Crosstalk between RNA Metabolism and Cellular Stress Responses during Zika Virus Replication

 ,
, 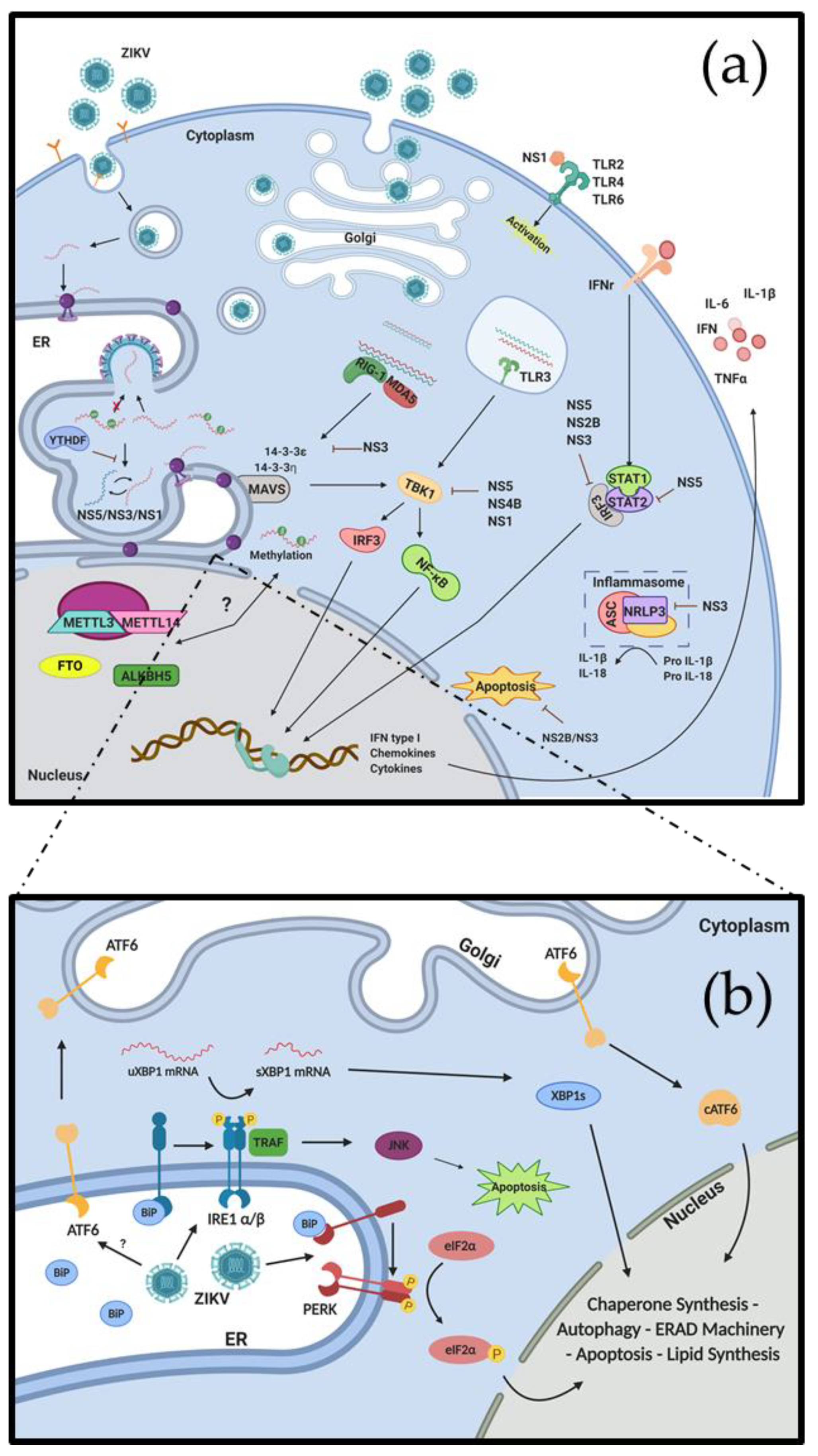{kind=link}
Abstract
1. Overview of ZIKV Pathogenesis and ZIKV-Associated Diseases
2. ZIKV Replication Cycle
3. Interactions between ZIKV and the Immune System
4. ZIKV Remodels the ER and Induces ER Stress
5. Regulation of Viral and Host Cell Gene Expression during ZIKV Replication
6. Interplay between RNA Modifications, ZIKV Replication and Cellular Responses
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Acronyms
References
- Dick, G.; Kitchen, S.; Haddow, A. Zika Virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Plourde, A.R.; Bloch, E.M. A Literature Review of Zika Virus. Emerg. Infect. Dis. 2016, 22, 1185–1192. [Google Scholar] [CrossRef]
- Noorbakhsh, F.; Abdolmohammadi, K.; Fatahi, Y.; Dalili, H.; Rasoolinejad, M.; Rezaei, F.; Salehi-Vaziri, M.; Shafiei-Jandaghi, N.Z.; Gooshki, E.S.; Zaim, M.; et al. Zika Virus Infection, Basic and Clinical Aspects: A Review Article. Iran. J. Public Health 2019, 48, 20–31. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://apps.who.int/iris/bitstream/10665/204421/1/WHO_ZIKV_MOC_16.1_eng.pdf (accessed on 20 January 2020).
- Hill, S.C.; Vasconcelos, J.; Neto, Z.; Jandondo, D.; Zé-Zé, L.; Aguiar, R.S.; Xavier, J.; Thézé, J.; Mirandela, M.; Cândido, A.L.M.; et al. Emergence of the Zika virus Asian lineage in Angola: Supplementary Materials and Methods. BioRxiv 2019, 520437. [Google Scholar] [CrossRef]
- Beaver, J.T.; Lelutiu, N.; Habib, R.; Skountzou, I. Evolution of Two Major Zika Virus Lineages: Implications for Pathology, Immune Response, and Vaccine Development. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Valderramos, S.G.; Wu, A.; Ouyang, S.; Li, C.; Brasil, P.; Bonaldo, M.; Coates, T.; Nielsen-Saines, K.; Jiang, T.; et al. From Mosquitos to Humans: Genetic Evolution of Zika Virus. Cell Host Microbe 2016, 19, 561–565. [Google Scholar] [CrossRef]
- Alonso-Palomares, L.A.; Moreno-García, M.; Lanz-Mendoza, H.; Salazar, M.I. Molecular Basis for Arbovirus Transmission by Aedes aegypti Mosquitoes. Intervirology 2019, 61, 255–264. [Google Scholar] [CrossRef]
- Matusali, G.; Houzet, L.; Satie, A.-P.; Mahé, D.; Aubry, F.; Couderc, T.; Frouard, J.; Bourgeau, S.; Bensalah, K.; Lavoué, S.; et al. Zika virus infects human testicular tissue and germ cells. J. Clin. Investig. 2018, 128, 4697–4710. [Google Scholar] [CrossRef]
- Shapiro-Mendoza, C.K.; Rice, M.E.; Galang, R.R.; Fulton, A.C.; VanMaldeghem, K.; Prado, M.V.; Ellis, E.; Anesi, M.S.; Simeone, R.M.; Petersen, E.E.; et al. Pregnancy Outcomes after Maternal Zika Virus Infection during Pregnancy—U.S. Territories, January 1, 2016–April 25, 2017. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 615–621. [Google Scholar] [CrossRef]
- Calvet, G.; Aguiar, R.; Melo, A.S.O.; Sampaio, S.A.; De Filippis, I.; Fabri, A.; Araujo, E.S.M.; De Sequeira, P.C.; De Mendonça, M.C.L.; De Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Martinez, M. Preventing Zika Virus Infection during Pregnancy Using a Seasonal Window of Opportunity for Conception. PLoS Boil. 2016, 14, e1002520. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Bowen, J.R.; Johnson, E.L.; McDonald, C.E.; Ma, H.; O’Neal, J.T.; Rajakumar, A.; Wrammert, J.; Rimawi, B.H.; Pulendran, B.; et al. Zika Virus Infects Human Placental Macrophages. Cell Host Microbe 2016, 20, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; De Noronha, L.; Dos Santos, C.N.D. Maternal-fetal transmission of the zika virus: An intriguing interplay. Tissue Barriers 2018, 6, e1402143. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.; Martín-Tapia, D.; Valdespino-Vázquez, Y.; Alarcón, L.; Nuñez, A.E.; Guzmán-Huerta, M.; Muñoz-Medina, J.E.; Shibayama, M.; Chávez-Munguía, B.; Estrada-Gutierrez, G.; et al. Syncytiotrophoblast of Placentae from Women with Zika Virus Infection Has Altered Tight Junction Protein Expression and Increased Paracellular Permeability. Cells 2019, 8, 1174. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Ventura, L.V. Ophthalmologic Manifestations Associated With Zika Virus Infection. Pediatrics 2018, 141, S161–S166. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.B.; De Moraes, C.G.; Petitto, M.; Yepez, J.B.; Sakuntabhai, A.; Simon-Lorière, E.; Prot, M.; Ruffie, C.; Kim, S.S.; Allikmets, R.; et al. Non-congenital severe ocular complications of Zika virus infection. JMM Case Rep. 2018, 5, e005152. [Google Scholar] [CrossRef]
- Roach, T.; Alcendor, D. Zika virus infection of cellular components of the blood-retinal barriers: Implications for viral associated congenital ocular disease. J. Neuroinflammation 2017, 14, 43. [Google Scholar] [CrossRef]
- Alcendor, D. Zika Virus Infection of the Human Glomerular Cells: Implications for Viral Reservoirs and Renal Pathogenesis. J. Infect. Dis. 2017, 216, 162–171. [Google Scholar] [CrossRef]
- Peralta-Aros, C.; García-Nieto, V. Does Zika virus infection induce prolonged remissions in children with idiopathic nephrotic syndrome? Pediatr. Nephrol. 2017, 32, 897–900. [Google Scholar] [CrossRef]
- Liu, T.; Tang, L.; Tang, H.; Pu, J.; Gong, S.; Fang, D.; Zhang, H.; Li, Y.-P.; Zhu, X.; Wang, W.; et al. Zika Virus Infection Induces Acute Kidney Injury Through Activating NLRP3 Inflammasome Via Suppressing Bcl-2. Front. Immunol. 2019, 10, 1925. [Google Scholar] [CrossRef]
- Sher, A.A.; Glover, K.K.M.; Coombs, K.M. Zika Virus Infection Disrupts Astrocytic Proteins Involved in Synapse Control and Axon Guidance. Front. Microbiol. 2019, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Cumberworth, S.L.; Barrie, J.A.; Cunningham, M.; De Figueiredo, D.P.G.; Schultz, V.; Wilder-Smith, A.J.; Brennan, B.; Pena, L.J.; Franca, R.F.D.O.; Linington, C.; et al. Zika virus tropism and interactions in myelinating neural cell cultures: CNS cells and myelin are preferentially affected. Acta Neuropathol. Commun. 2017, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, P.; Cochet, M.; Hamel, R.; Gladwyn-Ng, I.; Alfano, C.; Diop, F.; Garcia, D.; Talignani, L.; Montero-Menei, C.N.; Hamel, R.; et al. Zika Virus Differentially Infects Human Neural Progenitor Cells According to Their State of Differentiation and Dysregulates Neurogenesis through the Notch Pathway. Emerg. Microbes Infect. 2019, 8, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Dang, J.W.; Qin, Y.; Lichinchi, G.; Bansal, V.; Rana, T.M. Zika Virus Infection Reprograms Global Transcription of Host Cells to Allow Sustained Infection. Emerging Microbes & Infect. 2017, 6, 1–10. [Google Scholar] [CrossRef]
- Sun, G.; Larsen, C.N.; Baumgarth, N.; Klem, E.B.; Scheuermann, R.H. Comprehensive Annotation of Mature Peptides and Genotypes for Zika Virus. PLoS ONE 2017, 12, e0170462. [Google Scholar] [CrossRef]
- Lee, I.; Bos, S.; Li, G.; Wang, S.; Gadea, G.; Desprès, P.; Zhao, R.Y. Probing Molecular Insights into Zika Virus–Host Interactions. Viruses 2018, 10, 233. [Google Scholar] [CrossRef]
- Hastings, A.K.; Yockey, L.J.; Jagger, B.W.; Hwang, J.; Uraki, R.; Gaitsch, H.; Parnell, L.A.; Cao, B.; Mysorekar, I.U.; Rothlin, C.V.; et al. TAM Receptors Are Not Required for Zika Virus Infection in Mice. Cell Rep. 2017, 19, 558–568. [Google Scholar] [CrossRef]
- Sirohi, D.; Kuhn, R.J. Zika Virus Structure, Maturation, and Receptors. J. Infect. Dis. 2017, 216, S935–S944. [Google Scholar] [CrossRef]
- Kiermayr, S.; Kofler, R.M.; Mandl, C.W.; Messner, P.; Heinz, F.X. Isolation of Capsid Protein Dimers from the Tick-Borne Encephalitis Flavivirus and In Vitro Assembly of Capsid-Like Particles. J. Virol. 2004, 78, 8078–8084. [Google Scholar] [CrossRef]
- Sager, G.; Gabaglio, S.; Sztul, E.; Belov, G. Role of Host Cell Secretory Machinery in Zika Virus Life Cycle. Viruses 2018, 10, 559. [Google Scholar] [CrossRef]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, A.; Lindenmann, J. Pillars Article: Virus Interference. I. The Interferon. Proc R Soc Lond B Biol Sci. 1957. 147: 258-267. J. Immunol. 2015, 195, 258–267. [Google Scholar]
- Isaacs, A.; Lindenmann, J.; Valentine, R.C. Pillars Article: Virus Interference. II. Some Properties of Interferon. Proc R Soc Lond B Biol Sci. 1957. 147: 268-273. J. Immunol. 2015, 195, 268–273. [Google Scholar]
- Magoro, T.; Dandekar, A.; Jennelle, L.T.; Bajaj, R.; Lipkowitz, G.; Angelucci, A.; Bessong, P.O.; Hahn, Y.S. IL-1β/TNF-α/IL-6 inflammatory cytokines promote STAT1-dependent induction of CH25H in Zika virus-infected human macrophages. J. Boil. Chem. 2019, 294, 14591–14602. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Chang, D.C.; Hua, M.H.C.; Lim, S.P.; Chionh, Y.H.; Hia, F.; Lee, Y.H.; Kukkaro, P.; Lok, S.-M.; Dedon, P.C.; et al. 2′-O Methylation of Internal Adenosine by Flavivirus NS5 Methyltransferase. PLoS Pathog. 2012, 8, e1002642. [Google Scholar] [CrossRef] [PubMed]
- Bidet, K.; Garcia-Blanco, M.A. Flaviviral RNA Structures and Their Role in Replication and Immunity. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- De Borba, L.; Villordo, S.M.; Marsico, F.L.; Carballeda, J.M.; Filomatori, C.V.; Gebhard, L.G.; Pallarés, H.M.; Lequime, S.; Lambrechts, L.; Sánchez Vargas, I.; et al. RNA Structure Duplication in the Dengue Virus 3´ UTR: Redundancy or Host Specificity? mBio 2019. [Google Scholar] [CrossRef]
- Ooi, Y.S.; Majzoub, K.; Flynn, R.A.; Mata, M.A.; Diep, J.; Li, J.K.; Van Buuren, N.; Rumachik, N.; Johnson, A.; Puschnik, A.S.; et al. An RNA-centric dissection of host complexes controlling flavivirus infection. Nat. Microbiol. 2019, 4, 2369–2382. [Google Scholar] [CrossRef]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.; Shi, P.-Y.; Randall, R.; Khromykh, A.A. Inhibition of Interferon Signaling by the New York 99 Strain and Kunjin Subtype of West Nile Virus Involves Blockage of STAT1 and STAT2 Activation by Nonstructural Proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Bowen, J.R.; Quicke, K.M.; Maddur, M.S.; O’Neal, J.T.; McDonald, C.E.; Fedorova, N.B.; Puri, V.; Shabman, R.; Pulendran, B.; Suthar, M.S. Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog. 2017, 13, e1006164. [Google Scholar] [CrossRef]
- Gim, E.; Shim, D.-W.; Hwang, I.; Shin, O.S.; Yu, J.-W.; Kim, Y.U.; Kee, P.; Danila, D.; Teng, B.-B. Zika Virus Impairs Host NLRP3-mediated Inflammasome Activation in an NS3-dependent Manner. Immune Netw. 2019, 19, e40. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; Perreira, J.M.; Portmann, J.M.; Meraner, P.; Guo, Z.; Green, S.; Brass, A.L. The IFITMs Inhibit Zika Virus Replication. Cell Rep. 2016, 15, 2323–2330. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, S.; He, J.; Guest, J.; Ma, Z.; Yang, L.; Pierce, B.G.; Tang, Q.; Zhang, Y.-J. Zika virus NS5 protein antagonizes type I interferon production via blocking TBK1 activation. Virol. 2019, 527, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Hertzog, J.; Junior, A.G.D.; Rigby, R.; Donald, C.; Mayer, A.; Sezgin, E.; Song, C.; Jin, B.; Hublitz, P.; Eggeling, C.; et al. Infection with a Brazilian isolate of Zika virus generates RIG-I stimulatory RNA and the viral NS5 protein blocks type I IFN induction and signaling. Eur. J. Immunol. 2018, 48, 1120–1136. [Google Scholar] [CrossRef]
- Riedl, W.; Acharya, D.; Lee, J.-H.; Liu, G.; Serman, T.; Chiang, C.; Chan, Y.K.; Diamond, M.S.; Gack, M. Zika Virus NS3 Mimics a Cellular 14-3-3-Binding Motif to Antagonize RIG-I- and MDA5-Mediated Innate Immunity. Cell Host Microbe 2019, 26, 493–503.e6. [Google Scholar] [CrossRef]
- Hastings, A.K.; Hastings, K.; Uraki, R.; Hwang, J.; Gaitsch, H.; Dhaliwal, K.; Williamson, E.; Fikrig, E. Loss of the TAM Receptor Axl Ameliorates Severe Zika Virus Pathogenesis and Reduces Apoptosis in Microglia. iScience 2019, 13, 339–350. [Google Scholar] [CrossRef]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.-S.; Lee, S.-A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B Proteins Deregulate Akt-mTOR Signaling in Human Fetal Neural Stem Cells to Inhibit Neurogenesis and Induce Autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef]
- Avirutnan, P.; Punyadee, N.; Noisakran, S.; Komoltri, C.; Thiemmeca, S.; Auethavornanan, K.; Jairungsri, A.; Kanlaya, R.; Tangthawornchaikul, N.; Puttikhunt, C.; et al. Vascular Leakage in Severe Dengue Virus Infections: A Potential Role for the Nonstructural Viral Protein NS1 and Complement. J. Infect. Dis. 2006, 193, 1078–1088. [Google Scholar] [CrossRef]
- Chua, J.J.E.; Bhuvanakantham, R.; Chow, V.T.-K.; Ng, M.-L. Recombinant non-structural 1 (NS1) protein of dengue-2 virus interacts with human STAT3β protein. Virus Res. 2005, 112, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ng, M.M.-L.; Chu, J.J.H. Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection. PLoS Pathog. 2015, 11, e1005053. [Google Scholar] [CrossRef] [PubMed]
- Falconar, A.K.I. The dengue virus nonstructural-1 protein (NS1) generatesantibodies to common epitopes on human blood clotting, integrin/adhesin proteins and binds to humanendothelial cells: Potential implications in haemorrhagic fever pathogenesis. Arch. Virol. 1997, 142, 897–916. [Google Scholar] [CrossRef] [PubMed]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598–1613.e8. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.-P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS 1-caspase-1 axis. EMBO J. 2018, 37, e99347. [Google Scholar] [CrossRef]
- Sornjai, W.; Jaratsittisin, J.; Auewarakul, P.; Wikan, N.; Smith, D.R. Analysis of Zika virus neutralizing antibodies in normal healthy Thais. Sci. Rep. 2018, 8, 17193. [Google Scholar] [CrossRef]
- Bailey, M.; Duehr, J.; Dulin, H.; Broecker, F.; Brown, J.A.; Arumemi, F.O.; González, M.C.B.; Leyva-Grado, V.H.; Evans, M.J.; Simon, V.; et al. Human antibodies targeting Zika virus NS1 provide protection against disease in a mouse model. Nat. Commun. 2018, 9, 4560. [Google Scholar] [CrossRef]
- Gorman, M.J.; Caine, E.A.; Zaitsev, K.; Begley, M.; Weger-Lucarelli, J.; Uccellini, M.; Tripathi, S.; Morrison, J.; Yount, B.L.; Dinnon, K.H.; et al. An Immunocompetent Mouse Model of Zika Virus Infection. Cell Host Microbe 2018, 23, 672–685.e6. [Google Scholar] [CrossRef]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Pol, A.V.D.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256.e4. [Google Scholar] [CrossRef]
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018, 3, eaao1680. [Google Scholar] [CrossRef]
- Ngono, A.E.; Shresta, S. Immune Response to Dengue and Zika. Annu. Rev. Immunol. 2018, 36, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Yockey, L.J.; Wong, P.W.; Lee, S.; Huttner, A.J.; Iwasaki, A. Antiviral CD8 T cells induce Zika-virus-associated paralysis in mice. Nat. Microbiol. 2017, 3, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Winkler, C.W.; Myers, L.M.; Woods, T.A.; Messer, R.J.; Carmody, A.B.; McNally, K.L.; Scott, D.P.; Hasenkrug, K.J.; Best, S.M.; Peterson, K.E. Adaptive Immune Responses to Zika Virus Are Important for Controlling Virus Infection and Preventing Infection in Brain and Testes. J. Immunol. 2017, 198, 3526–3535. [Google Scholar] [CrossRef]
- Nguyen, S.; Antony, K.M.; Dudley, D.M.; Kohn, S.; Simmons, H.A.; Wolfe, B.; Salamat, M.S.; Teixeira, L.; Wiepz, G.J.; Thoong, T.H.; et al. Highly efficient maternal-fetal Zika virus transmission in pregnant rhesus macaques. PLoS Pathog. 2017, 13, e1006378. [Google Scholar] [CrossRef]
- Foo, S.-S.; Chen, W.; Chan, Y.; Lee, W.-S.; Lee, S.-A.; Cheng, G.; Nielsen-Saines, K.; Brasil, P.; Jung, J.U. Biomarkers and immunoprofiles associated with fetal abnormalities of ZIKV-positive pregnancies. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Rabelo, K.; Souza, L.J.; Salomão, N.G.; Oliveira, E.R.; Sentinelli, L.D.P.; Lacerda, M.S.; Saraquino, P.B.; Rosman, F.C.; Basílio-De-Oliveira, R.; Carvalho, J.; et al. Placental Inflammation and Fetal Injury in a Rare Zika Case Associated With Guillain-Barré Syndrome and Abortion. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Nagler, M.; Cuker, A. Profile of Instrumentation Laboratory’s HemosIL® AcuStar HIT-Ab(PF4-H) assay for diagnosis of heparin-induced thrombocytopenia. Expert Rev. Mol. Diagn. 2017, 17, 419–426. [Google Scholar] [CrossRef]
- Tonnerre, P.; Gil Melgaço, J.; Torres-Cornejo, A.; Pinto, M.A.; Yue, C.; Blümel, J.; De Sousa, P.S.F.; Mello, V.; Moran, J.; De Filippis, A.M.B.; et al. Evolution of the innate and adaptive immune response in women with acute Zika virus infection. Nat. Microbiol. 2019, 5, 76–83. [Google Scholar] [CrossRef]
- Grifoni, A.; Pham, J.; Sidney, J.; O’Rourke, P.H.; Paul, S.; Peters, B.; Martini, S.R.; De Silva, A.D.; Ricciardi, M.J.; Magnani, D.M.; et al. Prior Dengue Virus Exposure Shapes T Cell Immunity to Zika Virus in Humans. J. Virol. 2017, 91, e01469-17. [Google Scholar] [CrossRef]
- Ci, Y.; Liu, Z.-Y.; Zhang, N.-N.; Niu, Y.; Yang, Y.; Xu, C.; Yang, W.; Qin, C.-F.; Shi, L. Zika NS1-induced ER remodeling is essential for viral replication. J. Cell Boil. 2020, 219. [Google Scholar] [CrossRef] [PubMed]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural Characterization and Three-Dimensional Architecture of Replication Sites in Dengue Virus-Infected Mosquito Cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef] [PubMed]
- Thaker, S.K.; Chapa, T.; Garcia, G.; Gong, D.; Schmid, E.W.; Arumugaswami, V.; Sun, R.; Christofk, H. Differential Metabolic Reprogramming by Zika Virus Promotes Cell Death in Human versus Mosquito Cells. Cell Metab. 2019, 29, 1206–1216.e4. [Google Scholar] [CrossRef] [PubMed]
- Diop, F.; Vial, T.; Ferraris, P.; Wichit, S.; Bengue, M.; Hamel, R.; Talignani, L.; Liegeois, F.; Pompon, J.; Yssel, H.; et al. Zika virus infection modulates the metabolomic profile of microglial cells. PLoS ONE 2018, 13, e0206093. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, W.-P.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV infection activates the IRE1-XBP1 and ATF6 pathways of unfolded protein response in neural cells. J. Neuroinflammation 2018, 15, 275. [Google Scholar] [CrossRef]
- Alfano, C.; Gladwyn-Ng, I.; Couderc, T.; Lecuit, M.; Nguyen, L. The Unfolded Protein Response: A Key Player in Zika Virus-Associated Congenital Microcephaly. Front. Cell. Neurosci. 2019, 13, 94. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hänelt, I.; Hummer, A.G.; Ernst, R.; Hänelt, I. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol. Cell 2017, 67, 673–684.e8. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Rozpędek, W.; Pytel, D.; Mucha, B.; Leszczyńska, H.; Diehl, J.; Majsterek, I. The role of the PERK/eIF2α/ATF4/CHOP signaling pathway in tumor progression during Endoplasmic Reticulum stress. Curr. Mol. Med. 2016, 16, 1. [Google Scholar] [CrossRef]
- Gladwyn-Ng, I.; Cordón-Barris, L.; Alfano, C.; Creppe, C.; Couderc, T.; Morelli, G.; Thelen, N.; America, M.; Bessières, B.; Encha-Razavi, F.; et al. Stress-induced unfolded protein response contributes to Zika virus–associated microcephaly. Nat. Neurosci. 2017, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Hillary, R.F.; Fitzgerald, U. A lifetime of stress: ATF6 in development and homeostasis. J. Biomed. Sci. 2018, 25, 48. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the XBP-1 MRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Niwa, M.; Koong, A. Targeting the IRE1α-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Boil. 2015, 33, 48–56. [Google Scholar] [CrossRef]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized mRNAs During the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in Cell Fate Regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Bright, M.D.; Itzhak, D.; Wardell, C.; Morgan, G.J.; Davies, F.E. Cleavage of BLOC1S1 mRNA by IRE1 Is Sequence Specific, Temporally Separate from XBP1 Splicing, and Dispensable for Cell Viability under Acute Endoplasmic Reticulum Stress. Mol. Cell. Boil. 2015, 35, 2186–2202. [Google Scholar] [CrossRef]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef]
- Upton, J.-P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1 Cleaves Select microRNAs During ER Stress to Derepress Translation of Proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Walle, L.V.; Upton, J.-P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Kuo, S.-H.; Lin, C.-Y.; Fu, P.-J.; Lin, Y.-S.; Yeh, T.-M.; Liu, H.-S. Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Sci. Rep. 2018, 8, 489. [Google Scholar] [CrossRef] [PubMed]
- Perera, G.N.D.; Miller, J.L.; Zitzmann, N. The role of the unfolded protein response in dengue virus pathogenesis. Cell. Microbiol. 2017, 19, e12734. [Google Scholar] [CrossRef] [PubMed]
- Umareddy, I.; Pluquet, O.; Wang, Q.-Y.; Vasudevan, S.G.; Chevet, E.; Gu, F. Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol. J. 2007, 4, 91. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Hsu, Y.-W.; Liao, C.-L.; Lin, Y.-L. Flavivirus Infection Activates the XBP1 Pathway of the Unfolded Protein Response To Cope with Endoplasmic Reticulum Stress. J. Virol. 2006, 80, 11868–11880. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, R.L.; MacKenzie, J. West Nile Virus Differentially Modulates the Unfolded Protein Response To Facilitate Replication and Immune Evasion. J. Virol. 2010, 85, 2723–2732. [Google Scholar] [CrossRef]
- Medigeshi, G.; Lancaster, A.M.; Hirsch, A.J.; Briese, T.; Lipkin, W.I.; DeFilippis, V.; Früh, K.; Mason, P.W.; Nikolich-Zugich, J.; Nelson, J.A. West Nile Virus Infection Activates the Unfolded Protein Response, Leading to CHOP Induction and Apoptosis. J. Virol. 2007, 81, 10849–10860. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Sen, U.; Vrati, S. Regulated IRE1-dependent decay pathway is activated during Japanese encephalitis virus-induced unfolded protein response and benefits viral replication. J. Gen. Virol. 2013, 95, 71–79. [Google Scholar] [CrossRef]
- Carletti, T.; Zakaria, M.K.; Faoro, V.; Reale, L.; Kazungu, Y.; Licastro, D.; Marcello, A. Viral priming of cell intrinsic innate antiviral signaling by the unfolded protein response. Nat. Commun. 2019, 10, 3889–3899. [Google Scholar] [CrossRef]
- Tardif, K.D.; Kaufman, R.J.; Mori, K.; Siddiqui, A. Hepatitis C Virus Suppresses the IRE1-XBP1 Pathway of the Unfolded Protein Response. J. Boil. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C virus core protein activates autophagy through EIF2AK3 and ATF6 UPR pathway-mediated MAP1LC3B and ATG12 expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Egan, P.A.; Sobkowiak, M.; Chan, S.-W. Hepatitis C Virus Envelope Protein E1 Binds PERK and Represses the Unfolded Protein Response. Open Virol. J. 2013, 7, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.-P.; Tedbury, P.R.; Griffin, S.D.; Harris, M. Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J. Virol. 2012, 86, 10724–10732. [Google Scholar] [CrossRef] [PubMed]
- Funaoka, Y.; Sakamoto, N.; Suda, G.; Itsui, Y.; Nakagawa, M.; Kakinuma, S.; Watanabe, T.; Mishima, K.; Ueyama, M.; Onozuka, I.; et al. Analysis of Interferon Signaling by Infectious Hepatitis C Virus Clones with Substitutions of Core Amino Acids 70 and 91. J. Virol. 2011, 85, 5986–5994. [Google Scholar] [CrossRef]
- Chan, S.-W. Unfolded protein response in hepatitis C virus infection. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Amorim, R.; Temzi, A.; Griffin, B.D.; Mouland, A.J. Zika virus inhibits eIF2α-dependent stress granule assembly. PLoS Neglected Trop. Dis. 2017, 11, e0005775. [Google Scholar] [CrossRef]
- Turpin, J.; Frumence, E.; Wissal, H.; El Kalamouni, C.; Desprès, P.; Krejbich-Trotot, P.; Viranaicken, W. Crosstalk Between Endoplasmic Reticulum Stress and The Unfolded Protein Response During ZIKA Virus Infection. October 2019, 2. [Google Scholar] [CrossRef]
- Gaete-Argel, A.; Márquez, C.L.; Barriga, G.P.; Soto-Rifo, R.; Valiente-Echeverría, F. Strategies for Success. Viral Infections and Membraneless Organelles. Front. Microbiol. 2019, 9, 336. [Google Scholar] [CrossRef]
- Edgil, D.; Polacek, C.; Harris, E. Dengue Virus Utilizes a Novel Strategy for Translation Initiation When Cap-Dependent Translation Is Inhibited. J. Virol. 2006, 80, 2976–2986. [Google Scholar] [CrossRef]
- Song, Y.; Mugavero, J.; Stauft, C.B.; Wimmer, E.; Sarnow, P.; Rice, C. Dengue and Zika Virus 5’ Untranslated Regions Harbor Internal Ribosomal Entry Site Functions. mBio 2019, 10. [Google Scholar] [CrossRef]
- Roth, H.; Magg, V.; Uch, F.; Mutz, P.; Klein, P.; Haneke, K.; Lohmann, V.; Bartenschlager, R.; Fackler, O.T.; Locker, N.; et al. Flavivirus Infection Uncouples Translation Suppression from Cellular Stress Responses. mBio 2017, 8, e02150-16. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Götte, M.; Power, C.; et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J. Virol. 2017, 91, e00474-17. [Google Scholar] [CrossRef]
- Bonenfant, G.; Williams, N.; Netzband, R.; Schwarz, M.C.; Evans, M.J.; Pager, C.T. Zika Virus Subverts Stress Granules To Promote and Restrict Viral Gene Expression. J. Virol. 2019, 93, e00520-19. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Okamoto, T.; Fukuhara, T.; Kambara, H.; Morita, E.; Mori, Y.; Kamitani, W.; Matsuura, Y. Japanese Encephalitis Virus Core Protein Inhibits Stress Granule Formation through an Interaction with Caprin-1 and Facilitates Viral Propagation. J. Virol. 2012, 87, 489–502. [Google Scholar] [CrossRef]
- Dang, J.W.; Tiwari, S.K.; Qin, Y.; Rana, T.M. Genome-wide Integrative Analysis of Zika-Virus-Infected Neuronal Stem Cells Reveals Roles for MicroRNAs in Cell Cycle and Stemness. Cell Rep. 2019, 27, 3618–3628.e5. [Google Scholar] [CrossRef]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Baginski, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kötter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2017, 46, D303–D307. [Google Scholar] [CrossRef]
- Pereira-Montecinos, C.; Valiente-Echeverria, F.; Rifo, R.S. Epitranscriptomic regulation of viral replication. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1860, 460–471. [Google Scholar] [CrossRef]
- McIntyre, W.; Netzband, R.; Bonenfant, G.; Biegel, J.M.; Miller, C.; Fuchs, G.; Henderson, E.; Arra, M.; Canki, M.; Fabris, D.; et al. Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection. Nucleic Acids Res. 2018, 46, 5776–5791. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Meyer, K. m6A-mediated translation regulation. Biochim. Biophys. Acta (BBA) Bioenerg. 2019, 1862, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Lichinchi, G.; Zhao, B.; Wu, Y.; Lu, Z.; Qin, Y.; He, C.; Rana, T.M. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe 2016, 20, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.; McIntyre, A.; McFadden, M.J.; Roder, A.E.; Kennedy, E.M.; Gandara, J.A.; Hopcraft, S.E.; Quicke, K.M.; Vazquez, C.; Willer, J.; et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe 2016, 20, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, C. Reading RNA methylation codes through methyl-specific binding proteins. RNA Boil. 2014, 11, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, N.S.; McIntyre, A.B.; Mattocks, M.D.; Holley, C.L.; LaZear, H.M.; Mason, C.E.; Horner, S.M. Altered m6A Modification of Specific Cellular Transcripts Affects Flaviviridae Infection. Mol. Cell 2019, 77, 542–555.e8. [Google Scholar] [CrossRef] [PubMed]
- Mason, C.E.; McIntyre, A.B.; Gokhale, N.S.; Cerchietti, L.; Horner, S.M. Methods and Tools for MeRIP-Seq and Nanopore Detection of m6A(m) Changes. J. Biomol. Tech. JBT 2019, 30, S56. [Google Scholar]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmiedel, J.; Friedman, N.; Le-Trilling, V.T.K.; et al. M6A Modification Controls the Innate Immune Response to Infection by Targeting Type I Interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef]
- Rubio, R.M.; Depledge, D.; Bianco, C.; Thompson, L.; Mohr, I. RNA m6 A modification enzymes shape innate responses to DNA by regulating interferon β. Genes Dev. 2018, 32, 1472–1484. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Tang, H.; Shen, Y.; Gong, Z.; Xie, N.; Zhang, X.; Wang, W.; Kong, W.; Zhou, Y.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am. J. Physiol. Physiol. 2019, 317, C762–C775. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2013, 505, 117–120. [Google Scholar] [CrossRef]
- Ries, R.J.; Zaccara, S.; Klein, P.; Olarerin-George, A.; Namkoong, S.; Pickering, B.F.; Patil, D.P.; Kwak, H.; Lee, J.H.; Jaffrey, S.R. m6A enhances the phase separation potential of mRNA. Nature 2019, 571, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Göertz, G.; Abbo, S.R.; Fros, J.; Pijlman, G. Functional RNA during Zika virus infection. Virus Res. 2018, 254, 41–53. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oyarzún-Arrau, A.; Alonso-Palomares, L.; Valiente-Echeverría, F.; Osorio, F.; Soto-Rifo, R. Crosstalk between RNA Metabolism and Cellular Stress Responses during Zika Virus Replication. Pathogens 2020, 9, 158. https://doi.org/10.3390/pathogens9030158
Oyarzún-Arrau A, Alonso-Palomares L, Valiente-Echeverría F, Osorio F, Soto-Rifo R. Crosstalk between RNA Metabolism and Cellular Stress Responses during Zika Virus Replication. Pathogens. 2020; 9(3):158. https://doi.org/10.3390/pathogens9030158
Chicago/Turabian StyleOyarzún-Arrau, Aarón, Luis Alonso-Palomares, Fernando Valiente-Echeverría, Fabiola Osorio, and Ricardo Soto-Rifo. 2020. "Crosstalk between RNA Metabolism and Cellular Stress Responses during Zika Virus Replication" Pathogens 9, no. 3: 158. https://doi.org/10.3390/pathogens9030158
APA StyleOyarzún-Arrau, A., Alonso-Palomares, L., Valiente-Echeverría, F., Osorio, F., & Soto-Rifo, R. (2020). Crosstalk between RNA Metabolism and Cellular Stress Responses during Zika Virus Replication. Pathogens, 9(3), 158. https://doi.org/10.3390/pathogens9030158