Host Species Determines the Composition of the Prokaryotic Microbiota in Phlebotomus Sandflies

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Morphological Identification of Phlebotomus Species
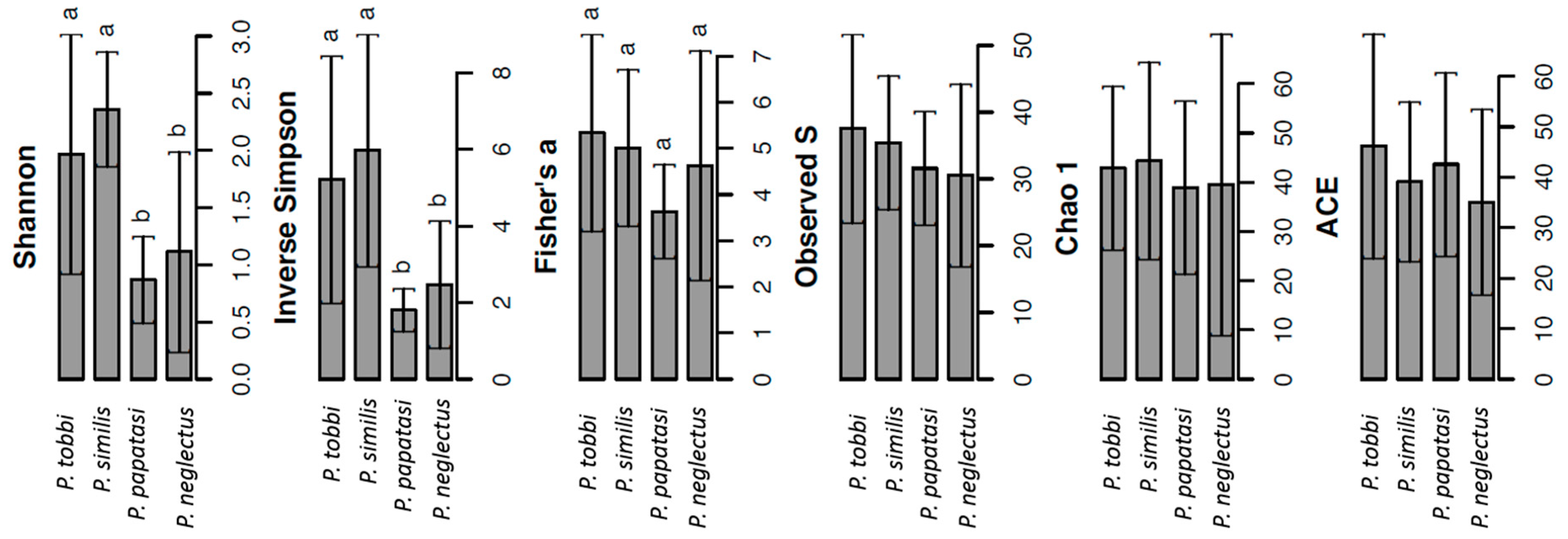
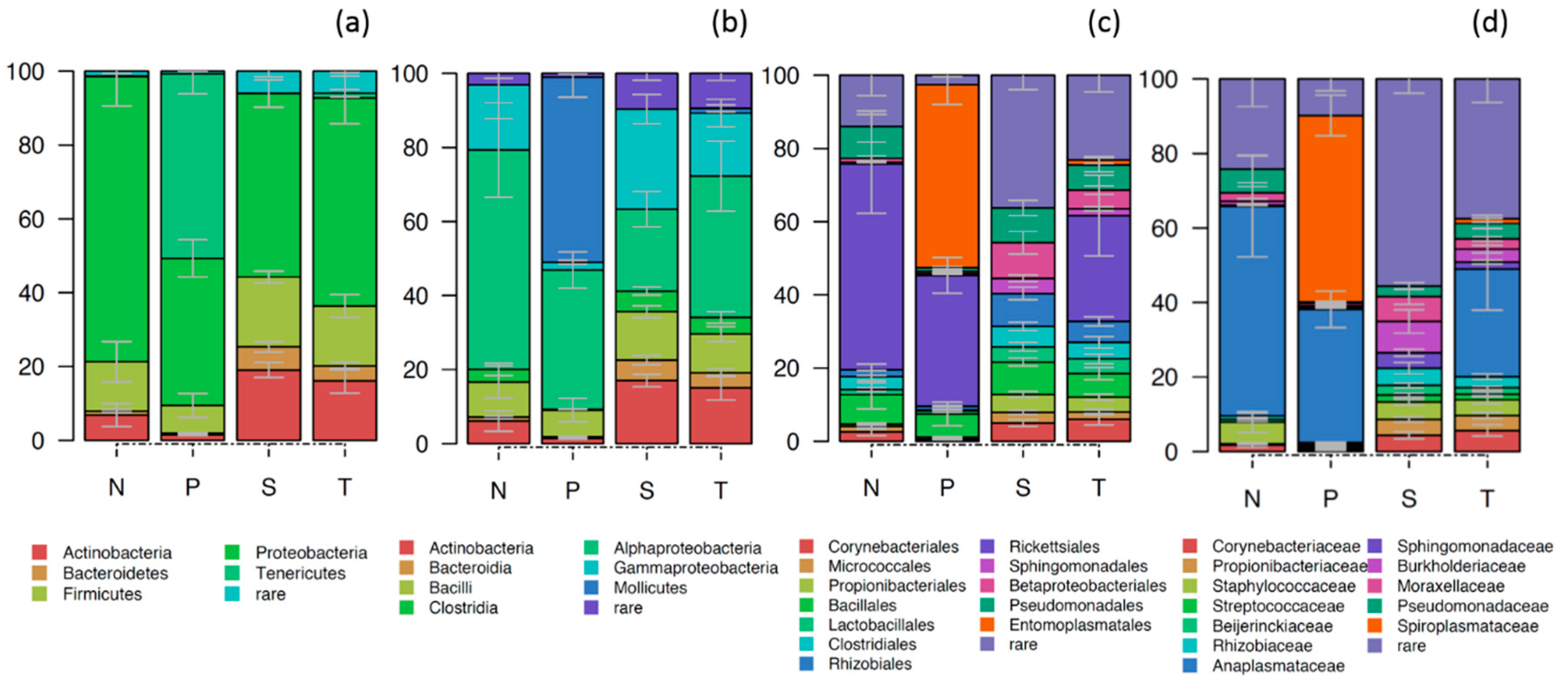
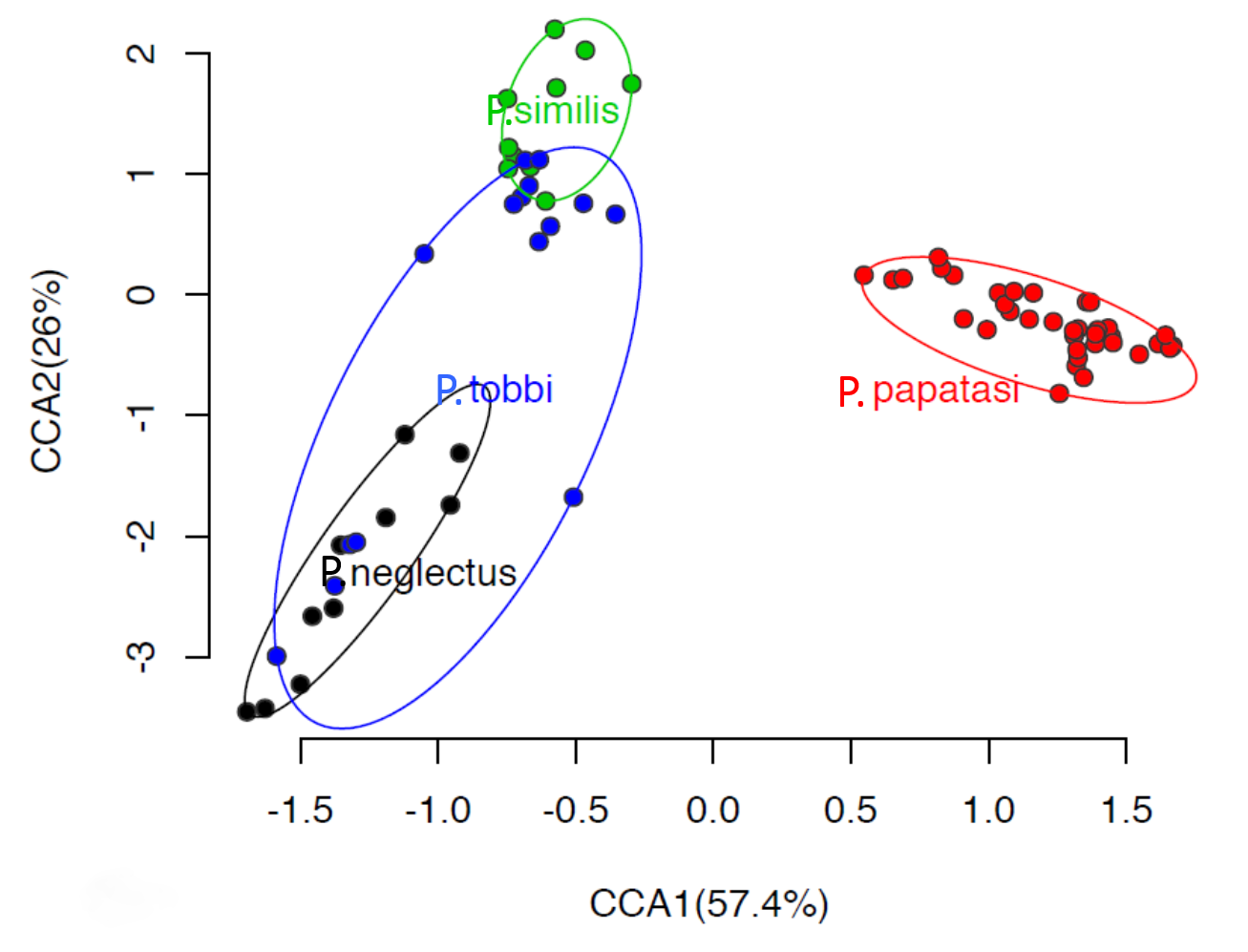
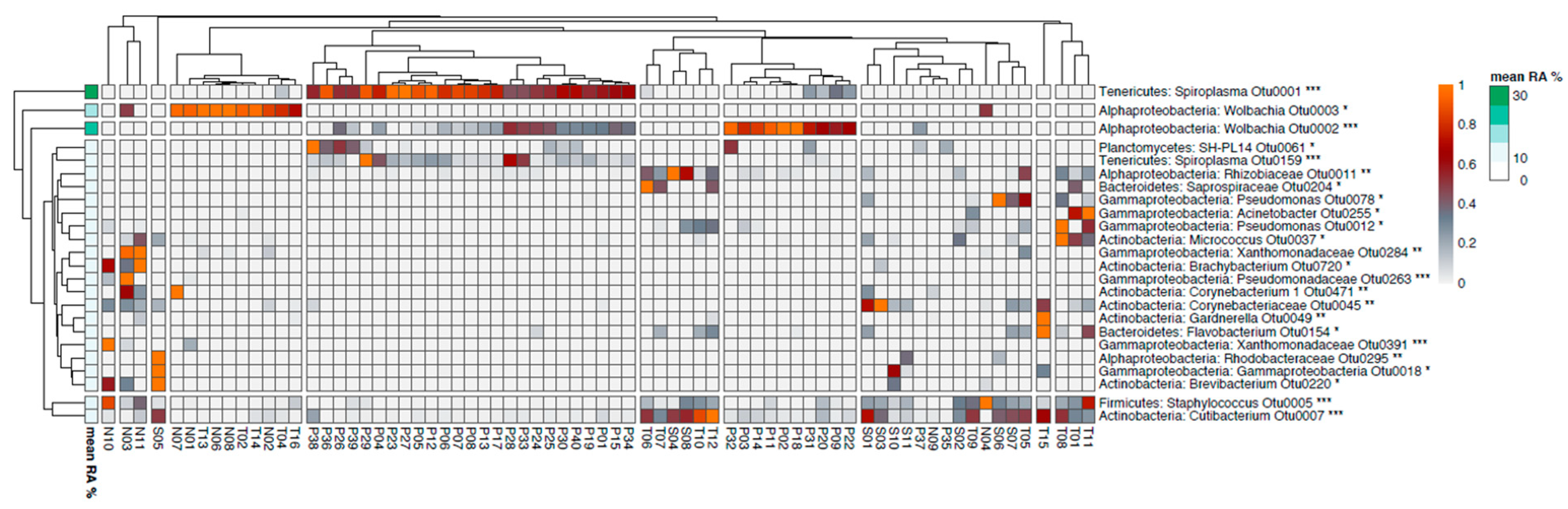
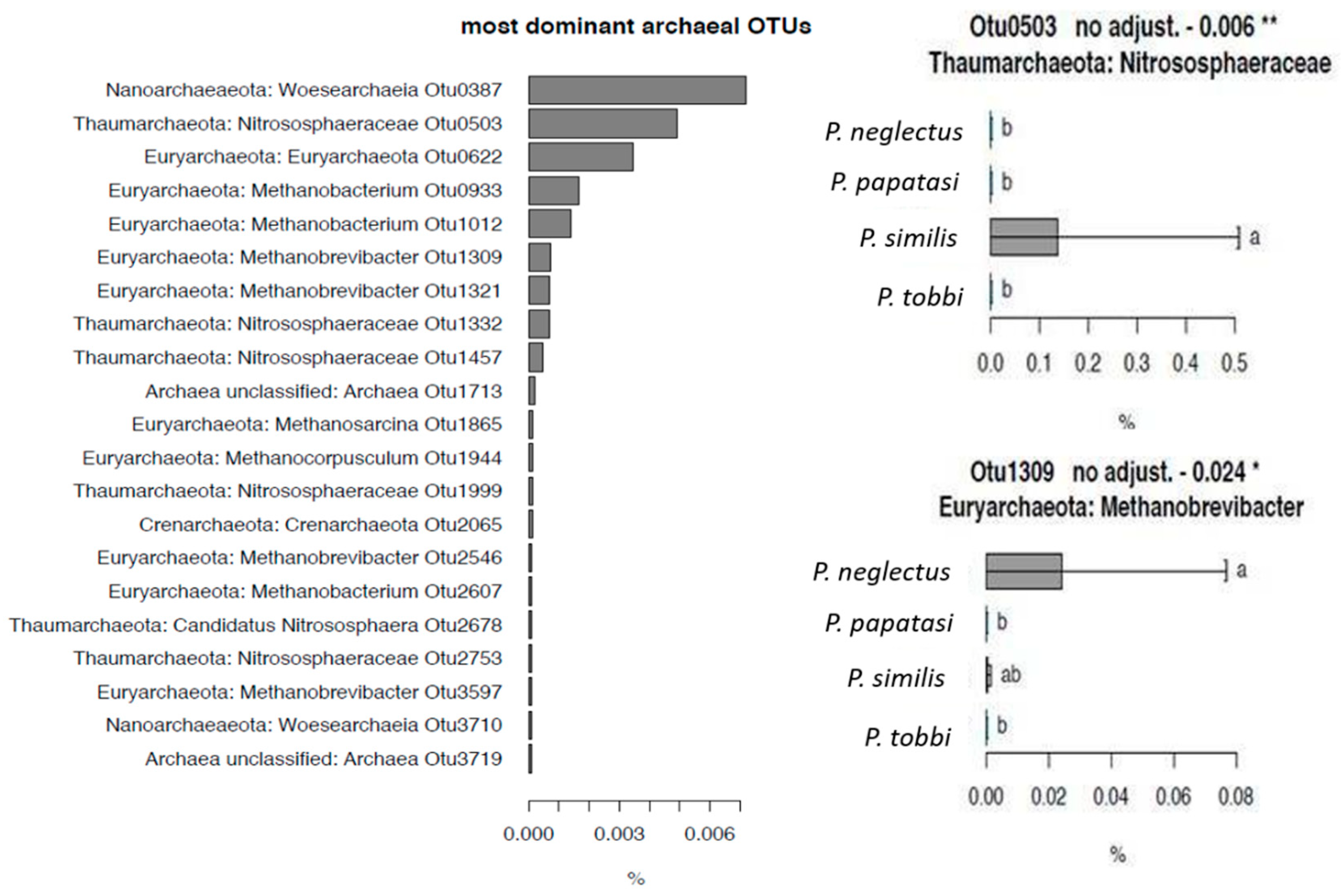
2.2. The Composition of the Bacterial and Archaeal Microbiome in Sand Flies
3. Discussion
4. Materials and Methods
4.1. Samples Collection
4.2. Species-Level Identification of Phlebotomus Sandflies
4.3. DNA Extraction
4.4. Quantitative Polymerase Chain Reaction (q-PCR) Detection of Leishmania spp. in Sandfly Samples
4.5. PCR Amplification of the 16S rRNA Gene and Amplicon Sequencing Analysis of the Microbiome
4.6. Bioinformatic Analysis of Amplicon Sequencing Data
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pappa, A.; Konstantinou, G.; Pavlidou, V.; Antoniadis, A. Sandfly fever virus outbreak in Cyprus. Clin. Microbiol. Infec. 2006, 12, 192–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alwassouf, S.; Christodoulou, V.; Bichaud, L.; Ntais, P.; Mazeris, A.; Antoniou, M.; Charel, R.N. Seroprevalence of sand fly-borne phleboviruses belonging to three serocomplexes (sand fly fever Naples, sand fly fever Sicilian and Salehabad) in dogs from Greece and Cyprus using neutralization test. PLoS Negl. Trop Dis. 2016, 10, e0005063. [Google Scholar] [CrossRef] [PubMed]
- Billeter, S.A.; Levy, M.G.; Chomel, B.B.; Breitschwerdt, E.B. Vector transmission of Bartonella species with emphasis on the potential for tick transmission. Med. Vet. Entomol. 2008, 22, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votypka, J.; Marty, P.; Delaunay, P.; Sereno, D. A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLOS Negl. Trop Dis 2016, 10, e0004770. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, M.; Gramiccia, M.; Molina, R.; Dvorak, V.; Volf, P. The role of indigenous phlebotomine sandflies and mammals in the spreading of leishmaniasis agents in the Mediterranean region. Euro Surveil. 2013, 18, e20540. [Google Scholar] [CrossRef] [Green Version]
- Bates, P.A.; Depquit, J.; Galati, E.A.B.; Kamhawi, S.; Maroli, M.; McDowell, M.A.; Picado, A.; Ready, P.D.; Salomon, O.D.; Shaw, J.J.; et al. Recent advances in phlebotomine sandfly research related to leishmaniasis control. Paras Vect 2015, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Alten, B.; Maia, C.; Afonso, M.O.; Campino, L.; Jiménez, M.; González, E.; Molina, R.; Banuls, A.-L.; Prudhomme, J.; Vergnes, B.; et al. Seasonal dynamics of Phlebotomine sand fly species proven vectors of Mediterranean Leishmaniasis caused by Leishmania infantum. PLoS Negl. Trop Dis. 2016, 10, e0004458. [Google Scholar] [CrossRef]
- Velo, E.; Bongiorno, G.; Kadriaj, P.; Myrseli, T.; Crilly, J.; Lika, A.; Mersini, K.; Di Muccio, T.; Bino, S.; Gramiccia, M.; et al. The current status of phlebotomine sand flies in Albania and incrimination of Phlebotomus neglectus (Diptera, Psychodidae) as the main vector of Leishmania infantum. PLoS ONE 2017, 12, e0179118. [Google Scholar] [CrossRef]
- Vaselek, S.; Ayhan, N.; Oguz, G.; Erisoz Kasap, O.; Savić, S.; Di Muccio, T.; Gradoni, L.; Ozbel, Y.; Alten, B.; Petric, D. Sand fly and Leishmania spp. survey in Vojvodina (Serbia): First detection of Leishmania infantum DNA in sand flies and the first record of Phlebotomus (Transphlebotomus) mascittii Grassi, 1908. Paras. Vect 2017, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Vaselek, S.; Dvorak, V.; Hlavackova, K.; Ayhan, N.; Halada, P.; Oguz, G.; Ivovic, V.; Ozbel, Y.; Charrel, R.N.; Alten, B.; et al. A survey of sand flies (Diptera, Phlebotominae) along recurrent transit routes in Serbia. Acta Tropica. 2019, 197, 105063. [Google Scholar] [CrossRef]
- Xanthopoulou, K.; Anagnostou, V.; Ivovic, V.; Djurkovic-Djiakovic, O.; Rogozi, E.; Sotiraki, S.; Papa, A. Distribution of sandflies (Diptera, Psychodidae) in two Ionian islands and northern Greece. Vector-Borne Zoon Dis. 2011, 11, 1591–1594. [Google Scholar] [CrossRef] [PubMed]
- Tsirigotakis, N.; Pavlou, C.; Christodoulou, V.; Dokianakis, E.; Kourouniotis, C.; Alten, B.; Antoniou, M. Phlebotomine sand flies (Diptera: Psychodidae) in the Greek Aegean Islands: Ecological approaches. Paras Vect. 2018, 11, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, A.E. Nutritional interactions in insect-microbial symbioses: Aphids and their symbiotic bacteria Buchnera. Ann. Rev. Entomol. 1998, 43, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Dedeine, F.; Vavre, F.; Fleury, F.; Loppin, B.; Hochberg, M.; Bouletreau, M. Removing symbiotic Wolbachia bacteria specifically inhibits oogenesis in a parasitic wasp. Proc. Natl. Acad. Sci. USA 2001, 98, 6247–6252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harumoto, T.; Lemaitre, B. Male-killing toxin in a Drosophila bacterial symbiont. Nature 2018, 557, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Paredes, J.C.; Herren, J.K.; Schüpfer, F.; Lemaitre, B. The role of lipid competition for endosymbiont-mediated protection against parasitoid wasps in Drosophila. mBio 2016, 7, e01006-16. [Google Scholar] [CrossRef] [Green Version]
- Moreira, L.A.; Iturbe-Ormaetxe, I.; Jeffery, J.A.; Lu, G.; Pyke, A.T.; Hedges, L.M.; Rocha, B.C.; Hall-Mendelin, S.; Day, A.; Riegler, M.; et al. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and Plasmodium. Cell 2009, 139, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, R.; Margullis, L.; Berlanga, M. Symbiogenesis: The holobiont as a unit of evolution. Int. Microbiol 2013, 16, 133–143. [Google Scholar]
- Cheng, D.; Guo, Z.; Riegler, M.; Xi, Z.; Liang, G.; Xu, Y. Gut symbiont enhances insecticide resistance in a significant pest, the oriental fruit fly Bactrocera dorsalis (Hendel). Microbiome 2017, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, R.S.; Teixeira, E.W.; Tauber, J.P.; Birke, J.M.; Martins, M.F.; Fonseca, I.; Evans, J.D. Honey bee colonies act as reservoirs for two Spiroplasma facultative symbionts and incur complex, multiyear infection dynamics. Microbiol. Open 2014, 3, 341–355. [Google Scholar] [CrossRef]
- Mason, C.J.; Ray, S.; Shikano, I.; Peiffer, M.; Jones, A.G.; Luthe, D.S.; Hoover, K.; Felton, G.W. Plant defenses interact with insect enteric bacteria by initiating a leaky gut syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 15991–15996. [Google Scholar] [CrossRef] [Green Version]
- Kolasa, M.; Scibior, R.; Mazur, M.A.; Kubisz, D.; Dudek, K.; Kajtoch, L. How hosts taxonomy, trophy, and endosymbionts shape microbiome diversity in beetles. Microb. Ecol. 2018, 78, 995–1013. [Google Scholar] [CrossRef] [Green Version]
- Muturi, E.J.; Lagos-Kutz, D.; Dunlap, C.; Ramirez, J.L.; Rooney, A.P. Mosquito microbiota cluster by host sampling location. Paras Vect 2018, 11, 468. [Google Scholar] [CrossRef] [Green Version]
- Sanders, J.G.; Powell, S.; Kronauer, D.J.; Vasconcelos, H.L.; Frederickson, M.E.; Pierce, N.E. Stability and phylogenetic correlation in gut microbiota: Lessons from ants and apes. Molec Ecol. 2014, 23, 1268–1283. [Google Scholar] [CrossRef]
- Yun, J.-H.; Roh, S.W.; Whon, T.W.; Jung, M.-J.; Kim, M.-S.; Park, D.-S.; Yoon, C.; Nam, Y.-D.; Kim, Y.-J.; Choi, J.-H.; et al. Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 2014, 80, 5254–5264. [Google Scholar] [CrossRef] [Green Version]
- Pires, A.C.A.M.; Villegas, L.E.M.; Campolina, D.B.; Orfano, A.S.; Pimenta, P.F.P.; Secundino, N.F.C. Bacterial diversity of wild-caught Lutzomyia longipalpis (a vector of zoonotic visceral leishmaniasis in Brazil) under distinct physiological conditions by metagenomics analysis. Paras Vect 2017, 10, 627. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.H.; Bahr, S.M.; Serafim, T.D.; Ajami, N.J.; Petrosino, J.F.; Meneses, C.; Kirby, J.R.; Valenzuela, J.G.; Kamhawi, S.; Wilson, M.E. The gut microbiome of the vector Lutzomyia longipalpis is essential for survival of Leishmania infantum. mBio 2017, 8, e01121-16. [Google Scholar] [CrossRef] [Green Version]
- Fraihi, W.; Fares, W.; Perrin, P.; Dorkeld, F.; Sereno, D.; Barhoumi, V.; Sbissi, I.; Cherni, S.; Chelbi, I.; Durvasula, R.; et al. An integrated overview of the midgut bacterial flora composition of Phlebotomus perniciosus, a vector of zoonotic visceral leishmaniasis in the Western Mediterranean Basin. PLoS Negl. Trop Dis. 2017, 11, e0005484. [Google Scholar] [CrossRef]
- Karimian, F.; Vatandoost, H.; Rassi, Y.; Maleki-Ravasan, N.; Mohebali, M.; Shirazi, M.H.; Koosha, M.; Choubdar, N.; Oshagi, M.A. Aerobic midgut microbiota of sand fly vectors of zoonotic visceral leishmaniasis from northern Iran, a step toward finding potential paratransgenic candidates. Paras Vect. 2019, 12, 10. [Google Scholar] [CrossRef]
- Li, K.; Chen, H.; Jiang, J.; Li, X.; Xu, J.; Ma, Y. Diversity of bacteriome associated with Phlebotomus chinensis (Diptera: Psychodidae) sand flies in two wild populations from China. Sci. Rep. 2016, 6, 36406. [Google Scholar] [CrossRef] [Green Version]
- Vivero, R.J.; Villegas-Plazas, M.; Cadavid-Restrepo, G.E.; Herrera, C.X.M.; Uribe, S.I.; Junca, H. Wild specimens of sand fly phlebotomine Lutzomyia evansi, vector of leishmaniasis, show high abundance of Methylobacterium and natural carriage of Wolbachia and Cardinium types in the midgut microbiome. Sci. Rep. 2019, 9, 17746. [Google Scholar] [CrossRef] [Green Version]
- Santana, R.H.; Pires Catao, E.C.; Cardoso Lopes, F.A.; Constantino, R.; Chaves Barreto, C.; Kruger, R.H. The gut microbiota of workers of the litter-feeding termite Syntermes wheeleri (Termitidae: Syntermitinae): Archaeal, bacterial, and fungal communities. Microb. Ecol. 2015, 70, 545–556. [Google Scholar] [CrossRef]
- Schlein, Y.; Polacheck, I.; Yuval, B. Mycoses, bacterial infections and antibacterial activity in sandflies (Psychodidae) and their possible role in the transmission of leishmaniasis. Parasitology 1985, 90, 57–66. [Google Scholar] [CrossRef]
- Louradour, I.; Monteiro, C.C.; Inbar, E.; Ghosh, K.; Merkhofer, R.; Lawyer, P.; Paun, A.; Smelkinson, M.; Secundino, N.; Lewis, M.; et al. The midgut microbiota plays an essential role in sand fly vector competence for Leishmania major. Cell Microbiol. 2017, 19, e12755. [Google Scholar] [CrossRef] [Green Version]
- Dey, R.; Joshi, A.B.; Oliveira, F.; Pereira, L.; Guimaraes-Costa, A.B.; Serafim, T.D.; de Castro, W.; Coutinho-Abreu, I.V.; Bhattacharya, P.; Townsend, S.; et al. Microbes egested during bites of infected sand flies augment severity of Leishmaniasis via inflammasome-derived IL-1b. Cell Host Microbe 2018, 23, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Gurung, K.; Wertheim, B.; Salles, J.F. The microbiome of pest insects: It is not just bacteria. Entomol Experim. Appl. 2019, 167, 156–170. [Google Scholar] [CrossRef] [Green Version]
- Vivero, J.; Jaramillo, G.; Cadavid-Restrepo, G.; Soto, S.; Herrera, C. Structural differences in gut bacteria communities in developmental stages of natural populations of Lutzomyia evansi from Colombia’s Caribbean coast. Paras Vect. 2016, 13, 496. [Google Scholar] [CrossRef] [Green Version]
- Paniagua Voirol, L.R.; Frago, E.; Kaltenpoth, M.; Hilker, M.; Fatouros, N.E. Bacterial symbionts in Lepidoptera: Their diversity, transmission, and impact on the host. Front. Microbiol. 2018, 9, 556. [Google Scholar] [CrossRef]
- Maleki-Ravasan, N.; Oshaghi, M.A.; Afshar, D.; Arandian, M.H.; Hajikhani, S.; Akhavan, A.A.; Yakhchali, B.; Shirazi, M.H.; Rassi, Y.; Jafari, R.; et al. Aerobic bacterial flora of biotic and abiotic compartments of a hyperendemic Zoonotic Cutaneous Leishmaniasis (ZCL) focus. Paras Vect. 2015, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, J.; Braig, H.R.; Rowton, E.D.; Ghosh, K. Naturally occurring culturable aerobic gut flora of adult Phlebotomus papatasi, vector of Leishmania major in the Old World. PLoS ONE 2012, 7, e35748. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.G.; Steen, A.D.; Ladau, J.; Yin, J.; Crosby, L. Phylogenetically novel uncultured microbial cells dominate Earth microbiomes. mSystems 2018, 3, e00055-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colman, D.R.; Toolson, E.C.; Takacs-Vesbach, C.D. Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 2012, 21, 5124–5137. [Google Scholar] [CrossRef] [PubMed]
- McLean, A.H.C.; Godfrey, H.C.J.; Ellers, J.; Henry, L.M. Host relatedness influences the composition of aphid Microbiomes. Environ. Microbiol. Rep. 2019, 11, 808–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karatepe, B.; Aksoy, S.; Karatepe, M. Investigation of Wolbachia spp. and Spiroplasma spp. in Phlebotomus species by molecular methods. Sci. Rep. 2018, 8, 10616. [Google Scholar]
- Duron, O.; Bouchon, D.; Boutin, S.; Bellamy, L.; Zhou, L.; Engelstadter, J.; Hurst, G.D. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 2008, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Hedges, L.M.; Brownlie, J.C.; O’Neill, S.L.; Johnson, K.N. Wolbachia and virus protection in insects. Science 2008, 322, 702. [Google Scholar] [CrossRef]
- Xie, J.; Vilchez, I.; Mateos, M. Spiroplasma bacteria enhance survival of Drosophila hydei attacked by the parasitic wasp Leptopilina heterotoma. PLoS ONE 2010, 5, e12149. [Google Scholar] [CrossRef]
- Ballinger, M.J.; Moore, L.D.; Perlman, S.J. Evolution and diversity of inherited Spiroplasma symbionts in Myrmica ants. Appl. Environ. Microbiol. 2018, 84, e02299-17. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Rastogi, G.; Nayduch, D.; Sawant, S.S.; Bhonde, R.R.; Shouche, Y.S. Molecular phylogenetic profiling of gut-associated bacteria in larvae and adults of flesh flies. Med. Vet. Entomol. 2014, 28, 345–354. [Google Scholar] [CrossRef]
- Ngo, C.T.; Romano-Bertrand, S.; Manguin, S.; Jumas-Bilak, E. Diversity of the bacterial microbiota of anopheles mosquitoes from Binh Phuoc Province, Vietnam. Front. Microbiol. 2016, 7, 2095. [Google Scholar] [CrossRef] [Green Version]
- Haloi, K.; Kankana, M.; Nath, R.; Devi, D. Characterization and pathogenicity assessment of gut-associated microbes of muga silkworm Antheraea assamensis Helfer (Lepidoptera: Saturniidae). J. Invertebr. Pathol. 2016, 138, 73–85. [Google Scholar] [CrossRef]
- Sant’Anna, M.R.; Diaz-Albiter, H.; Aguiar-Martins, K.; Al Salem, W.S.; Cavalcante, R.R.; Dilon, V.M.; Bates, P.A.; Genta, F.A.; Dilon, R.J. Colonisation resistance in the sand fly gut: Leishmania protects Lutzomyia longipalpis from bacterial infection. Paras Vect. 2014, 7, 329. [Google Scholar] [CrossRef] [Green Version]
- Koskinioti, P.; Ras, E.; Augustinos, A.; Tsiamis, G.; Beukeboom, L.W.; Caceres, C.; Bourtzis, K. The effects of geographic origin and antibiotic treatment on the gut symbiotic communities of Bactrocera oleae populations. Entomol. Experim. Appl. 2019, 167, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Martin, V.N.R.; Schwab, C.; Krych, L.; Voney, E.; Geirnaert, A.; Braegger, C.; Lacroix, C. Colonization of Cutibacterium avidum during infant gut microbiota establishment. FEMS Microbiol. Ecol. 2019, 95, fiy215. [Google Scholar]
- Jones, A.G.; Mason, C.J.; Felton, G.W.; Hoover, K. Host plant and population source drive diversity of microbial gut communities in two polyphagous insects. Sci. Rep. 2019, 9, 2792. [Google Scholar] [CrossRef] [Green Version]
- Muratore, M.; Prather, C.; Sun, Y. The gut bacterial communities across six grasshopper species from a coastal tallgrass prairie. PLoS ONE 2020, 15, e0228406. [Google Scholar] [CrossRef] [Green Version]
- Telleria, E.L.; Martins-da-Silva, A.; Tempone, A.J.; Traub-Csekö, Y.M. Leishmania, microbiota and sand fly immunity. Parasitology 2018, 145, 1336–1353. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.A.; Parks, D.H.; Willner, D.L.; Engelbrektson, A.L.; Goffredi, S.K.; Warnecke, F.; Scheffrahn, R.H.; Hugenholtz, P. A molecular survey of Australian and North American termite genera indicates that vertical inheritance is the primary force shaping termite gut microbiomes. Microbiome 2015, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Koskinen, K.; Pausan, M.-R.; Perras, A.K.; Beck, M.; Bang, C.; Mora, M.; Schilhabel, A.; Schmitz, R.; Moissl-Eichinger, C. First insights into the diverse human archaeome: Specific detection of Archaea in the gastrointestinal tract, lung, and nose and on skin. mBio 2017, 8, e00824-17. [Google Scholar] [CrossRef] [Green Version]
- Pausan, M.R.; Csorba, C.; Singer, G.; Till, H.; Schöpf, V.; Santigli, E.; Klug, B.; Högenauer, C.; Blohs, M.; Moissl-Eichinger, C. Exploring the archaeome: Detection of archaeal signatures in the human body. Front. Microbiol. 2019, 10, 2796. [Google Scholar] [CrossRef] [Green Version]
- Ziganshina, E.; Mohammed, W.; Shagimardanova, E.I.; Vankov, P.Y.; Gogoleva, N.E.; Ziganshin, A.M. Fungal, bacterial, and archaeal diversity in the digestive tract of several beetle larvae (Coleoptera). Biomed. Res. Intern. 2018. [Google Scholar] [CrossRef]
- Tourna, M.; Stieglmeier, M.; Spang, A.; Koenneke, M.; Schintlmeister, A.; Urich, T.; Engel, M.; Schloter, M.; Wagner, M.; Richter, A.; et al. Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc. Natl. Acad. Sci. USA 2011, 108, 8420–8425. [Google Scholar] [CrossRef] [Green Version]
- Huber, H.; Hohn, M.J.; Rachel, R.; Fuchs, T.; Wimmer, V.C.; Stetter, K.O. A new phylum of Archaea represented by a nanosized hyperthermophilic symbiont. Nature 2002, 417, 63–67. [Google Scholar] [CrossRef]
- Jarett, J.K.; Nayfach, S.; Podar, M.L.; Inskeep, W.P.; Ivanova, N.N.; Munson-McGee, J.; Schulz, F.; Young, M.; Jay, Z.J.; Beam, J.P.; et al. Single-cell genomics of co-sorted Nanoarchaeota suggests novel putative host associations and diversification of proteins involved in symbiosis. Microbiome 2018, 6, 161. [Google Scholar] [CrossRef] [Green Version]
- Waters, E.; Hohn, M.J.; Ahel, I.; Graham, D.E.; Adams, M.D.; Barnstead, M.; Beeson, K.Y.; Bibbs, L.; Bolanos, R.; Keller, M.; et al. The genome of Nanoarchaeum equitans: Insights into early archaeal evolution and derived parasitism. Proc. Natl. Acad. Sci. USA 2003, 100, 12984–12988. [Google Scholar] [CrossRef] [Green Version]
- McCliment, E.A.; Voglesonger, K.M.; O’Day, P.A.; Dunn, E.E.; Holloway, J.R.; Cary, C. Colonization of nascent, deep-sea hydrothermal vents by a novel Archaeal and Nanoarchaeal assemblage. Environ. Microbiol. 2006, 8, 114–125. [Google Scholar] [CrossRef]
- Casanueva, A.; Galada, N.; Baker, G.C.; Grant, W.D.; Heaphy, S.; Jones, B.; Yanhe, M.; Ventosa, A.; Blamey, J.; Cowan, D.A. Nanoarchaeal 16S rRNA gene sequences are widely dispersed in hyperthermophilic and mesophilic halophilic environments. Extremophiles 2008, 12, 651–656. [Google Scholar] [CrossRef]
- Clingenpeel, S.; Kan, J.; Macur, R.E.; Woyke, T.; Lovalvo, D.; Varley, J.; Inskeep, W.P.; Nealson, K.; McDermott, T.R. Yellowstone lake nanoarchaeota. Front. Microbiol. 2013, 4, 274. [Google Scholar] [CrossRef] [Green Version]
- Wurch, L.; Giannone, R.J.; Belisle, B.S.; Swift, C.; Utturkar, S.; Hettich, R.L.; Reysenbach, A.L.; Podar, M. Genomics informed isolation and characterization of a symbiotic Nanoarchaeota system from a terrestrial geothermal environment. Nat. Commun. 2016, 7, 12115. [Google Scholar] [CrossRef] [Green Version]
- Giannone, R.J.; Wurch, L.L.; Heimerl, T.; Martin, S.; Yang, Z.; Huber, H.; Rachel, R.; Hettich, R.L.; Podar, M. Life on the edge: Functional genomic response of Ignicoccus hospitalis to the presence of Nanoarchaeum equitans. ISME J. 2015, 9, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Feliciangeli, M.D. Natural breeding places of phlebotomine sandflies. Med. Vet. Entomol. 2004, 18, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodor, O. Psychodidae-Phlebotominae; Lindner, E., Ed.; Schweizerbart’sche Verlagsbuchhandlung: Stuttgart, Germany, 1958. [Google Scholar]
- Killick-Kendrick, R.; Tang, Y.; Killick-kendrick, M.; Sang, D.K.; Sirdar, M.K.; Ke, L.; Ashford, R.W.; Schorscher, J.; Johnson, R.W. The identification of female sandflies, of the subgenus Larroussius by the morphology of the spermathecal ducts. Parasitologia 1991, 33, 335–347. [Google Scholar]
- Francino, O.; Altet, L.; Sánchez-Robert, E.; Rodriguez, A.; Solano-Gallego, L.; Alberola, J.; Ferrer, L.; Sanchez, A.; Roura, X. Advantages of real-time PCR assay for diagnosis and monitoring of canine leishmaniosis. Vet. Parasitol. 2006, 137, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 2015, 1, e00009-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Ackermann, G.; Apprill, A.; Bauer, M.; Berg-Lyons, D.; Betley, J.; Fierer, N.; Fraser, L.; Gilbert, J.A.; Gormley, M.; et al. Earth Microbiome Project: EMP 16S Illumina Amplicon Protocol. 2018. Available online: https://www.protocols.io/view/emp-16s-illumina-amplicon-protocol-nuudeww (accessed on 29 May 2020).
- Katsoula, A.; Vasileiadis, S.; Sapountzi, M.; Karpouzas, D.G. The response of soil and phyllosphere microbial communities to repeated application of the fungicide iprodione: Accelerated biodegradation or toxicity? FEMS Microbiol. Ecol. 2020, fiaa056. [Google Scholar] [CrossRef]
- Dodt, M.; Roehr, J.T.; Ahmed, R.; Dieterich, C. FLEXBAR—Flexible barcode and adapter processing for next-generation sequencing platforms. Biology 2012, 1, 895–905. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Genome analysis Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 647, 1–5. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauswedell, H.; Singer, J.; Reinert, K. Lambda: The local aligner for massive biological data. Bioinformatics 2014, 30, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, G.; Ludwig, W.; Glockner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Jost, L. Entropy and diversity. Opinion 2006, 113, 363–375. [Google Scholar] [CrossRef]
- Pielou, E.C. Ecological Diversity, 8th ed.; John Wiley & Sons: Hoboken, NJ, USA, 1975. [Google Scholar]
- Martinez Arbizu, P. PairwiseAdonis: Pairwise Multilevel Comparison Using Adonis. R Package Version 0.3. 2019. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 29 May 2020).
- R Core Team. R: A Language and Environment for Statistical Computing, Reference Index Version 3.3.3. 2017. Available online: http://www.r-project.org/ (accessed on 29 May 2020).






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulos, C.; Karas, P.A.; Vasileiadis, S.; Ligda, P.; Saratsis, A.; Sotiraki, S.; Karpouzas, D.G. Host Species Determines the Composition of the Prokaryotic Microbiota in Phlebotomus Sandflies. Pathogens 2020, 9, 428. https://doi.org/10.3390/pathogens9060428
Papadopoulos C, Karas PA, Vasileiadis S, Ligda P, Saratsis A, Sotiraki S, Karpouzas DG. Host Species Determines the Composition of the Prokaryotic Microbiota in Phlebotomus Sandflies. Pathogens. 2020; 9(6):428. https://doi.org/10.3390/pathogens9060428
Chicago/Turabian StylePapadopoulos, Christos, Panagiotis A. Karas, Sotirios Vasileiadis, Panagiota Ligda, Anastasios Saratsis, Smaragda Sotiraki, and Dimitrios G. Karpouzas. 2020. "Host Species Determines the Composition of the Prokaryotic Microbiota in Phlebotomus Sandflies" Pathogens 9, no. 6: 428. https://doi.org/10.3390/pathogens9060428
APA StylePapadopoulos, C., Karas, P. A., Vasileiadis, S., Ligda, P., Saratsis, A., Sotiraki, S., & Karpouzas, D. G. (2020). Host Species Determines the Composition of the Prokaryotic Microbiota in Phlebotomus Sandflies. Pathogens, 9(6), 428. https://doi.org/10.3390/pathogens9060428