAGATAGAATGGGTGTGAG [21] | 125999–125979 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garvey, M.; Lyons, R.; Hector, R.D.; Walsh, C.; Arkins, S.; Cullinane, A. Molecular Characterisation of Equine Herpesvirus 1 Isolates from Cases of Abortion, Respiratory and Neurological Disease in Ireland between 1990 and 2017. Pathogens 2019, 8, 7. https://doi.org/10.3390/pathogens8010007
Garvey M, Lyons R, Hector RD, Walsh C, Arkins S, Cullinane A. Molecular Characterisation of Equine Herpesvirus 1 Isolates from Cases of Abortion, Respiratory and Neurological Disease in Ireland between 1990 and 2017. Pathogens. 2019; 8(1):7. https://doi.org/10.3390/pathogens8010007
Chicago/Turabian StyleGarvey, Marie, Rachel Lyons, Ralph D Hector, Cathal Walsh, Sean Arkins, and Ann Cullinane. 2019. "Molecular Characterisation of Equine Herpesvirus 1 Isolates from Cases of Abortion, Respiratory and Neurological Disease in Ireland between 1990 and 2017" Pathogens 8, no. 1: 7. https://doi.org/10.3390/pathogens8010007
APA StyleGarvey, M., Lyons, R., Hector, R. D., Walsh, C., Arkins, S., & Cullinane, A. (2019). Molecular Characterisation of Equine Herpesvirus 1 Isolates from Cases of Abortion, Respiratory and Neurological Disease in Ireland between 1990 and 2017. Pathogens, 8(1), 7. https://doi.org/10.3390/pathogens8010007