The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Viral Genome, Proteins, and Lifecycle
3. Estrogen, Estrogen Receptors, and Estrogen-Receptor Signaling
4. HPV and Estrogen in Carcinogenesis
4.1. Cell Culture-Related Evidence for Estrogen Involvement in HPV Carcinogenesis
4.2. Animal Model Evidence for Estrogen Involvement in HPV Carcinogenesis
4.3. Clinical Evidence for Estrogen Involvement in HPV Carcinogenesis
5. HPV and Estrogen as a Possible Treatment Paradigm
5.1. Cell Culture-Related Evidence for Estrogen as an Hpv Treatment
5.2. Animal Model Evidence for Estrogen as an HPV Treatment
5.3. Clinical Evidence for Estrogen as an HPV Treatment
6. Estrogen, HPV, and Immune Function
7. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chesson, H.W.; Dunne, E.F.; Hariri, S.; Markowitz, L.E. The estimated lifetime probability of acquiring human papillomavirus in the United States. Sex. Transm. Dis. 2014, 41, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Brianti, P.; De Flammineis, E.; Mercuri, S.R. Review of HPV-related diseases and cancers. New Microbiol. 2017, 40, 80–85. [Google Scholar] [PubMed]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.D.; Chatterjee, S.; Alam, S.; Salzberg, A.C.; Milici, J.; van der Burg, S.H.; Meyers, C. Effect of Productive Human Papillomavirus 16 Infection on Global Gene Expression in Cervical Epithelium. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin-Drubin, M.E.; Meyers, C. Evidence for the coexistence of two genital HPV types within the same host cell in vitro. Virology 2004, 321, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin-Drubin, M.E.; Meyers, J.; Munger, K. Cancer associated human papillomaviruses. Curr. Opin. Virol. 2012, 2, 459–466. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A.; Münger, K. Expert Views on HPV Infection. Viruses 2018, 10, 94. [Google Scholar] [CrossRef] [Green Version]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Psyrri, A.; DiMaio, D. Human papillomavirus in cervical and head-and-neck cancer. Nat. Clin. Pract. Oncol. 2008, 5, 24–31. [Google Scholar] [CrossRef]
- Huh, W.K.; Joura, E.A.; Giuliano, A.R.; Iversen, O.-E.; de Andrade, R.P.; Ault, K.A.; Bartholomew, D.; Cestero, R.M.; Fedrizzi, E.N.; Hirschberg, A.L.; et al. Final efficacy, immunogenicity, and safety analyses of a nine-valent human papillomavirus vaccine in women aged 16-26 years: A randomised, double-blind trial. Lancet 2017, 390, 2143–2159. [Google Scholar] [CrossRef]
- Kavanagh, K.; Pollock, K.G.; Cuschieri, K.; Palmer, T.; Cameron, R.L.; Watt, C.; Bhatia, R.; Moore, C.; Cubie, H.; Cruickshank, M.; et al. Changes in the prevalence of human papillomavirus following a national bivalent human papillomavirus vaccination programme in Scotland: A 7-year cross-sectional study. Lancet Infect. Dis. 2017, 17, 1293–1302. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Human Papillomavirus (HPV) Infection. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 2007. [Google Scholar]
- Graham, S.V. The human papillomavirus replication cycle, and its links to cancer progression: A comprehensive review. Clin. Sci. 2017, 131, 2201–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S. Human papillomavirus: Gene expression, regulation and prospects for novel diagnostic methods and antiviral therapies. Future Microbiol. 2010, 5, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.-M.; Baker, C.C. Papillomavirus Genome Structure, Expression, and Post-Transcriptional Regulation. Front. Biosci. 2006, 11, 2286–2302. [Google Scholar] [CrossRef] [Green Version]
- Stünkel, W.; Bernard, H.-U. The Chromatin Structure of the Long Control Region of Human Papillomavirus Type 16 Represses Viral Oncoprotein Expression. J. Virol. 1999, 73, 1918–1930. [Google Scholar] [CrossRef] [Green Version]
- Kurvinen, K.; Yliskoski, M.; Saarikoski, S.; Syrjänen, K.; Syrjänen, S. Variants of the long control region of human papillomavirus type 16. Eur. J. Cancer 2000, 36, 1402–1410. [Google Scholar] [CrossRef]
- Ribeiro, A.L.; Caodaglio, A.S.; Sichero, L. Regulation of HPV transcription. Clinics 2018, 73. [Google Scholar] [CrossRef]
- Woodman, C.B.J.; Collins, S.I.; Young, L.S. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef]
- Chen, X.S.; Garcea, R.L.; Goldberg, I.; Casini, G.; Harrison, S.C. Structure of Small Virus-like Particles Assembled from the L1 Protein of Human Papillomavirus 16. Mol. Cell 2000, 5, 557–567. [Google Scholar] [CrossRef]
- Gravitt, P.E.; Peyton, C.L.; Apple, R.J.; Wheeler, C.M. Genotyping of 27 Human Papillomavirus Types by Using L1 Consensus PCR Products by a Single-Hybridization, Reverse Line Blot Detection Method. J. Clin. Microbiol. 1998, 36, 3020–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.J.; Park, J.S. Clinical significance of human papillomavirus genotyping. J. Gynecol. Oncol. 2016, 27. [Google Scholar] [CrossRef] [Green Version]
- Melendy, T.; Sedman, J.; Stenlund, A. Cellular factors required for papillomavirus DNA replication. J. Virol. 1995, 69, 7857–7867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narahari, J.; Fisk, J.C.; Melendy, T.; Roman, A. Interactions of the cellular CCAAT displacement protein and human papillomavirus E2 protein with the viral origin of replication can regulate DNA replication. Virology 2006, 350, 302–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clower, R.V.; Hu, Y.; Melendy, T. Papillomavirus E2 protein interacts with and stimulates human topoisomerase I. Virology 2006, 348, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Lehoux, M.; Fradet-Turcotte, A.; Archambault, J. Methods to assess the nucleocytoplasmic shuttling of the HPV E1 helicase and its effects on cellular proliferation and induction of a DNA damage response. Methods Mol. Biol. 2015, 1249, 67–80. [Google Scholar] [CrossRef]
- McBride, A.A. The Papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, M.D.; Lilley, C.E.; Chaurushiya, M.S. Genomes in conflict: Maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 2010, 64, 61–81. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [Green Version]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef]
- Kadaja, M.; Sumerina, A.; Verst, T.; Ojarand, M.; Ustav, E.; Ustav, M. Genomic instability of the host cell induced by the human papillomavirus replication machinery. EMBO J. 2007, 26, 2180–2191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardenas-Mora, J.; Spindler, J.E.; Jang, M.K.; McBride, A.A. Dimerization of the papillomavirus E2 protein is required for efficient mitotic chromosome association and Brd4 binding. J. Virol. 2008, 82, 7298–7305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [Green Version]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Suprynowicz, F.A.; Krawczyk, E.; Hebert, J.D.; Sudarshan, S.R.; Simic, V.; Kamonjoh, C.M.; Schlegel, R. The Human Papillomavirus Type 16 E5 Oncoprotein Inhibits Epidermal Growth Factor Trafficking Independently of Endosome Acidification. J. Virol. 2010, 84, 10619–10629. [Google Scholar] [CrossRef] [Green Version]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [Green Version]
- Songock, W.K.; Kim, S.-M.; Bodily, J.M. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res. 2017, 231, 56–75. [Google Scholar] [CrossRef] [Green Version]
- Hoppe-Seyler, K.; Honegger, A.; Bossler, F.; Sponagel, J.; Bulkescher, J.; Lohrey, C.; Hoppe-Seyler, F. Viral E6/E7 oncogene and cellular hexokinase 2 expression in HPV-positive cancer cell lines. Oncotarget 2017, 8, 106342–106351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2017, 772, 23–35. [Google Scholar] [CrossRef] [PubMed]
- White, E.A. Manipulation of Epithelial Differentiation by HPV Oncoproteins. Viruses 2019, 11, 369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajitani, N.; Satsuka, A.; Kawate, A.; Sakai, H. Productive Lifecycle of Human Papillomaviruses that Depends Upon Squamous Epithelial Differentiation. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Stanley, M. Pathology and epidemiology of HPV infection in females. Gynecol. Oncol. 2010, 117, S5–S10. [Google Scholar] [CrossRef]
- Li, V.C.; Kirschner, M.W. Molecular ties between the cell cycle and differentiation in embryonic stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 9503–9508. [Google Scholar] [CrossRef] [Green Version]
- Reinson, T.; Henno, L.; Toots, M.; Ustav, M.; Ustav, M. The Cell Cycle Timing of Human Papillomavirus DNA Replication. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Pyeon, D.; Lambert, P.F.; Ahlquist, P. Production of infectious human papillomavirus independently of viral replication and epithelial cell differentiation. Proc. Natl. Acad. Sci. USA 2005, 102, 9311–9316. [Google Scholar] [CrossRef] [Green Version]
- Buitrago-Pérez, Á.; Garaulet, G.; Vázquez-Carballo, A.; Paramio, J.M.; García-Escudero, R. Molecular Signature of HPV-Induced Carcinogenesis: pRb, p53 and Gene Expression Profiling. Curr. Genom. 2009, 10, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Yim, E.-K.; Park, J.-S. The Role of HPV E6 and E7 Oncoproteins in HPV-associated Cervical Carcinogenesis. Cancer Res. Treat. 2005, 37, 319–324. [Google Scholar] [CrossRef] [Green Version]
- Yugawa, T.; Kiyono, T. Molecular mechanisms of cervical carcinogenesis by high-risk human papillomaviruses: Novel functions of E6 and E7 oncoproteins. Rev. Med. Virol. 2009, 19, 97–113. [Google Scholar] [CrossRef]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef] [Green Version]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pett, M.; Coleman, N. Integration of high-risk human papillomavirus: A key event in cervical carcinogenesis? J. Pathol. 2007, 212, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Arias-Pulido, H.; Peyton, C.L.; Joste, N.E.; Vargas, H.; Wheeler, C.M. Human Papillomavirus Type 16 Integration in Cervical Carcinoma In Situ and in Invasive Cervical Cancer. J. Clin. Microbiol. 2006, 44, 1755–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.P.; Friedman, C.L.; Bryant, E.M.; McDougall, J.K. Viral integration and fragile sites in human papillomavirus-immortalized human keratinocyte cell lines. Genes Chromosomes Cancer 1992, 5, 150–157. [Google Scholar] [CrossRef]
- Ferber, M.J.; Thorland, E.C.; Brink, A.A.; Rapp, A.K.; Phillips, L.A.; McGovern, R.; Gostout, B.S.; Cheung, T.H.; Chung, T.K.H.; Fu, W.Y.; et al. Preferential integration of human papillomavirus type 18 near the c- myc locus in cervical carcinoma. Oncogene 2003, 22, 7233–7242. [Google Scholar] [CrossRef] [Green Version]
- Peter, M.; Rosty, C.; Couturier, J.; Radvanyi, F.; Teshima, H.; Sastre-Garau, X. MYC activation associated with the integration of HPV DNA at the MYC locus in genital tumors. Oncogene 2006, 25, 5985–5993. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Homaei, A.; Raju, A.B.; Meher, B.R. Estrogen: The necessary evil for human health, and ways to tame it. Biomed. Pharmacother. 2018, 102, 403–411. [Google Scholar] [CrossRef]
- Jia, M.; Dahlman-Wright, K.; Gustafsson, J.-Å. Estrogen receptor alpha and beta in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 557–568. [Google Scholar] [CrossRef]
- Cauley, J.A. Estrogen and bone health in men and women. Steroids 2015, 99, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 54–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The role of estrogens in control of energy balance and glucose homeostasis. Endocr. Rev. 2013, 34, 309–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-L.; Herndon, C. New roles for neuronal estrogen receptors. Neurogastroenterol. Motil. 2017, 29. [Google Scholar] [CrossRef]
- Simpson, E.R. Sources of estrogen and their importance. J. Steroid Biochem. Mol. Biol. 2003, 86, 225–230. [Google Scholar] [CrossRef]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef]
- Hariri, L.; Rehman, A. Estradiol. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Wasada, T.; Akamine, Y.; Kato, K.; Ibayashi, H.; Nomura, Y. Adrenal contribution to circulating estrogens in woman. Endocrinol. Jpn. 1978, 25, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Caroccia, B.; Seccia, T.; Barton, M.; Rossi, G.P. Estrogen Signaling in the Adrenal Cortex. Hypertension 2016, 68, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Nelson, L.R.; Bulun, S.E. Estrogen production and action. J. Am. Acad. Dermatol. 2001, 45, S116–S124. [Google Scholar] [CrossRef]
- Barakat, R.; Oakley, O.; Kim, H.; Jin, J.; Ko, C.J. Extra-gonadal sites of estrogen biosynthesis and function. BMB Rep. 2016, 49, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during ageing: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, R.F.; Bonnar, J. Clinical uses of estrogens. Pharmacol. Ther. 1980, 11, 451–467. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Itonaga, T.; Ikegawa, K.; Nishigaki, S.; Kawai, M.; Koga, E.; Sakakibara, H.; Ross, J.L. Ultra-low-dose estrogen therapy for female hypogonadism. Clin. Pediatr. Endocrinol. 2020, 29, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Cannarella, R.; Condorelli, R.A.; Mongioì, L.M.; Barbagallo, F.; Calogero, A.E.; La Vignera, S. Effects of the selective estrogen receptor modulators for the treatment of male infertility: A systematic review and meta-analysis. Expert Opin. Pharmacother. 2019, 20, 1517–1525. [Google Scholar] [CrossRef]
- Marquardt, R.M.; Kim, T.H.; Shin, J.-H.; Jeong, J.-W. Progesterone and Estrogen Signaling in the Endometrium: What Goes Wrong in Endometriosis? Int. J. Mol. Sci. 2019, 20, 3800. [Google Scholar] [CrossRef] [Green Version]
- Regidor, P.-A. Clinical relevance in present day hormonal contraception. Horm. Mol. Biol. Clin. Investig. 2018, 37. [Google Scholar] [CrossRef]
- Dobbs, R.W.; Malhotra, N.R.; Greenwald, D.T.; Wang, A.Y.; Prins, G.S.; Abern, M.R. Estrogens and prostate cancer. Prostate Cancer Prostatic Dis. 2019, 22, 185–194. [Google Scholar] [CrossRef]
- Nakano, T.; Kadono, Y.; Iwamoto, H.; Yaegashi, H.; Iijima, M.; Kawaguchi, S.; Nohara, T.; Shigehara, K.; Izumi, K.; Mizokami, A. Therapeutic Effect of Ethinylestradiol in Castration-resistant Prostate Cancer. Anticancer Res. 2020, 40, 2291–2296. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. Estrogen Biology: New Insights into GPER Function and Clinical Opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Xia, W.; Nie, M.; Sun, Y.; Jiang, Y.; Zhao, J.; He, S.; Xu, L. Association of ESR1 and C6orf97 gene polymorphism with osteoporosis in postmenopausal women. Mol. Biol. Rep. 2014, 41, 3235–3243. [Google Scholar] [CrossRef] [PubMed]
- ESR1 Gene—GeneCards|ESR1 Protein|ESR1 Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ESR1 (accessed on 28 April 2020).
- ESR2 Gene—GeneCards|ESR2 Protein|ESR2 Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=ESR2&keywords=esr2 (accessed on 28 April 2020).
- Pirskanen, M.; Hiltunen, M.; Mannermaa, A.; Helisalmi, S.; Lehtovirta, M.; Hänninen, T.; Soininen, H. Estrogen receptor beta gene variants are associated with increased risk of Alzheimer’s disease in women. Eur. J. Hum. Genet. 2005, 13, 1000–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GPER1 Gene—GeneCards|GPER1 Protein|GPER1 Antibody. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=GPER1 (accessed on 28 April 2020).
- Vajaria, R.; Vasudevan, N. Is the membrane estrogen receptor, GPER1, a promiscuous receptor that modulates nuclear estrogen receptor-mediated functions in the brain? Horm. Behav. 2018, 104, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Paterni, I.; Granchi, C.; Katzenellenbogen, J.A.; Minutolo, F. Estrogen Receptors Alpha (ERα) and Beta (ERβ): Subtype-Selective Ligands and Clinical Potential. Steroids 2014, 90, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Helguero, L.A.; Faulds, M.H. Estrogen receptors alfa (ERa) and beta (ERb) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene 2005, 24, 6605–6616. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, L.; James, T.; Jung, Y.; Kim, I.; Tan, R.; Hoffmann, F.M.; Xu, W. Reciprocal Regulation of ERα and ERβ Stability and Activity by Diptoindonesin G. Chem. Biol. 2015, 22, 1608–1621. [Google Scholar] [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685. [Google Scholar] [CrossRef]
- deGraffenried, L.A.; Hilsenbeck, S.G.; Fuqua, S.A.W. Sp1 is essential for estrogen receptor alpha gene transcription. J. Steroid Biochem. Mol. Biol. 2002, 82, 7–18. [Google Scholar] [CrossRef]
- Hong, K.; Choi, Y. Role of estrogen and RAS signaling in repeated implantation failure. BMB Rep. 2018, 51, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; He, L.; Zhang, Y.; Zhao, J.; Liu, Z.; Xing, F.; Liu, M.; Feng, Z.; Li, W.; Zhang, J. Estrogen receptor alpha and beta regulate actin polymerization and spatial memory through an SRC-1/mTORC2-dependent pathway in the hippocampus of female mice. J. Steroid Biochem. Mol. Biol. 2017, 174, 96–113. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.J.; Daniel, J.M. Nuclear estrogen receptor activation by insulin-like growth factor-1 in Neuro-2A neuroblastoma cells requires endogenous estrogen synthesis and is mediated by mutually repressive MAPK and PI3K cascades. Mol. Cell. Endocrinol. 2019, 490, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Tanjak, P.; Thiantanawat, A.; Watcharasit, P.; Satayavivad, J. Genistein reduces the activation of AKT and EGFR, and the production of IL6 in cholangiocarcinoma cells involving estrogen and estrogen receptors. Int. J. Oncol. 2018, 53, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Auborn, K.; James, C. Altered growth and viral gene expression in human papillomavirus type 16-containing cancer cell lines treated with progesterone. Cancer Investig. 1999, 17, 19–29. [Google Scholar] [CrossRef]
- c-Jun/AP-1 Overexpression Reprograms ERα Signaling Related to Tamoxifen Response in ERα-Positive Breast Cancer. Available online: https://www.ncbi.nlm.nih.gov/pubmed/29467493 (accessed on 13 April 2020).
- Fiorillo, A.A.; Heier, C.R.; Huang, Y.-F.; Tully, C.B.; Punga, T.; Punga, A.R. Estrogen Receptor, Inflammatory, and FOXO Transcription Factors Regulate Expression of Myasthenia Gravis-Associated Circulating microRNAs. Front. Immunol. 2020, 11, 151. [Google Scholar] [CrossRef] [Green Version]
- Shimamoto, K.; Tanimoto, K.; Fukazawa, T.; Nakamura, H.; Kanai, A.; Bono, H.; Ono, H.; Eguchi, H.; Hirohashi, N. GLIS1, a novel hypoxia-inducible transcription factor, promotes breast cancer cell motility via activation of WNT5A. Carcinogenesis 2020. [Google Scholar] [CrossRef]
- Dai, R.; Phillips, R.A.; Karpuzoglu, E.; Khan, D.; Ahmed, S.A. Estrogen regulates transcription factors STAT-1 and NF-kappaB to promote inducible nitric oxide synthase and inflammatory responses. J. Immunol. 2009, 183, 6998–7005. [Google Scholar] [CrossRef] [Green Version]
- Rudraraju, B.; Droog, M.; Abdel-Fatah, T.M.A.; Zwart, W.; Giannoudis, A.; Malki, M.I.; Moore, D.; Patel, H.; Shaw, J.; Ellis, I.O.; et al. Phosphorylation of activating transcription factor-2 (ATF-2) within the activation domain is a key determinant of sensitivity to tamoxifen in breast cancer. Breast Cancer Res. Treat. 2014, 147, 295–309. [Google Scholar] [CrossRef]
- Li, S.; Xie, L.; Du, M.; Xu, K.; Zhu, L.; Chu, H.; Chen, J.; Wang, M.; Zhang, Z.; Gu, D. Association study of genetic variants in estrogen metabolic pathway genes and colorectal cancer risk and survival. Arch. Toxicol. 2018, 92, 1991–1999. [Google Scholar] [CrossRef]
- Zeng, C.; Xu, J.-N.; Zhou, Y.; Yang, H.-X.; Zhou, Y.-F.; Xue, Q. C-Jun NH2-Terminal Kinase and p38 Inhibition Suppresses Prostaglandin E2-Stimulated Aromatase and Estrogen Receptor Levels in Human Endometriosis. J. Clin. Endocrinol. Metab. 2015, 100, E1404–E1414. [Google Scholar] [CrossRef]
- Barreto-Andrade, J.N.; de Fátima, L.A.; Campello, R.S.; Guedes, J.A.C.; de Freitas, H.S.; Machado, M.M.O.U.F. Estrogen Receptor 1 (ESR1) Enhances Slc2a4/GLUT4 Expression by a SP1 Cooperative Mechanism. Int. J. Med. Sci. 2018, 15, 1320–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas, A.Z.; Nascimento, A.M.; Medeiros, A.R.S.; Caliman, I.F.; Dalpiaz, P.L.M.; Firmes, L.B.; Sousa, G.J.; Oliveira, P.W.C.; Andrade, T.U.; Reis, A.M.; et al. The selective estrogen receptor modulators (SERMs) raloxifene and tamoxifen improve ANP levels and decrease nuclear translocation of NF-kB in estrogen-deficient rats. Pharmacol. Rep. 2017, 69, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Cavalcanti, F.N.; Lucas, T.F.G.; Lazari, M.F.M.; Porto, C.S. Estrogen receptor ESR1 mediates activation of ERK1/2, CREB, and ELK1 in the corpus of the epididymis. J. Mol. Endocrinol. 2015, 54, 339–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourguignon, L.Y.W.; Gilad, E.; Rothman, K.; Peyrollier, K. Hyaluronan-CD44 interaction with IQGAP1 promotes Cdc42 and ERK signaling, leading to actin binding, Elk-1/estrogen receptor transcriptional activation, and ovarian cancer progression. J. Biol. Chem. 2005, 280, 11961–11972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciocca, D.R.; Fanelli, M.A. Estrogen receptors and cell proliferation in breast cancer. Trends Endocrinol. Metab. 1997, 8, 313–321. [Google Scholar] [CrossRef]
- Chalbos, D.; Vignon, F.; Keydar, I.; Rochefort, H. Estrogens stimulate cell proliferation and induce secretory proteins in a human breast cancer cell line (T47D). J. Clin. Endocrinol. Metab. 1982, 55, 276–283. [Google Scholar] [CrossRef]
- Tan, H.; Zhong, Y.; Pan, Z. Autocrine regulation of cell proliferation by estrogen receptor-alpha in estrogen receptor-alpha-positive breast cancer cell lines. BMC Cancer 2009, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Katzenellenbogen, B.S.; Kendra, K.L.; Norman, M.J.; Berthois, Y. Proliferation, Hormonal Responsiveness, and Estrogen Receptor Content of MCF-7 Human Breast Cancer Cells Grown in the Short-Term and Long-Term Absence of Estrogens. Cancer Res. 1987, 47, 4355–4360. [Google Scholar]
- Foster, J.S.; Henley, D.C.; Ahamed, S.; Wimalasena, J. Estrogens and cell-cycle regulation in breast cancer. Trends Endocrinol. Metab. 2001, 12, 320–327. [Google Scholar] [CrossRef]
- Foster, J.S.; Henley, D.C.; Bukovsky, A.; Seth, P.; Wimalasena, J. Multifaceted Regulation of Cell Cycle Progression by Estrogen: Regulation of Cdk Inhibitors and Cdc25A Independent of Cyclin D1-Cdk4 Function. Mol. Cell. Biol. 2001, 21, 794–810. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.-H.; Franceschi, S.; Lambert, P.F. Estrogen and ERα: Culprits in Cervical Cancer? Trends Endocrinol. Metab. 2010, 21, 504–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auborn, K.J.; Woodworth, C.; DiPaolo, J.A.; Bradlow, H.L. The interaction between HPV infection and estrogen metabolism in cervical carcinogenesis. Int. J. Cancer 1991, 49, 867–869. [Google Scholar] [CrossRef] [PubMed]
- Caldon, C.E.; Sutherland, R.L.; Musgrove, E.A. Cell cycle proteins in epithelial cell differentiation: Implications for breast cancer. Cell Cycle 2010, 9, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef]
- Liang, J.; Shang, Y. Estrogen and Cancer. Ann. Rev. Physiol. 2013, 75, 225–240. [Google Scholar] [CrossRef] [Green Version]
- Hellberg, D. Sex Steroids and Cervical Cancer. Anticancer Res. 2012, 32, 3045–3054. [Google Scholar]
- Mitrani-Rosenbaum, S.; Tsvieli, R.; Tur-Kaspa, R. Oestrogen Stimulates Differential Transcription of Human Papillomavirus Type 16 in SiHa Cervical Carcinoma Cells. J. Gen. Virol. 1989, 70 Pt 8, 2227–2232. [Google Scholar] [CrossRef]
- Sun, Q.; Liang, Y.; Zhang, T.; Wang, K.; Yang, X. ER-α36 mediates estrogen-stimulated MAPK/ERK activation and regulates migration, invasion, proliferation in cervical cancer cells. Biochem. Biophys. Res. Commun. 2017, 487, 625–632. [Google Scholar] [CrossRef]
- Tomaszewska, A.; Roszak, A.; Pawlik, P.; Sajdak, S.; Jagodziński, P.P. Increased 17ß-hydroxysteroid dehydrogenase type 1 levels in primary cervical cancer. Biomed. Pharmacother. 2015, 72, 179–183. [Google Scholar] [CrossRef]
- Jabbar, S.F.; Abrams, L.; Glick, A.; Lambert, P.F. Persistence of high-grade cervical dysplasia and cervical cancer requires the continuous expression of the human papillomavirus type 16 E7 oncogene. Cancer Res. 2009, 69, 4407–4414. [Google Scholar] [CrossRef] [Green Version]
- Riley, R.R.; Duensing, S.; Brake, T.; Münger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of Human Papillomavirus E6 and E7 Function in Transgenic Mouse Models of Cervical Carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar] [PubMed]
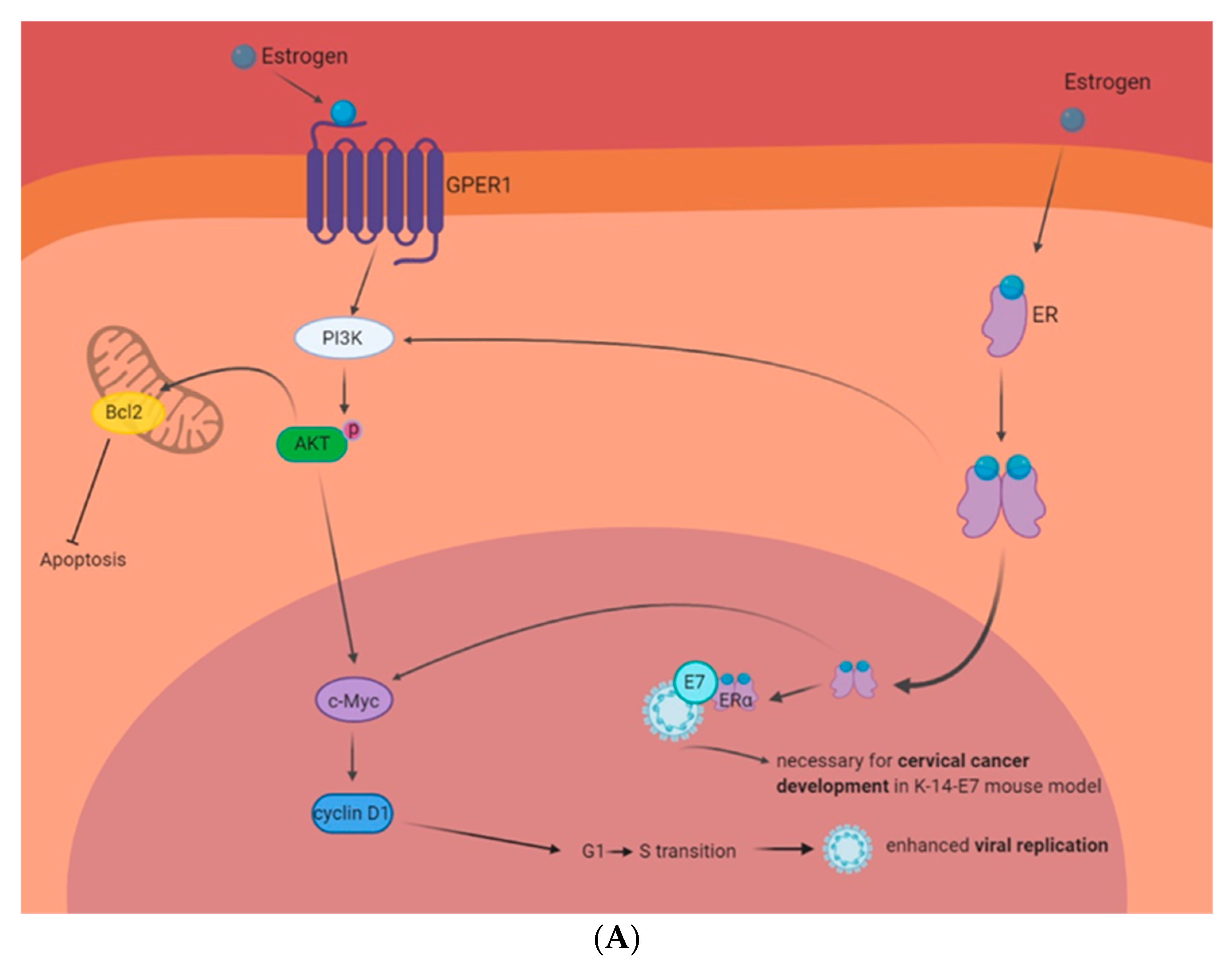
- Chung, S.-H.; Wiedmeyer, K.; Shai, A.; Korach, K.S.; Lambert, P.F. Requirement for Estrogen Receptor Alpha in a Mouse Model for Human Papillomavirus-Associated Cervical Cancer. Cancer Res. 2008, 68, 9928–9934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, J.; Park, J.W.; Lambert, P.F.; Chung, S.-H. Requirement of estrogen receptor alpha DNA-binding domain for HPV oncogene-induced cervical carcinogenesis in mice. Carcinogenesis 2014, 35, 489–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.-H.; Shin, M.K.; Korach, K.S.; Lambert, P.F. Requirement for stromal estrogen receptor alpha in cervical neoplasia. Horm. Cancer 2013, 4, 50–59. [Google Scholar] [CrossRef]
- Maufort, J.P.; Shai, A.; Pitot, H.; Lambert, P.F. A Role for HPV 16 E5 in Cervical Carcinogenesis. Cancer Res. 2010, 70, 2924–2931. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, M.E.; den Boon, J.A.; Horswill, M.; Barthakur, S.; Forouzan, O.; Rader, J.S.; Beebe, D.J.; Roopra, A.; Ahlquist, P.; Lambert, P.F. Human papillomavirus oncogenes reprogram the cervical cancer microenvironment independently of and synergistically with estrogen. Proc. Natl. Acad. Sci. USA 2017, 114, E9076–E9085. [Google Scholar] [CrossRef] [Green Version]
- Women Exposed to DES in the Womb Face Increased Cancer Risk. Available online: https://www.nih.gov/news-events/news-releases/women-exposed-des-womb-face-increased-cancer-risk (accessed on 10 April 2020).
- Conlon, J.L. Diethylstilbestrol: Potential health risks for women exposed in utero and their offspring. JAAPA 2017, 30, 49–52. [Google Scholar] [CrossRef]
- Samir, R.; Asplund, A.; Tot, T.; Pekar, G.; Hellberg, D. High-Risk HPV Infection and CIN Grade Correlates to the Expression of c-myc, CD4+, FHIT, E-cadherin, Ki-67, and p16INK4a. J. Low. Genit. Tract Dis. 2011, 15, 280–286. [Google Scholar] [CrossRef]
- Silins, I.; Kallings, I.; Dillner, J. Correlates of the Spread of Human Papillomavirus Infection. Cancer Epidemiol. Biomark. Prev. 2000, 9, 953–959. [Google Scholar]
- Monsonego, J.; Magdelenat, H.; Catalan, F.; Coscas, Y.; Zerat, L.; Sastre, X. Estrogen and progesterone receptors in cervical human papillomavirus related lesions. Int. J. Cancer 1991, 48, 533–539. [Google Scholar] [CrossRef]
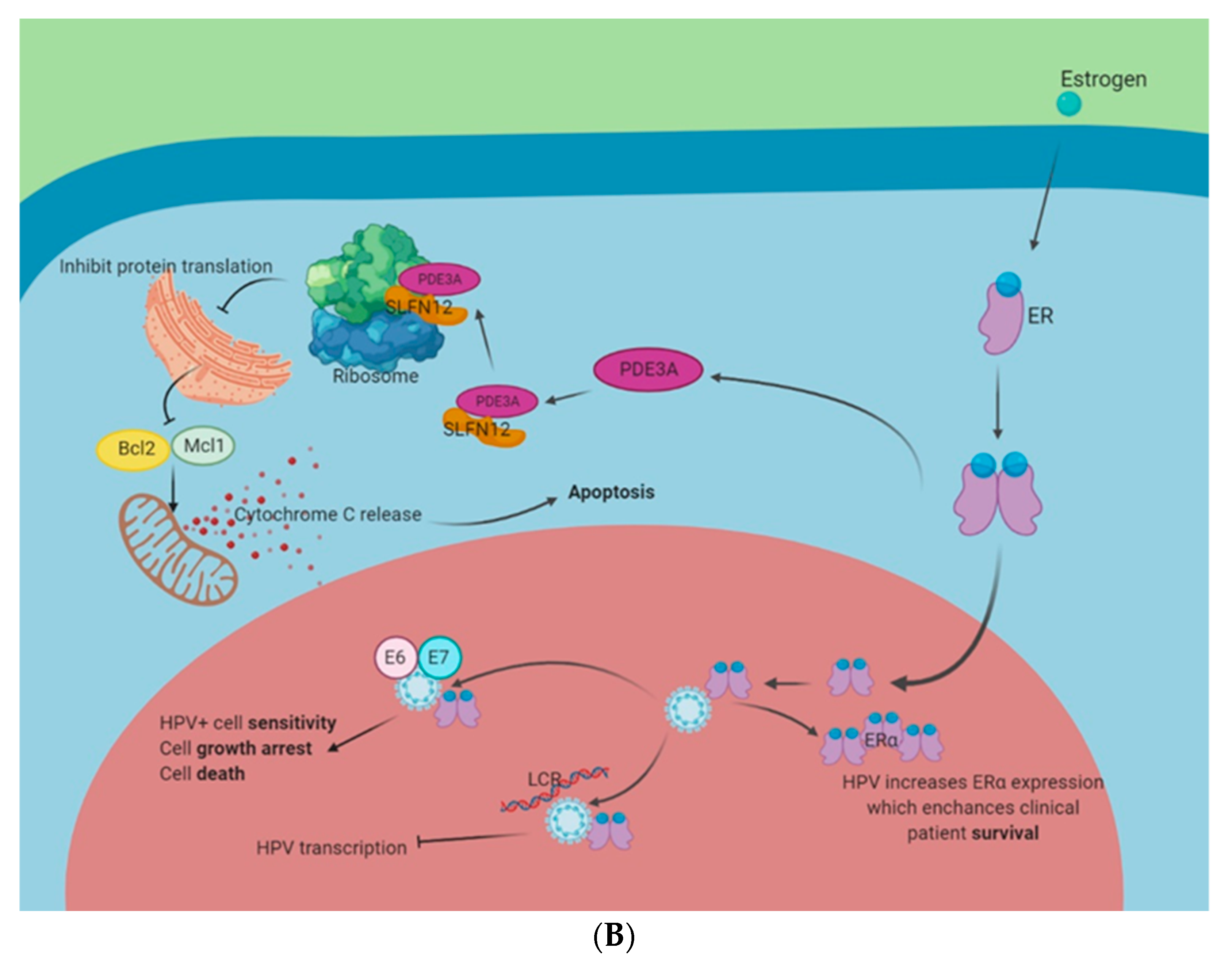
- Li, D.; Chen, J.; Ai, Y.; Gu, X.; Li, L.; Che, D.; Jiang, Z.; Li, L.; Chen, S.; Huang, H.; et al. Estrogen-Related Hormones Induce Apoptosis by Stabilizing Schlafen-12 Protein Turnover. Mol. Cell 2019, 75, 1103–1116.e9. [Google Scholar] [CrossRef] [PubMed]
- Bristol, M.L.; James, C.D.; Wang, X.; Fontan, C.T.; Morgan, I.M. Estrogen Attenuates the Growth of Human Papillomavirus-Positive Epithelial Cells. mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, H.M.; Hardwick, J.M. The Dark Side of Estrogen Stops Translation to Induce Apoptosis. Mol. Cell 2019, 75, 1087–1089. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Schwartz, N.; Cohen, D.J.; Patel, V.; Nageris, B.; Bachar, G.; Boyan, B.D.; Schwartz, Z. Loss of Estrogen Receptors is Associated with Increased Tumor Aggression in Laryngeal Squamous Cell Carcinoma. Sci. Rep. 2020, 10, 4227. [Google Scholar] [CrossRef] [PubMed]
- Arjomandnejad, M.; Muhammadnejad, A.; Haddadi, M.; Sherkat-Khameneh, N.; Rismanchi, S.; Amanpour, S.; Muhammadnejad, S. HeLa cell line xenograft tumor as a suitable cervical cancer model: Growth kinetic characterization and immunohistochemistry array. Arch. Iran. Med. 2014, 17, 273–277. [Google Scholar] [PubMed]
- Luedke, E.; Jaime-Ramirez, A.C.; Bhave, N.; Roda, J.; Choudhary, M.M.; Kumar, B.; Teknos, T.N.; Carson, W.E. Cetuximab therapy in head and neck cancer: Immune modulation with interleukin-12 and other natural killer cell-activating cytokines. Surgery 2012, 152, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.L.; Davis, S.J.; Owen, J.H.; Graham, M.P.; Czerwinski, M.J.; Park, J.J.; Walline, H.; Stoerker, J.; McHugh, J.B.; Chepeha, D.; et al. UM-SCC-104: A new human papillomavirus-16 containing head and neck squamous cell carcinoma cell line. Head Neck 2012, 34, 1480–1491. [Google Scholar] [CrossRef] [Green Version]
- Identifying Predictors of HPV-Related Head and Neck Squamous Cell Carcinoma Progression and Survival through Patient-Derived Models|bioRxiv. Available online: https://www.biorxiv.org/content/10.1101/652537v2.full (accessed on 15 April 2020).
- Kano, M.; Kondo, S.; Wakisaka, N.; Wakae, K.; Aga, M.; Moriyama-Kita, M.; Ishikawa, K.; Ueno, T.; Nakanishi, Y.; Hatano, M.; et al. Expression of estrogen receptor alpha is associated with pathogenesis and prognosis of human papillomavirus-positive oropharyngeal cancer. Int. J. Cancer 2019, 145, 1547–1557. [Google Scholar] [CrossRef]
- Koenigs, M.B.; Lefranc-Torres, A.; Bonilla-Velez, J.; Patel, K.B.; Hayes, D.N.; Glomski, K.; Busse, P.M.; Chan, A.W.; Clark, J.R.; Deschler, D.G.; et al. Association of Estrogen Receptor Alpha Expression With Survival in Oropharyngeal Cancer Following Chemoradiation Therapy. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef]
- Zhai, Y.; Bommer, G.T.; Feng, Y.; Wiese, A.B.; Fearon, E.R.; Cho, K.R. Loss of Estrogen Receptor 1 Enhances Cervical Cancer Invasion. Am. J. Pathol. 2010, 177, 884–895. [Google Scholar] [CrossRef]
- The Significance of Atypical Cervical Cytology in an Older Population. Available online: https://europepmc.org/article/med/2535763 (accessed on 15 April 2020).
- Piccoli, R.; Mandato, V.D.; Lavitola, G.; Acunzo, G.; Bifulco, G.; Tommaselli, G.A.; Attianese, W.; Nappi, C. Atypical squamous cells and low squamous intraepithelial lesions in postmenopausal women: Implications for management. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 140, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, H.E.; Chenevert, L.; Munsell, M. Vaginal intraepithelial neoplasia (VaIN 2/3): Comparing clinical outcomes of treatment with intravaginal estrogen. J. Low. Genit. Tract Dis. 2014, 18, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Guidozzi, F. Estrogen therapy in gynecological cancer survivors. Climacteric 2013, 16, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Schön, H.J.; Grgurin, M.; Szekeres, T.; Schurz, B. A new mode of treatment of human papilloma virus associated anogenital lesions using a nonsteroid estrogen analogue. Wien. Klin. Wochenschr. 1996, 108, 45–47. [Google Scholar] [PubMed]
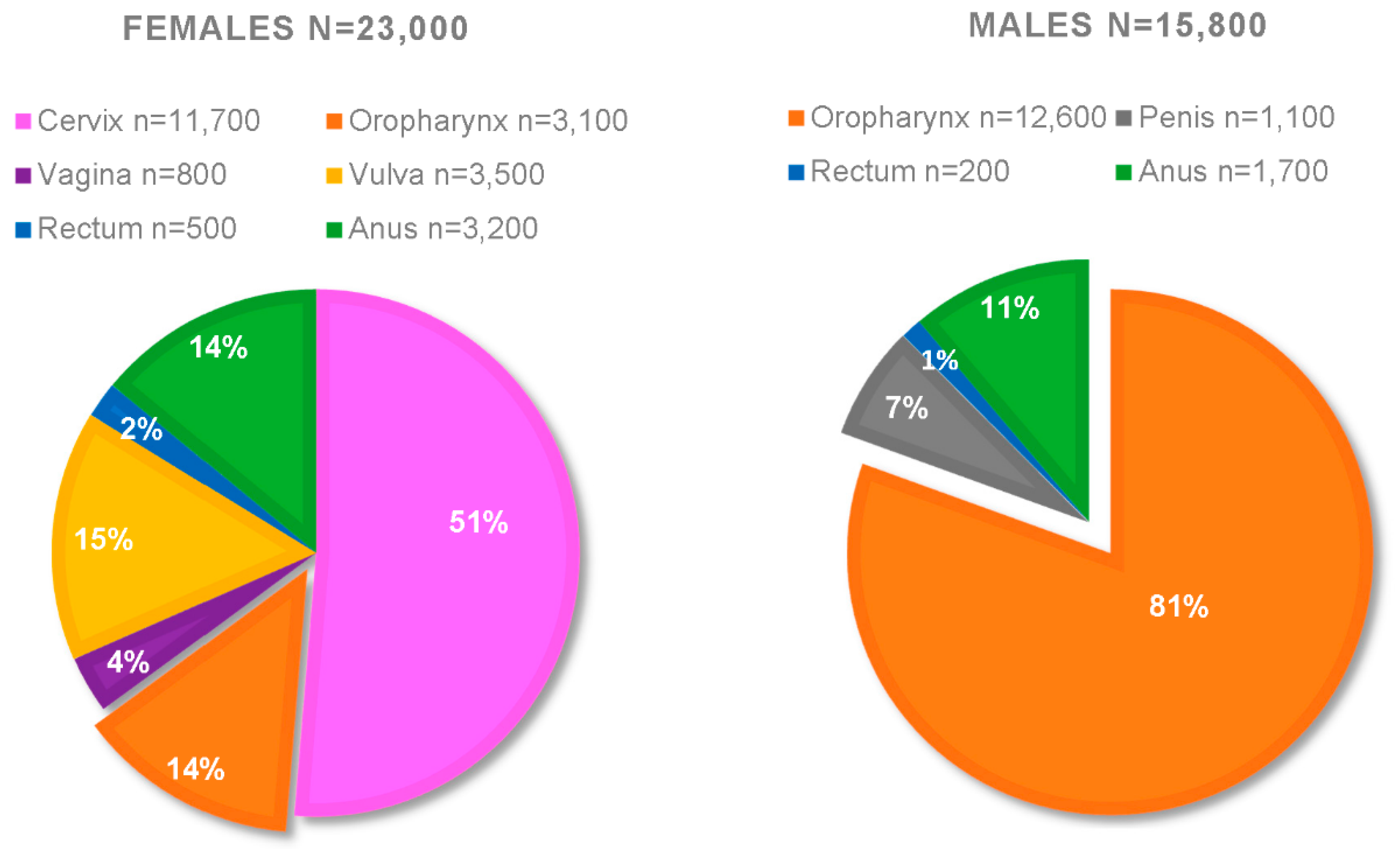
- Viens, L.J.; Henley, S.J.; Watson, M.; Markowitz, L.E.; Thomas, C.C.; Thompson, T.D.; Razzaghi, H.; Saraiya, M. Human Papillomavirus–Associated Cancers—United States, 2008–2012. Morb. Mortal. Wkly. Rep. 2016, 65, 24. [Google Scholar] [CrossRef] [PubMed]
- USCS Data Visualizations. Available online: https://gis.cdc.gov/grasp/USCS/DataViz.html (accessed on 17 April 2020).
- Fakhry, C.; Westra, W.H.; Wang, S.J.; van Zante, A.; Zhang, Y.; Rettig, E.; Yin, L.X.; Ryan, W.R.; Ha, P.K.; Wentz, A.; et al. The prognostic role of sex, race, and human papillomavirus in oropharyngeal and nonoropharyngeal head and neck squamous cell cancer. Cancer 2017, 123, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- HPV|Understanding HPV Coverage|CDC. Available online: https://www.cdc.gov/hpv/partners/outreach-hcp/hpv-coverage.html (accessed on 17 April 2020).
- HPV-Associated Cancer Statistics|CDC. Available online: https://www.cdc.gov/cancer/hpv/statistics/index.htm (accessed on 29 April 2020).
- Mathews, L.; Subramanya, V.; Zhao, D.; Ouyang, P.; Vaidya, D.; Guallar, E.; Yeboah, J.; Herrington, D.; Hays, A.G.; Budoff, M.J.; et al. Endogenous Sex Hormones and Endothelial Function in Postmenopausal Women and Men: The Multi-Ethnic Study of Atherosclerosis. J. Women’s Health 2019, 28, 900–909. [Google Scholar] [CrossRef]
- Ding, E.L.; Song, Y.; Malik, V.S.; Liu, S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: A systematic review and meta-analysis. JAMA 2006, 295, 1288–1299. [Google Scholar] [CrossRef]
- Stanhewicz, A.E.; Wenner, M.M.; Stachenfeld, N.S. Sex differences in endothelial function important to vascular health and overall cardiovascular disease risk across the lifespan. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1569–H1588. [Google Scholar] [CrossRef]
- den Boon, J.A.; Pyeon, D.; Wang, S.S.; Horswill, M.; Schiffman, M.; Sherman, M.; Zuna, R.E.; Wang, Z.; Hewitt, S.M.; Pearson, R.; et al. Molecular transitions from papillomavirus infection to cervical precancer and cancer: Role of stromal estrogen receptor signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E3255–E3264. [Google Scholar] [CrossRef] [Green Version]
- Goyal p16 Promoter Methylation, Expression, and Its Association with Estrogen Receptor, Progesterone Receptor, and Human Epidermal Growth Factor Receptor 2 Subtype of Breast Carcinoma. Available online: http://www.cancerjournal.net/article.asp?issn=0973-1482;year=2019;volume=15;issue=5;spage=1147;epage=1154;aulast=Goyal (accessed on 21 April 2020).
- Taneja, V. Sex Hormones Determine Immune Response. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Woodworth, C.D. HPV innate immunity. Front. Biosci. 2002, 7, d2058–d2071. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.R.; James, C.D.; Bristol, M.L.; Nulton, T.J.; Wang, X.; Kaur, N.; White, E.A.; Windle, B.; Morgan, I.M. Human Papillomavirus 16 E2 Regulates Keratinocyte Gene Expression Relevant to Cancer and the Viral Life Cycle. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amador-Molina, A.; Hernández-Valencia, J.F.; Lamoyi, E.; Contreras-Paredes, A.; Lizano, M. Role of Innate Immunity against Human Papillomavirus (HPV) Infections and Effect of Adjuvants in Promoting Specific Immune Response. Viruses 2013, 5, 2624–2642. [Google Scholar] [CrossRef] [Green Version]
- Khan, D.; Ansar Ahmed, S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2016, 6. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
James, C.D.; Morgan, I.M.; Bristol, M.L. The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens 2020, 9, 403. https://doi.org/10.3390/pathogens9050403
James CD, Morgan IM, Bristol ML. The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens. 2020; 9(5):403. https://doi.org/10.3390/pathogens9050403
Chicago/Turabian StyleJames, Claire D., Iain M. Morgan, and Molly L. Bristol. 2020. "The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers" Pathogens 9, no. 5: 403. https://doi.org/10.3390/pathogens9050403
APA StyleJames, C. D., Morgan, I. M., & Bristol, M. L. (2020). The Relationship between Estrogen-Related Signaling and Human Papillomavirus Positive Cancers. Pathogens, 9(5), 403. https://doi.org/10.3390/pathogens9050403