β-HPV 8E6 Attenuates ATM and ATR Signaling in Response to UV Damage



Abstract
1. Introduction
2. Results
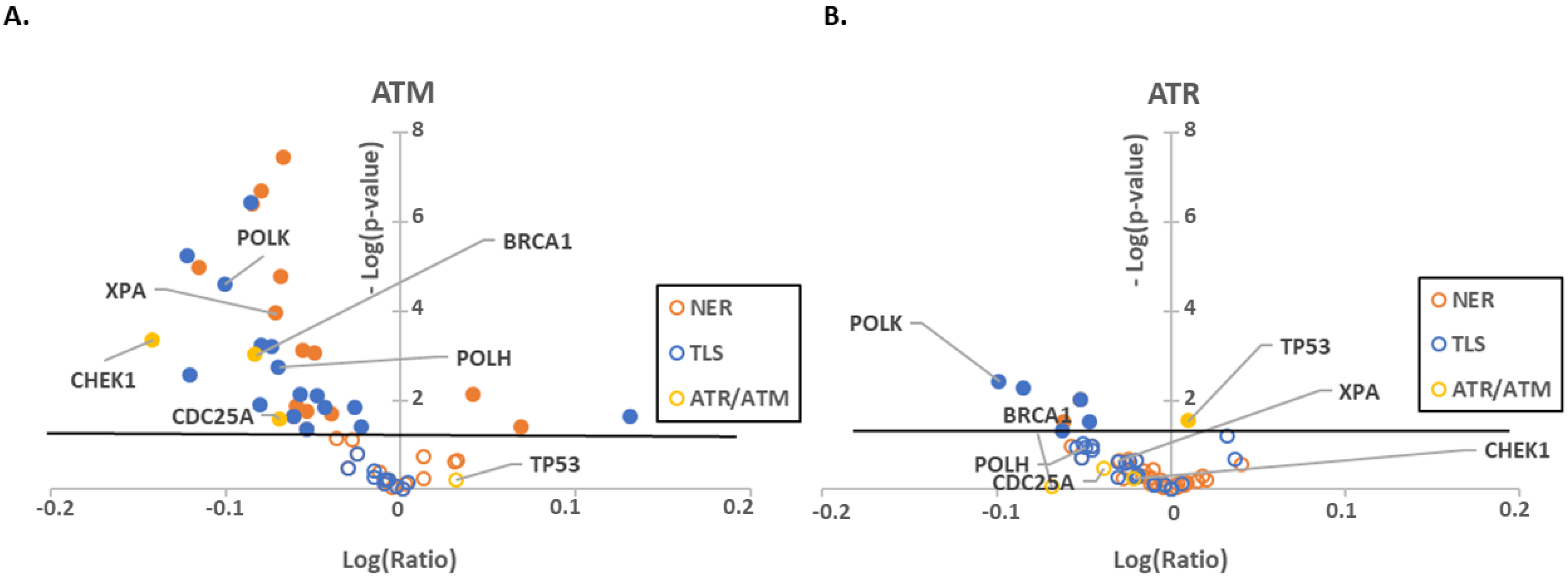
2.1. ATR, ATM and p53 Have Distinct Transcription Effector Profiles
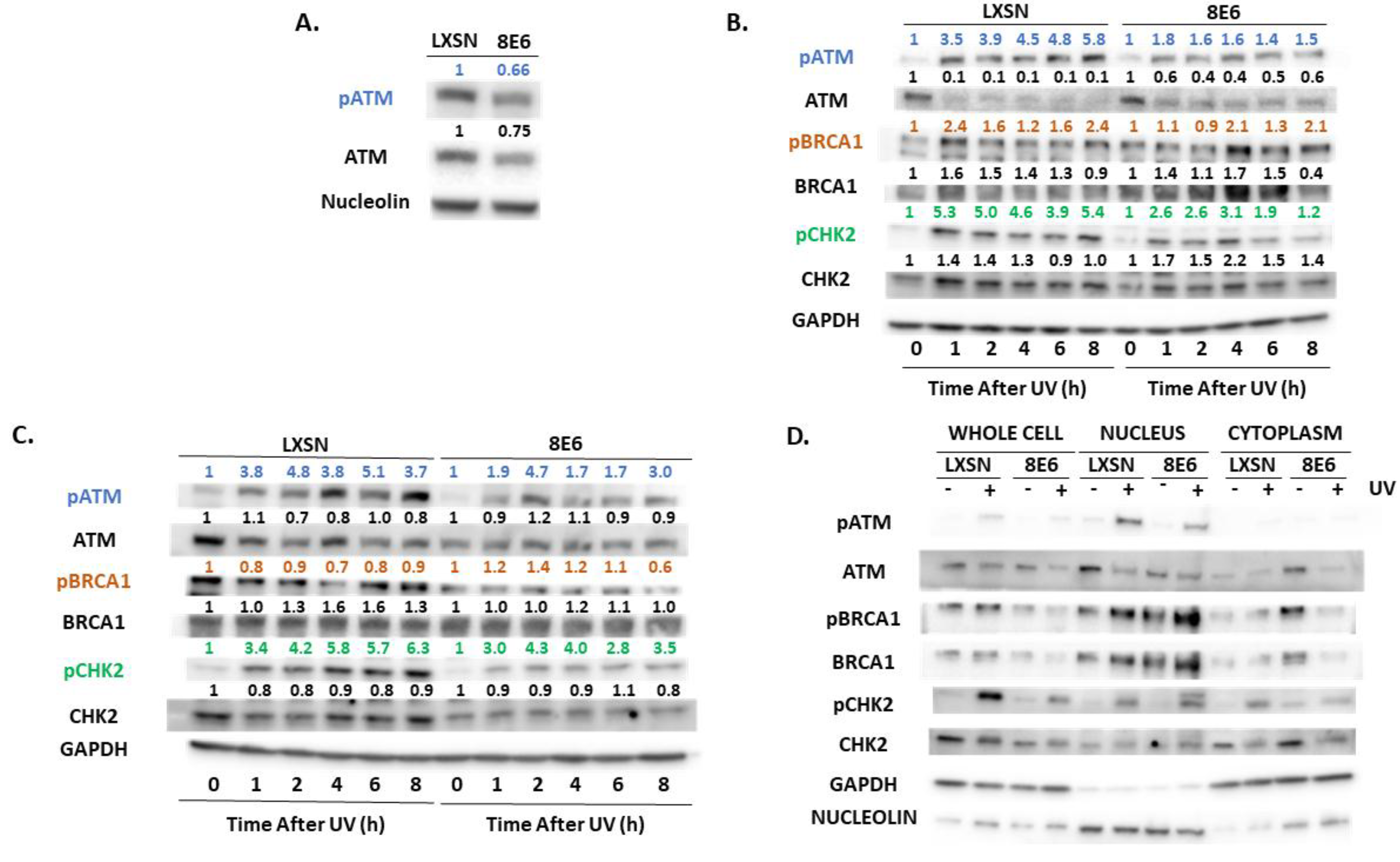
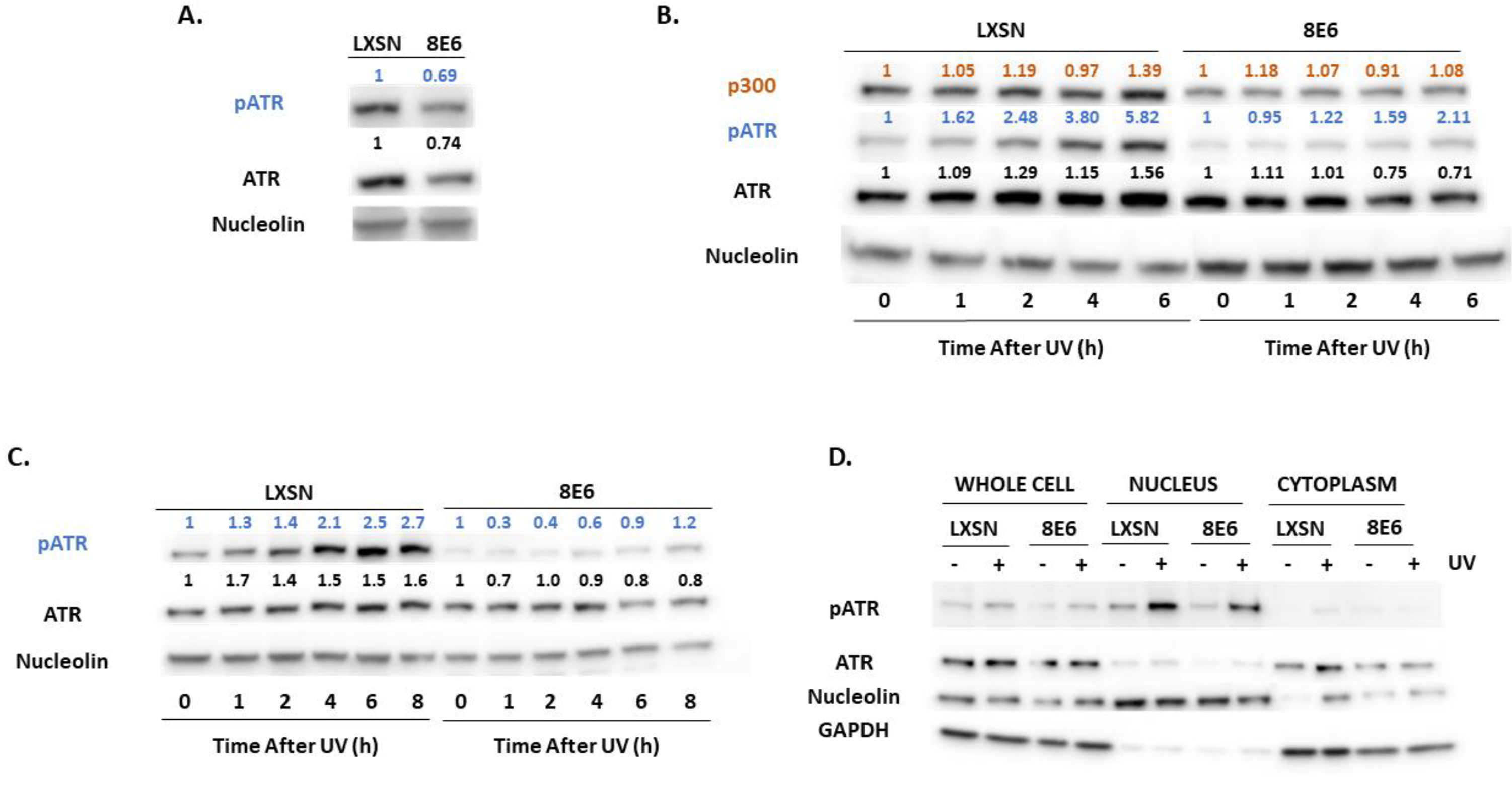
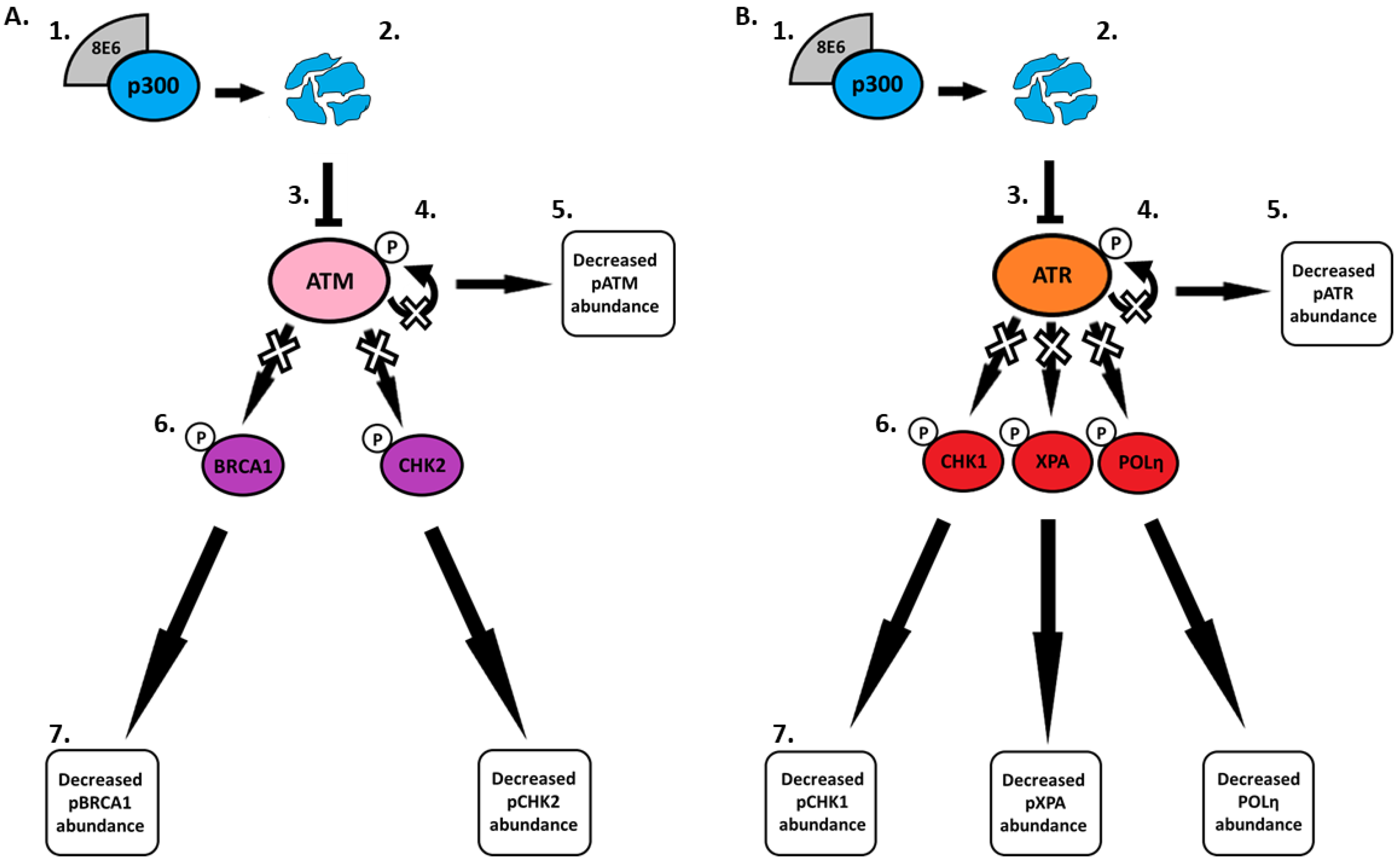
2.2. β-HPV E6 Decreases ATM and ATR Activation
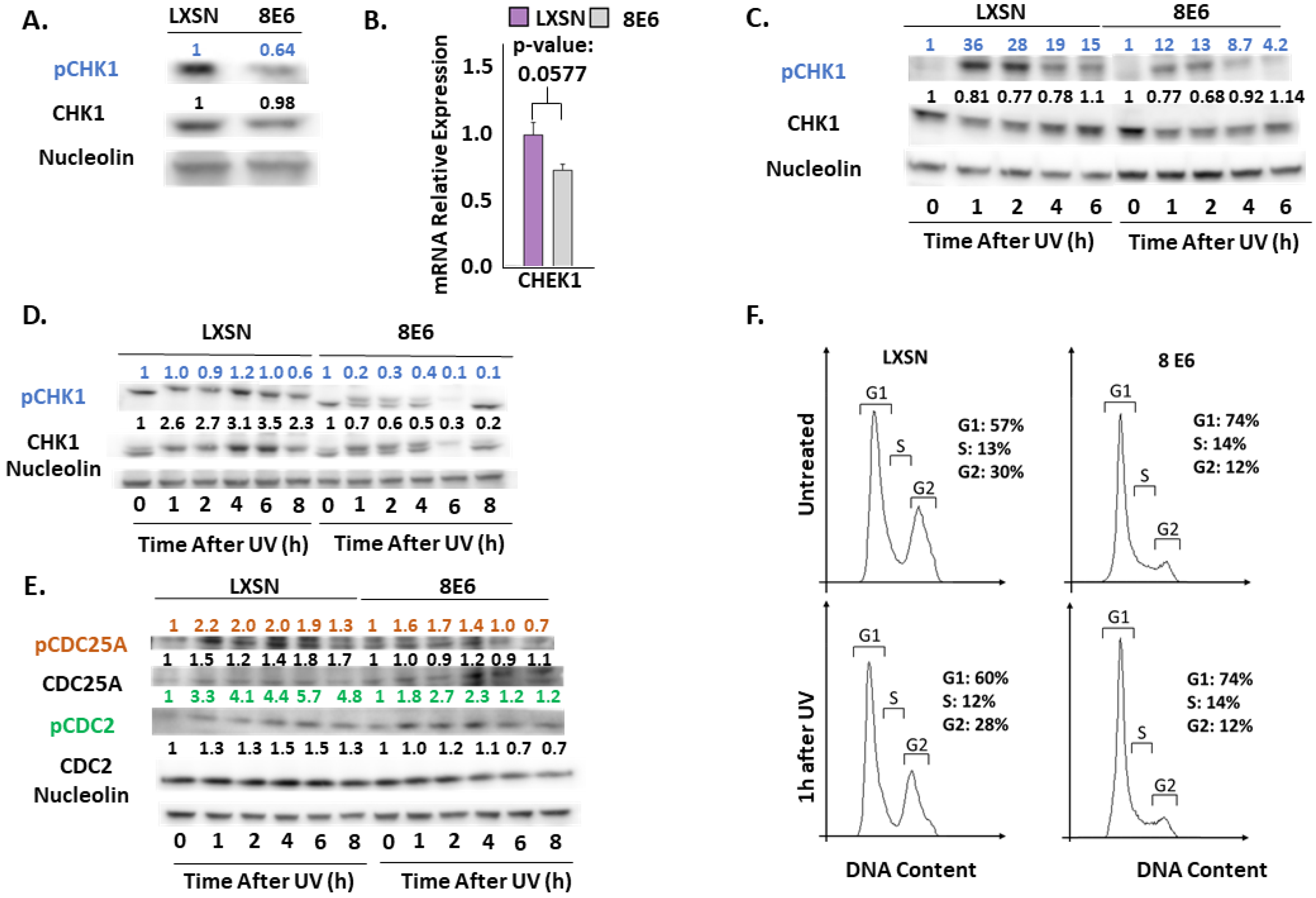
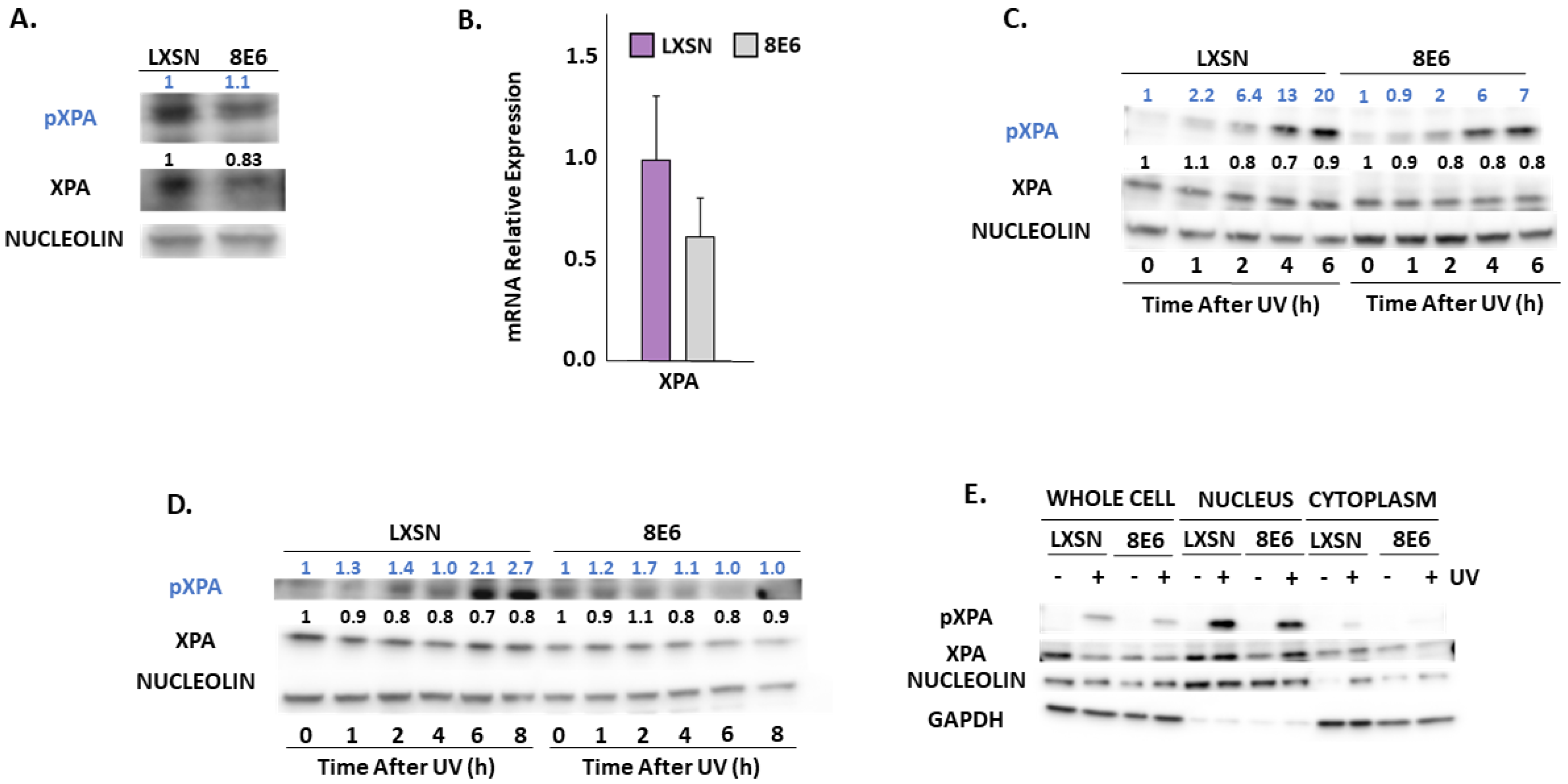
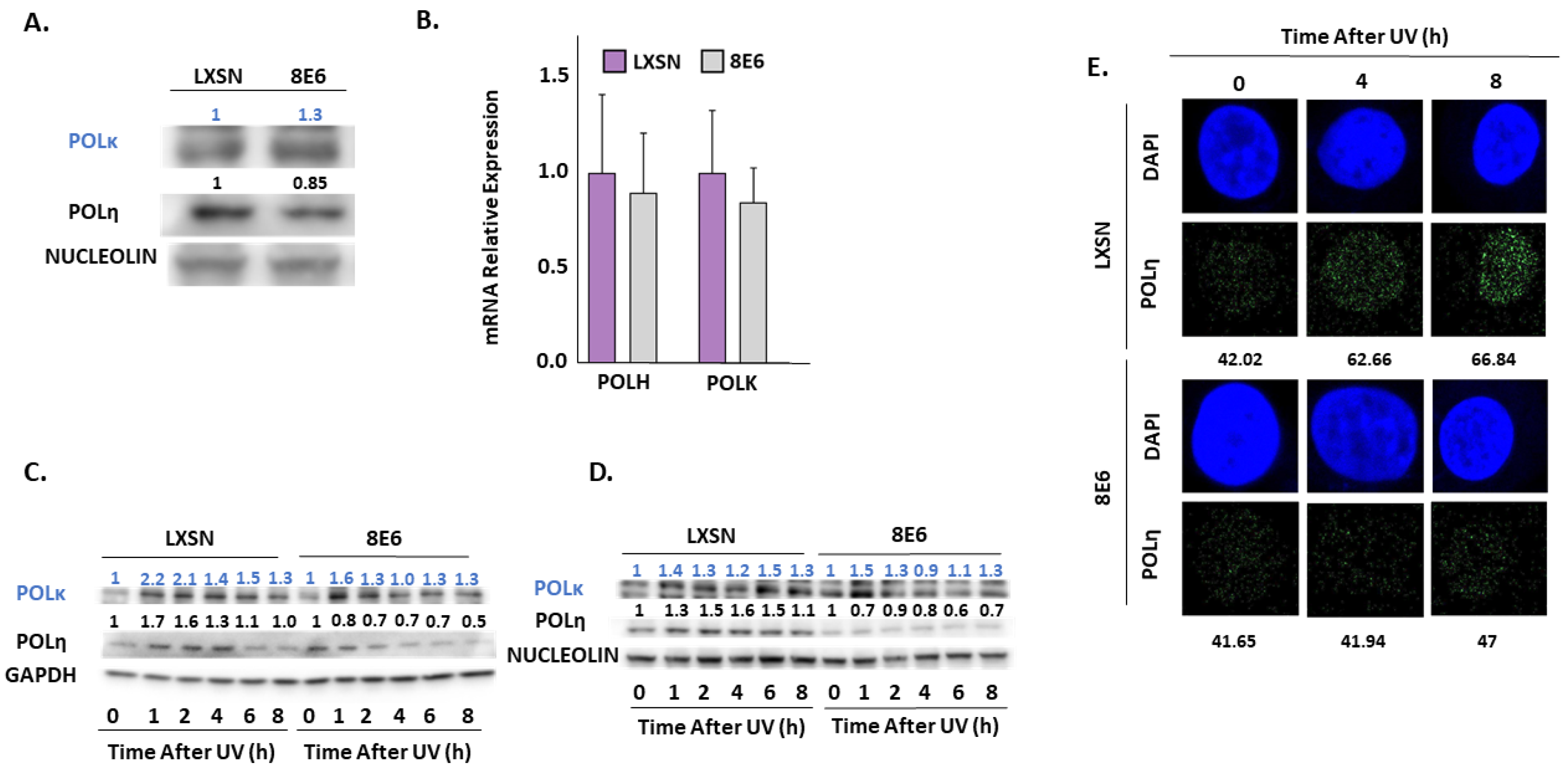
2.3. β-HPV 8E6 Decreases Phosphorylation of ATR Target Proteins
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Cycle Analysis
4.3. Comparative Transcriptomic Analysis
4.4. Immunoblot
4.5. Antibodies
4.6. Immunofluorescent Microscopy
4.7. Subcellular Fractionation
4.8. mRNA Quantification
4.9. UV Radiation
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Villiers, E.-M. Cross-roads in the classification of papillomaviruses. Virology 2013, 445, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.-U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.-M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, E.-M.; Fauquet, C.; Broker, T.R.; Bernard, H.-U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, D.; Eklund, C.; Dillner, J. International standardization and classification of human papillomavirus types. Virology 2015, 476, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Gheit, T. Mucosal and cutaneous human papillomavirus infections and cancer biology. Front. Oncol. 2019, 9, 355. [Google Scholar] [CrossRef]
- Brianti, P.; Flammineis, E.D.; Mercuri, S.R. Review of HPV-related diseases and cancers. New Microbiol. 2007, 40, 80–85. [Google Scholar]
- Egawa, N.; Doorbar, J. The low-risk papillomaviruses. Virus Res. 2017, 231, 119–127. [Google Scholar] [CrossRef]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef]
- Muñoz, N.; Castellsagué, X.; de González, A.B.; Gissmann, L. Chapter 1: HPV in the etiology of human cancer. Vaccine 2006, 24, S1–S10. [Google Scholar] [CrossRef]
- Duensing, S.; Münger, K. Mechanisms of genomic instability in human cancer: Insights from studies with human papillomavirus oncoproteins: Genomic Instability and Cervical Cancer. Int. J. Cancer 2004, 109, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Wendel, S.O.; Wallace, N.A. Loss of genome fidelity: Beta HPVs and the DNA damage response. Front. Microbiol. 2017, 8, 2250. [Google Scholar] [CrossRef] [PubMed]
- Pfister, H. Chapter 8: Human papillomavirus and skin cancer. JNCI Monogr. 2003, 2003, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Akgül, B. Molecular mechanisms of human papillomavirus induced skin carcinogenesis. Viruses 2017, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, S.; You, J.X. Targeting persistent human papillomavirus infection. Viruses 2017, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef]
- Weissenborn, S.J.; Nindl, I.; Purdie, K.; Harwood, C.; Proby, C.; Breuer, J.; Majewski, S.; Pfister, H.; Wieland, U. Human papillomavirus-DNA loads in actinic keratoses exceed those in non-melanoma skin cancers. J. Investig. Dermatol. 2005, 125, 93–97. [Google Scholar] [CrossRef]
- Ramasamy, K.; Shanmugam, M.; Balupillai, A.; Govindhasamy, K.; Gunaseelan, S.; Muthusamy, G.; Robert, B.; Nagarajan, R. Ultraviolet radiation-induced carcinogenesis: Mechanisms and experimental models. J. Radiat. Cancer Res. 2017, 8, 4–19. [Google Scholar]
- Rollison, D.E.; Viarisio, D.; Amorrortu, R.P.; Gheit, T.; Tommasino, M. An emerging issue in oncogenic virology: The role of beta human papillomavirus types in the development of cutaneous squamous cell carcinoma. J. Virol. 2019, 93, e01003-18. [Google Scholar] [CrossRef]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer: Worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef]
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Ioannides, D. Epidemiological trends in skin cancer. Dermatol. Pract. Concept. 2017, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.; Morrison, L.K.; Rady, P.; Tyring, S. Epidermodysplasia verruciformis and susceptibility to HPV. Dis. Markers 2010, 29, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Nindl, I.; Rösl, F. Molecular concepts of virus infections causing skin cancer in organ transplant recipients. Am. J. Transplant. 2008, 8, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Bavinck, J.N.B.; Feltkamp, M.; Struijk, L.; ter Schegget, J. Human papillomavirus infection and skin cancer risk in organ transplant recipients. J. Investig. Dermatol. Symp. Proc. 2001, 6, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V. Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers 2016, 8, 95. [Google Scholar] [CrossRef]
- Meyers, J.M.; Spangle, J.M.; Munger, K. The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol. 2013, 87, 4762–4767. [Google Scholar] [CrossRef]
- Meyers, J.M.; Uberoi, A.; Grace, M.; Lambert, P.F.; Munger, K. Cutaneous HPV8 and MmuPV1 E6 proteins target the NOTCH and TGF-β tumor suppressors to inhibit differentiation and sustain keratinocyte proliferation. PLoS Pathog. 2017, 13, e1006171. [Google Scholar] [CrossRef]
- Taute, S.; Pfister, H.J.; Steger, G. Induction of tyrosine phosphorylation of UV-activated EGFR by the beta-human papillomavirus type 8 E6 leads to papillomatosis. Front. Microbiol. 2017, 8, 2197. [Google Scholar] [CrossRef]
- Howie, H.L.; Koop, J.I.; Weese, J.; Robinson, K.; Wipf, G.; Kim, L.; Galloway, D.A. Beta-HPV 5 and 8 E6 promote p300 degradation by blocking AKT/p300 association. PLoS Pathog. 2011, 7, e1002211. [Google Scholar] [CrossRef]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef]
- Wallace, N.A.; Gasior, S.L.; Faber, Z.J.; Howie, H.L.; Deininger, P.L.; Galloway, D.A. HPV 5 and 8 E6 expression reduces ATM protein levels and attenuates LINE-1 retrotransposition. Virology 2013, 443, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, D.; Chang, B.; Huang, J.; Dunn, A.; Thayer, M. p300/CREB-binding protein interacts with ATR and is required for the DNA replication checkpoint. J. Biol. Chem. 2007, 282, 9678–9687. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Hufbauer, M.; Cooke, J.; van der Horst, G.T.J.; Pfister, H.; Storey, A.; Akgül, B. Human papillomavirus mediated inhibition of DNA damage sensing and repair drives skin carcinogenesis. Mol. Cancer 2015, 14, 183. [Google Scholar] [CrossRef]
- Sugitani, N.; Sivley, R.M.; Perry, K.E.; Capra, J.A.; Chazin, W.J. XPA: A key scaffold for human nucleotide excision repair. DNA Repair 2016, 44, 123–135. [Google Scholar] [CrossRef]
- Park, J.-M.; Kang, T.-H. Transcriptional and posttranslational regulation of nucleotide excision repair: The guardian of the genome against ultraviolet radiation. Int. J. Mol. Sci. 2016, 17, 1840. [Google Scholar] [CrossRef]
- Göhler, T.; Sabbioneda, S.; Green, C.M.; Lehmann, A.R. ATR-mediated phosphorylation of DNA polymerase η is needed for efficient recovery from UV damage. J. Cell Biol. 2011, 192, 219–227. [Google Scholar] [CrossRef]
- Tonzi, P.; Huang, T.T. Role of Y-family translesion DNA polymerases in replication stress: Implications for new cancer therapeutic targets. DNA Repair 2019, 78, 20–26. [Google Scholar] [CrossRef]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy: Chk1 review. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Chen, Z.; Gunasekera, A.H.; Sowin, T.J.; Rosenberg, S.H.; Fesik, S.; Zhang, H. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J. Biol. Chem. 2003, 278, 21767–21773. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data: Figure 1. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef]
- Cortez, D. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef]
- Canman, C.E. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.-Y.; Taya, Y.; Prives, C.; Abraham, R.T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999, 13, 152–157. [Google Scholar] [CrossRef]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Galloway, D.A. Beta human papillomavirus E6 expression inhibits stabilization of p53 and increases tolerance of genomic instability. J. Virol. 2014, 88, 6112–6127. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Möller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT promoter mutations are frequent in cutaneous basal cell carcinoma and squamous cell carcinoma. PLoS ONE 2013, 8, e80354. [Google Scholar] [CrossRef]
- Gatei, M.; Scott, S.P.; Filippovitch, I.; Soronika, N.; Lavin, M.F.; Weber, B.; Khanna, K.K. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000, 60, 3299–3304. [Google Scholar]
- Liu, S.; Shiotani, B.; Lahiri, M.; Maréchal, A.; Tse, A.; Leung, C.C.Y.; Glover, J.N.M.; Yang, X.H.; Zou, L. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 2011, 43, 192–202. [Google Scholar] [CrossRef]
- Stokes, M.P.; Rush, J.; MacNeill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef]
- Iyer, D.; Rhind, N. The intra-S checkpoint responses to DNA damage. Genes 2017, 8, 74. [Google Scholar] [CrossRef]
- Shen, T.; Huang, S. The role of Cdc25A in the regulation of cell proliferation and apoptosis. Anticancer Agents Med. Chem. 2012, 12, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Park, J.-M.; Leem, S.-H.; Kang, T.-H. Coordinated regulation of XPA stability by ATR and HERC2 during nucleotide excision repair. Oncogene 2014, 33, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Shell, S.M.; Li, Z.; Shkriabai, N.; Kvaratskhelia, M.; Brosey, C.; Serrano, M.A.; Chazin, W.J.; Musich, P.R.; Zou, Y. Checkpoint kinase ATR promotes nucleotide excision repair of UV-induced DNA damage via physical interaction with xeroderma pigmentosum group A. J. Biol. Chem. 2009, 284, 24213–24222. [Google Scholar] [CrossRef] [PubMed]
- Tonzi, P.; Yin, Y.; Lee, C.W.T.; Rothenberg, E.; Huang, T.T. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. ELife 2018, 7, e41426. [Google Scholar] [CrossRef] [PubMed]
- Hasche, D.; Vinzón, S.E.; Rösl, F. Cutaneous papillomaviruses and non-melanoma skin cancer: Causal agents or innocent bystanders? Front. Microbiol. 2018, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Nindl, I.; Gottschling, M.; Stockfleth, E. Human papillomaviruses and non-melanoma skin cancer: Basic virology and clinical manifestations. Dis. Markers 2007, 23, 247–259. [Google Scholar] [CrossRef]
- Webb, C.J.; Wu, Y.; Zakian, V.A. DNA repair at telomeres: Keeping the ends intact. Cold Spring Harb. Perspect. Biol. 2013, 5, a012666. [Google Scholar] [CrossRef]
- Forment, J.V.; Walker, R.V.; Jackson, S.P. A high-throughput, flow cytometry-based method to quantify DNA-end resection in mammalian cells. Cytom. Part A 2012, 81, 922–928. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p53 | ATR | ATM | |
|---|---|---|---|
| BRCA1 | n.s. | ++ | +++ |
| CDC25A | n.s. | n.s. | ++ |
| CHEK1 | − | n.s. | +++ |
| POLH | +++ | n.s. | ++ |
| POLK | + | ++ | +++ |
| XPA | − | n.s. | +++ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Snow, J.A.; Murthy, V.; Dacus, D.; Hu, C.; Wallace, N.A. β-HPV 8E6 Attenuates ATM and ATR Signaling in Response to UV Damage. Pathogens 2019, 8, 267. https://doi.org/10.3390/pathogens8040267
Snow JA, Murthy V, Dacus D, Hu C, Wallace NA. β-HPV 8E6 Attenuates ATM and ATR Signaling in Response to UV Damage. Pathogens. 2019; 8(4):267. https://doi.org/10.3390/pathogens8040267
Chicago/Turabian StyleSnow, Jazmine A., Vaibhav Murthy, Dalton Dacus, Changkun Hu, and Nicholas A. Wallace. 2019. "β-HPV 8E6 Attenuates ATM and ATR Signaling in Response to UV Damage" Pathogens 8, no. 4: 267. https://doi.org/10.3390/pathogens8040267
APA StyleSnow, J. A., Murthy, V., Dacus, D., Hu, C., & Wallace, N. A. (2019). β-HPV 8E6 Attenuates ATM and ATR Signaling in Response to UV Damage. Pathogens, 8(4), 267. https://doi.org/10.3390/pathogens8040267