Uncovering the First Atypical DS-1-like G1P[8] Rotavirus Strains That Circulated during Pre-Rotavirus Vaccine Introduction Era in South Africa


 , and
, and 
Abstract
:1. Introduction
2. Results
2.1. Nucleotide Sequencing
2.2. Full-Genome Constellation Analysis
2.3. Sequence and Phylogenetic Analysis
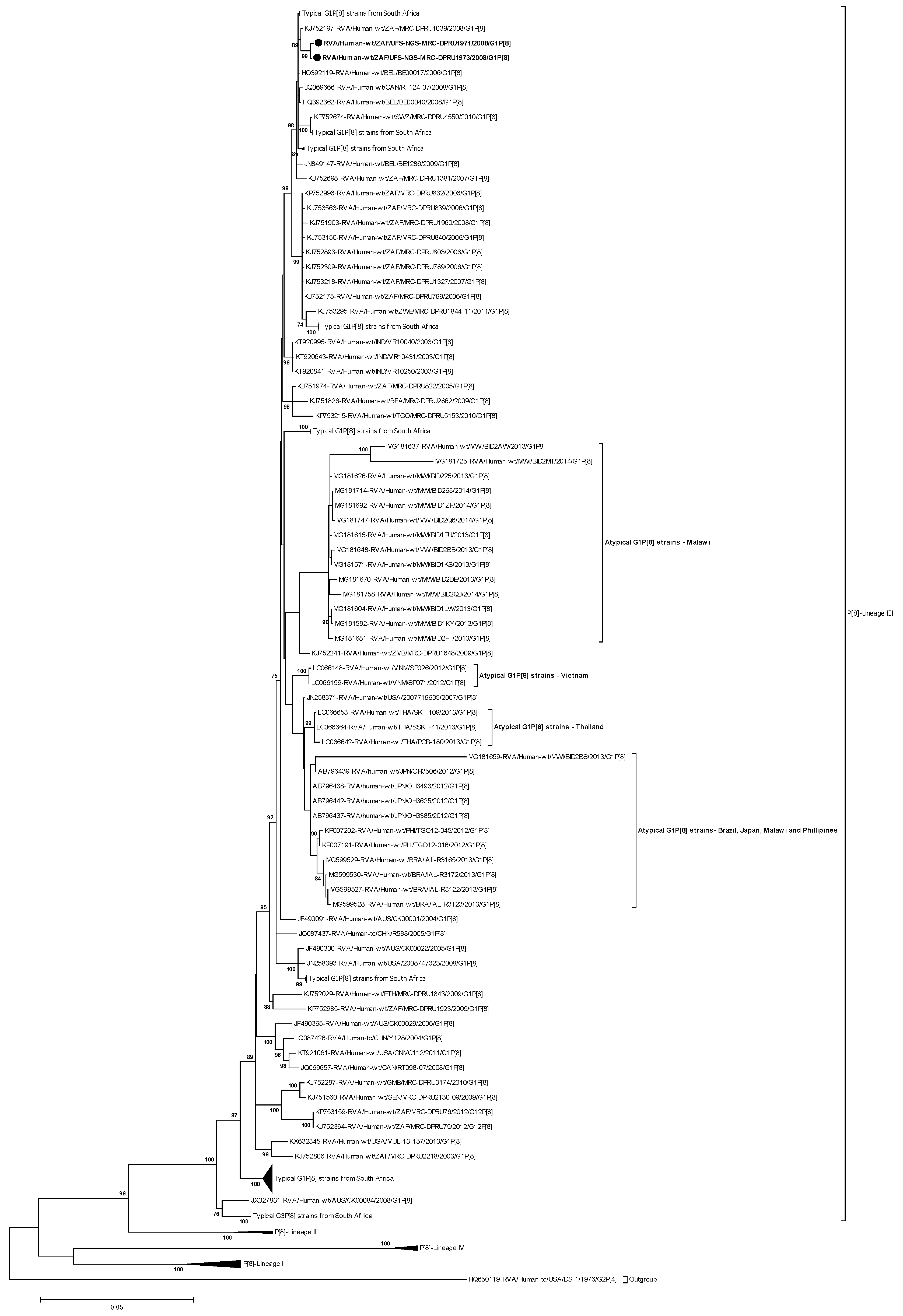
2.3.1. Phylogenetic Analysis of VP7
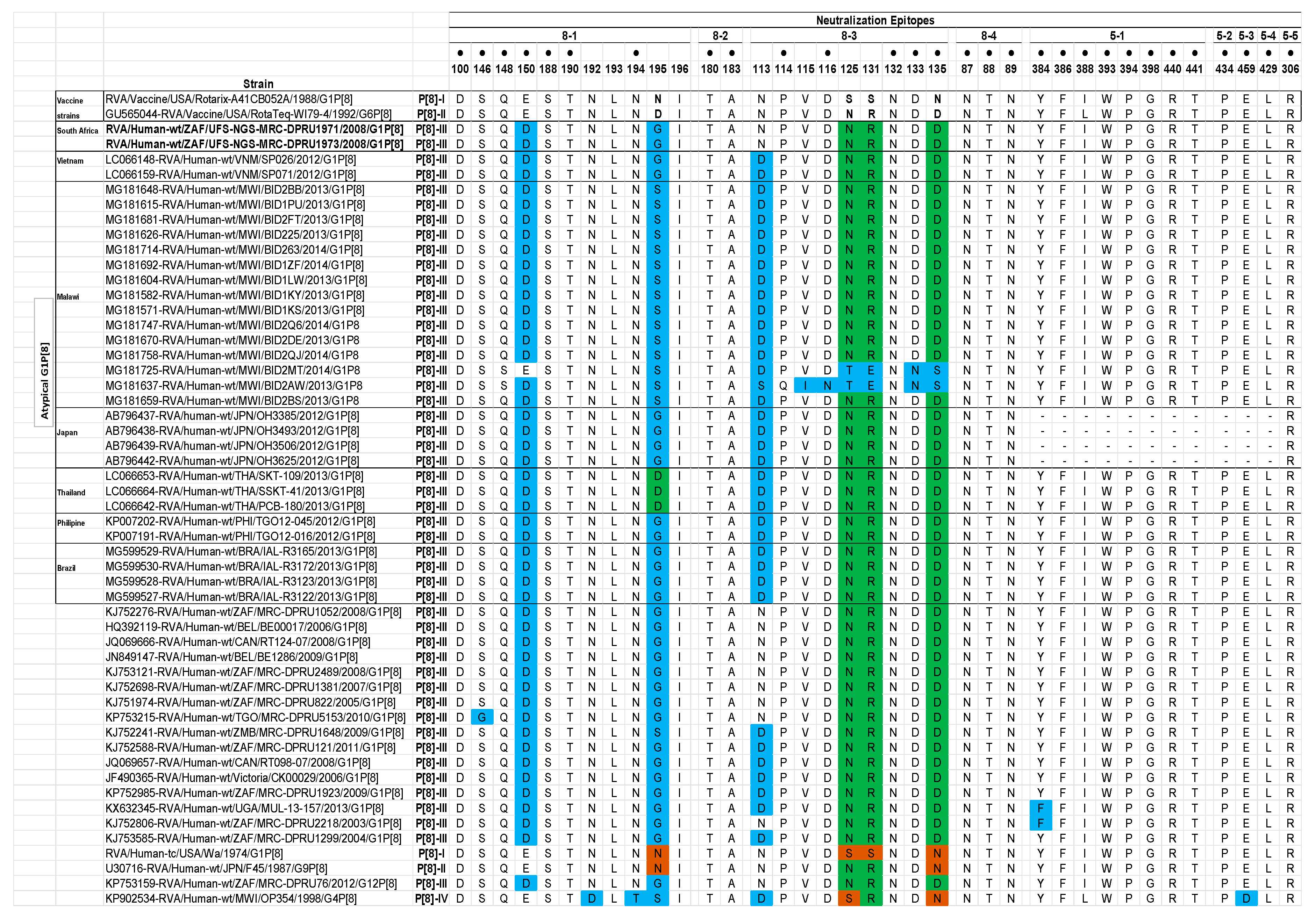
Analysis of the VP7 Neutralization Epitopes
2.3.2. Phylogenetic analysis of VP4
Analysis of the VP4 Neutralization Epitopes
2.3.3. Phylogenetic Analysis of VP1–VP3 and VP6
2.3.4. Phlylogenetic Analysis of NSP1–NSP5
2.4. Reassortment Analysis
3. Discussion
4. Materials and Methods
4.1. Ethics Approval
4.2. Sample Collection
4.3. Extraction and Purification of Double-Stranded RNA
4.4. Synthesis and Purification of Complementary DNA (cDNA)
4.5. DNA Library Preparation and Whole-Genome Sequencing
4.6. Genome Assembly
4.7. Determination of Rotavirus Whole-Genotype Constellations
4.8. Phylogenetic Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization Causes of Mild Mortality. (WHO 2019). Available online: https://www.who.int/gho/child_health/mortality/causes/en/ (accessed on 20 August 2019).
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus vaccination and the global burden of rotavirus diarrhea among children younger than 5 years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Tate, J.E.; Burton, A.H.; Boschi-Pinto, C.; Parashar, U.D.; World Health Organization–Coordinated Global Rotavirus Surveillance Network; Agocs, M.; Serhan, F.; de Oliveira, L.; Mwenda, J.M.; Mihigo, R.; et al. Global, regional, and national estimates of rotavirus mortality in children <5 years of age, 2000–2013. Clin. Infect. Dis. 2016, 62 (Suppl. 2), S96–S105. [Google Scholar]
- World Health Organization. Meeting of the immunization Strategic Advisory Group of Experts. Wkly. Epidemiol. Rec. 2009, 84, 220–236. [Google Scholar]
- World Health Organization. WHO Prequalifies New Rotavirus Vaccine. Available online: https://www.who.int/medicines/news/2018/prequalified_new-rotavirus_vaccine/en/ (accessed on 19 December 2019).
- Anh, D.D.; Van Trang, N.; Thiem, V.D.; Anh, N.T.H.; Mao, N.D.; Wang, Y.; Jiang, B.; Hien, N.D.; Rotavin-M1 Vaccine Trial Group. A dose-escalation safety and immunogenicity study of a new live attenuated human rotavirus vaccine (Rotavin-M1) in Vietnamese children. Vaccine 2012, 30, A114–A121. [Google Scholar] [CrossRef]
- Fu, C.; Wang, M.; Liang, J.; He, T.; Wang, D.; Xu, J. Effectiveness of Lanzhou lamb rotavirus vaccine against rotavirus gastroenteritis requiring hospitalization: A matched case-control study. Vaccine 2007, 25, 8756–8761. [Google Scholar] [CrossRef]
- Kirkwood, C.D.; Steele, A.D. Rotavirus Vaccines in China. Jama Netw. Open 2018, 1, e181579. [Google Scholar] [CrossRef]
- Madhi, S.A.; Cunliffe, N.A.; Steele, D.; Witte, D.; Kirsten, M.; Louw, C.; Ngwira, B.; Victor, J.C.; Gillard, P.H.; Cheuvart, B.B.; et al. Effect of human rotavirus vaccine on severe diarrhea in African infants. N. Engl. J. Med. 2010, 362, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Madhi, S.A.; Kirsten, M.; Louw, C.; Bos, P.; Aspinall, S.; Bouckenooghe, A.; Neuzil, K.M.; Steele, A.D. Efficacy and immunogenicity of two or three dose rotavirus-vaccine regimen in South African children over two consecutive rotavirus-seasons: A randomized, double-blind, placebo-controlled trial. Vaccine 2012, 30, A44–A51. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Ciarlet, M.; McDonald, S.M.; Attoui, H.; Bányai, K.; Brister, J.R.; Buesa, J.; Esona, M.D.; Estes, M.K.; Gentsch, J.R.; et al. Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 2011, 156, 1397–1413. [Google Scholar] [CrossRef] [Green Version]
- Maes, P.; Matthijnssens, J.; Rahman, M.; Van Ranst, M. RotaC: A web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol. 2009, 9, 238. [Google Scholar] [CrossRef] [Green Version]
- Dóró, R.; László, B.; Martella, V.; Leshem, E.; Gentsch, J.; Parashar, U.; Bányai, K. Review of global rotavirus strain prevalence data from six years post vaccine licensure surveillance: Is there evidence of strain selection from vaccine pressure? Infect. Genet. Evol. 2014, 28, 446–461. [Google Scholar] [CrossRef] [PubMed]
- Banyai, K.; Mijatovic-Rustempasic, S.; Hull, J.J.; Esona, M.D.; Freeman, M.M.; Frace, A.M.; Bowen, M.D.; Gentsch, J.R. Sequencing and phylogenetic analysis of the coding region of six common rotavirus strains: Evidence for intragenogroup reassortment among co-circulating G1P[8] and G2P[4] strains from the United States. J. Med. Virol. 2011, 83, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seheri, L.M.; Magagula, N.B.; Peenze, I.; Rakau, K.; Ndadza, A.; Mwenda, J.M.; Weldegebriel, G.; Steele, A.D.; Mphahlele, M.J. Rotavirus strain diversity in Eastern and Southern African countries before and after vaccine introduction. Vaccine 2018, 36, 7222–7230. [Google Scholar] [CrossRef]
- Mwenda, J.M.; Ntoto, K.M.; Abebe, A.; Enweronu-Laryea, C.; Amina, I.; Mchomvu, J.; Kisakye, A.; Mpabalwani, E.M.; Pazvakavambwa, I.; Armah, G.E.; et al. Burden and epidemiology of rotavirus diarrhea in selected African countries: Preliminary results from the African Rotavirus Surveillance Network. J. Infect. Dis. 2010, 202 (Suppl. 1), S5–S11. [Google Scholar] [CrossRef] [Green Version]
- Seheri, L.M.; Ngomane, G.; Page, N.A.; Mokomane, M.; Maphalala, G.P.; Weldegebriel, G.; Peenze, I.; Magagula, N.B.; Nyaga, M.M.; Lisoga, J.; et al. Outbreak investigation of diarrheal disease in Botswana and Eswatini in 2018. In Proceedings of the 12th African Rotavirus Symposium, Johannesburg, South Africa, 30 July–1 August 2019. [Google Scholar]
- Matthijnssens, J.; Van Ranst, M. Genotype constellation and evolution of group A rotaviruses infecting humans. Curr. Opin. Virol. 2012, 2, 426–433. [Google Scholar] [CrossRef]
- Do, L.P.; Nakagomi, T.; Otaki, H.; Agbemabiese, C.A.; Nakagomi, O.; Tsunemitsu, H. Phylogenetic inference of the porcine Rotavirus A origin of the human G1 VP7 gene. Infect. Genet. Evol. 2016, 40, 205–213. [Google Scholar] [CrossRef]
- Santos, F.S.; Junior, E.S.; Guerra, S.F.S.; Lobo, P.S.; Junior, E.P.; Lima, A.B.F.; Vinente, C.B.G.; Chagas, E.H.N.; Justino, M.C.A.; Linhares, A.C.; et al. G1P[8] Rotavirus in children with severe diarrhea in the post-vaccine introduction era in Brazil: Evidence of reassortments and structural modifications of the antigenic VP7 and VP4 regions. Infect. Genet. Evol. 2019, 69, 255–266. [Google Scholar] [CrossRef]
- Kirkwood, C.D. Genetic and antigenic diversity of human rotaviruses: Potential impact on vaccination programs. J. Infect. Dis. 2010, 202 (Suppl. 1), S43–S48. [Google Scholar] [CrossRef]
- Luchs, A.; da Costa, A.C.; Cilli, A.; Komninakis, S.C.V.; Carmona, R.D.C.C.; Morillo, S.G.; Sabino, E.C.; Timenetsky, M.D.C.S.T. First Detection of DS-1-like G1P[8] Double-gene Reassortant Rotavirus Strains on The American Continent, Brazil, 2013. Sci. Rep. 2019, 9, 2210. [Google Scholar] [CrossRef]
- Jere, K.C.; Chaguza, C.; Bar-Zeev, N.; Lowe, J.; Peno, C.; Kumwenda, B.; Nakagomi, O.; Tate, J.E.; Parashar, U.D.; Heyderman, R.S.; et al. Emergence of double-and triple-gene reassortant G1P[8] rotaviruses possessing a DS-1-like backbone after rotavirus vaccine introduction in Malawi. J. Virol. 2018, 92, e01246-17. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Nakagomi, T.; Nishimura, N.; Noguchi, A.; Miura, S.; Ito, H.; Doan, Y.H.; Takahashi, T.; Ozaki, T.; Katayama, K.; et al. Spread and predominance in Japan of novel G1P[8] double-reassortant rotavirus strains possessing a DS-1-like genotype constellation typical of G2P[4] strains. Infect. Genet. Evol. 2014, 28, 426–433. [Google Scholar] [CrossRef]
- Kuzuya, M.; Fujii, R.; Hamano, M.; Kida, K.; Mizoguchi, Y.; Kanadani, T.; Nishimura, K.; Kishimoto, T. Prevalence and molecular characterization of G1P[8] human rotaviruses possessing DS-1-like VP6, NSP4, and NSP5/6 in Japan. J. Med. Virol. 2014, 86, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.P.; Kaida, A.; Kubo, H.; Iritani, N. Gastroenteritis Outbreaks Caused by a DS-1–like G1P[8] Rotavirus Strain, Japan, 2012–2013. Emerg. Infect. Dis. 2014, 20, 1030. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Tacharoenmuang, R.; Guntapong, R.; Ide, T.; Tsuji, T.; Yoshikawa, T.; Tharmaphornpilas, P.; Sangkitporn, S.; Taniguchi, K. Reassortment of human and animal rotavirus gene segments in emerging DS-1-like G1P[8] rotavirus strains. PLoS ONE 2016, 11, e0148416. [Google Scholar] [CrossRef] [PubMed]
- Komoto, S.; Tacharoenmuang, R.; Guntapong, R.; Ide, T.; Haga, K.; Katayama, K.; Kato, T.; Ouchi, Y.; Kurahashi, H.; Tsuji, T.; et al. Emergence and characterization of unusual DS-1-like G1P[8] rotavirus strains in children with diarrhea in Thailand. PLoS ONE 2015, 10, e0141739. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, T.; Nguyen, M.Q.; Gauchan, P.; Agbemabiese, C.A.; Kaneko, M.; Do, L.P.; Vu, T.D.; Nakagomi, O. Evolution of DS-1-like G1P[8] double-gene reassortant rotavirus A strains causing gastroenteritis in children in Vietnam in 2012/2013. Arch. Virol. 2017, 162, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeller, M.; Heylen, E.; Tamim, S.; McAllen, J.K.; Kirkness, E.F.; Akopov, A.; De Coster, S.; Van Ranst, M.; Matthijnssens, J. Comparative analysis of the Rotarix™ vaccine strain and G1P[8] rotaviruses detected before and after vaccine introduction in Belgium. PeerJ 2017, 5, e2733. [Google Scholar] [CrossRef] [Green Version]
- Magagula, N.B.; Esona, M.D.; Nyaga, M.M.; Stucker, K.M.; Halpin, R.A.; Stockwell, T.B.; Seheri, M.L.; Steele, A.D.; Wentworth, D.E.; Mphahlele, M.J. Whole genome analyses of G1P[8] rotavirus strains from vaccinated and non-vaccinated South African children presenting with diarrhea. J. Med. Virol. 2015, 87, 79–101. [Google Scholar] [CrossRef] [Green Version]
- Shintani, T.; Ghosh, S.; Wang, Y.H.; Zhou, X.; Zhou, D.J.; Kobayashi, N. Whole genomic analysis of human G1P[8] rotavirus strains from different age groups in China. Viruses 2012, 4, 1289–1304. [Google Scholar] [CrossRef] [Green Version]
- Arora, R.; Chitambar, S.D. Full genomic analysis of Indian G1P[8] rotavirus strains. Infect. Genet. Evol. 2011, 11, 504–511. [Google Scholar] [CrossRef]
- Rahman, M.; Matthijnssens, J.; Saiada, F.; Hassan, Z.; Heylen, E.; Azim, T.; Van Ranst, M. Complete genomic analysis of a Bangladeshi G1P[8] rotavirus strain detected in 2003 reveals a close evolutionary relationship with contemporary human Wa-like strains. Infect. Genet. Evol. 2010, 10, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Arista, S.; Giammanco, G.M.; De Grazia, S.; Ramirez, S.; Biundo, C.L.; Colomba, C.; Cascio, A.; Martella, V. Heterogeneity and temporal dynamics of evolution of G1 human rotaviruses in a settled population. J. Virol. 2006, 80, 10724–10733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, S.T.; Settembre, E.C.; Trask, S.D.; Greenberg, H.B.; Harrison, S.C.; Dormitzer, P.R. Structure of rotavirus outer-layer protein VP7 bound with a neutralizing Fab. Science 2009, 324, 1444–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, V.P.; Chung, Y.C.; Kim, K.; Chung, S.I.; Lim, I.; Kim, W. Genetic variation of prevalent G1P[8] human rotaviruses in South Korea. J. Med. Virol. 2010, 82, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Ciarlet, M.; Hyser, J.M.; Estes, M.K. Sequence Analysis of the VP4, VP6, VP7, and NSP4 Gene Products of the Bovine Rotavirus WC3. Virus Genes 2002, 24, 107–118. [Google Scholar] [CrossRef]
- Zeller, M.; Patton, J.T.; Heylen, E.; De Coster, S.; Ciarlet, M.; Van Ranst, M.; Matthijnssens, J. Genetic analyses reveal differences in the VP7 and VP4 antigenic epitopes between human rotaviruses circulating in Belgium and rotaviruses in Rotarix and RotaTeq. J. Clin. Microbiol. 2012, 50, 966–976. [Google Scholar] [CrossRef] [Green Version]
- Guntapong, R.; Tacharoenmuang, R.; Singchai, P.; Upachai, S.; Sutthiwarakom, K.; Komoto, S.; Tsuji, T.; Tharmaphornpilas, P.; Yoshikawa, T.; Sangkitporn, S.; et al. Predominant prevalence of human rotaviruses with the G1P[8] and G8P[8] genotypes with a short RNA profile in 2013 and 2014 in Sukhothai and Phetchaboon provinces, Thailand. J. Med. Virol. 2017, 89, 615–620. [Google Scholar] [CrossRef]
- Arana, A.; Montes, M.; Jere, K.C.; Alkorta, M.; Iturriza-Gómara, M.; Cilla, G. Emergence and spread of G3P[8] rotaviruses possessing an equine-like VP7 and a DS-1-like genetic backbone in the Basque Country (North of Spain), 2015. Infect. Genet. Evol. 2016, 44, 137–144. [Google Scholar] [CrossRef]
- Cowley, D.; Donato, C.M.; Roczo-Farkas, S.; Kirkwood, C.D. Emergence of a novel equine-like G3P[8] inter-genogroup reassortant rotavirus strain associated with gastroenteritis in Australian children. J. Gen. Virol. 2016, 97, 403–410. [Google Scholar] [CrossRef]
- Guerra, S.F.S.; Soares, L.S.; Lobo, P.S.; Júnior, E.T.P.; Júnior, E.C.S.; Bezerra, D.A.M.; Vaz, L.R.; Linhares, A.C.; Mascarenhas, J.D.A.P. Detection of a novel equine-like G3 rotavirus associated with acute gastroenteritis in Brazil. J. Gen. Virol. 2016, 97, 3131–3138. [Google Scholar] [CrossRef]
- Abdel-Haq, N.M.; Thomas, R.A.; Asmar, B.I.; Zacharova, V.; Lyman, W.D. Increased prevalence of G1P[4] genotype among children with rotavirus-associated gastroenteritis in metropolitan Detroit. J. Clin. Microbiol. 2003, 41, 2680–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, M.E.S.; Pires, I.D.C.; Gouvea, V. 1998-1999 rotavirus seasons in Juiz de Fora, Minas Gerais, Brazil: Detection of an unusual G3P[4] epidemic strain. J. Clin. Microbiol. 2002, 40, 2837–2842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asmah, R.H.; Green, J.; Armah, G.E.; Gallimore, C.I.; Gray, J.J.; Iturriza-Gómara, M.; Anto, F.; Oduro, A.; Binka, F.N.; Brown, D.W.; et al. Rotavirus G and P genotypes in rural Ghana. J. Clin. Microbiol. 2001, 39, 1981–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulson, B.S.; Kirkwood, C. Relation of VP7 amino acid sequence to monoclonal antibody neutralization of rotavirus and rotavirus monotype. J. Virol. 1991, 65, 5968–5974. [Google Scholar] [CrossRef] [Green Version]
- Betts, M.J.; Russell, R.B. Amino acid properties and consequences of substitutions. Bioinform. Genet. 2003, 317, 289. [Google Scholar]
- Caust, J.; Dyall-Smith, M.L.; Lazdins, I.; Holmes, I.H. Glycosylation, an important modifier of rotavirus antigenicity. Arch. Virol. 1987, 96, 123–134. [Google Scholar] [CrossRef]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; Van Dijk, A.A. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 2009, 90, 1423–1432. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Esona, M.D.; Roy, S.; Rungsrisuriyachai, K.; Gautam, R.; Hermelijn, S.; Rey-Benito, G.; Bowen, M.D. Molecular characterization of a human G20P[28] rotavirus a strain with multiple genes related to bat rotaviruses. Infect. Genet. Evol. 2018, 57, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Esona, M.D.; Roy, S.; Rungsrisuriyachai, K.; Sanchez, J.; Vasquez, L.; Gomez, V.; Rios, L.A.; Bowen, M.D.; Vazquez, M. Characterization of a triple-recombinant, reassortant rotavirus strain from the Dominican Republic. J. Gen. Virol. 2017, 98, 134. [Google Scholar] [CrossRef]
- Ward, M.L.; Mijatovic-Rustempasic, S.; Roy, S.; Rungsrisuriyachai, K.; Boom, J.A.; Sahni, L.C.; Baker, C.J.; Rench, M.A.; Wikswo, M.E.; Payne, D.C.; et al. Molecular characterization of the first G24P[14] rotavirus strain detected in humans. Infect. Genet. Evol. 2016, 43, 338–342. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Rapp, B.A.; Wheeler, D.L. GenBank. Nucleic Acids Res. 2000, 28, 15–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Segment | VP7 | VP4 | VP6 | VP1 | VP2 | VP3 | NSP1 | NSP2 | NSP3 | NSP4 | NSP5 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Base pair size for full length sequences | 1062 | 2359 | 1355 | 3302 | 2684 | 2591 | 1566 | 1059 | 1066 | 751 | 810 |
| Base pair size for the complete study strain ORF | 978 | 2325 | 1191 | 3264 | 2637 | 2505 | 1458 | 951 | 939 | 525 | 600 |
| RVA/Human-wt/ZAF/UFS-NGS-MRC-DPRU1971/2008/G1P[8] | G1 | P[8] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
| RVA/Human-wt/ZAF/UFS-NGS-MRC-DPRU1973/2008/G1P[8] | G1 | P[8] | I2 | R2 | C2 | M2 | A2 | N2 | T2 | E2 | H2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mwangi, P.N.; Mogotsi, M.T.; Rasebotsa, S.P.; Seheri, M.L.; Mphahlele, M.J.; Ndze, V.N.; Dennis, F.E.; Jere, K.C.; Nyaga, M.M. Uncovering the First Atypical DS-1-like G1P[8] Rotavirus Strains That Circulated during Pre-Rotavirus Vaccine Introduction Era in South Africa. Pathogens 2020, 9, 391. https://doi.org/10.3390/pathogens9050391
Mwangi PN, Mogotsi MT, Rasebotsa SP, Seheri ML, Mphahlele MJ, Ndze VN, Dennis FE, Jere KC, Nyaga MM. Uncovering the First Atypical DS-1-like G1P[8] Rotavirus Strains That Circulated during Pre-Rotavirus Vaccine Introduction Era in South Africa. Pathogens. 2020; 9(5):391. https://doi.org/10.3390/pathogens9050391
Chicago/Turabian StyleMwangi, Peter N., Milton T. Mogotsi, Sebotsana P. Rasebotsa, Mapaseka L. Seheri, M. Jeffrey Mphahlele, Valantine N. Ndze, Francis E. Dennis, Khuzwayo C. Jere, and Martin M. Nyaga. 2020. "Uncovering the First Atypical DS-1-like G1P[8] Rotavirus Strains That Circulated during Pre-Rotavirus Vaccine Introduction Era in South Africa" Pathogens 9, no. 5: 391. https://doi.org/10.3390/pathogens9050391